
The Post-Translational Regulation of Epithelial–Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis


Abstract
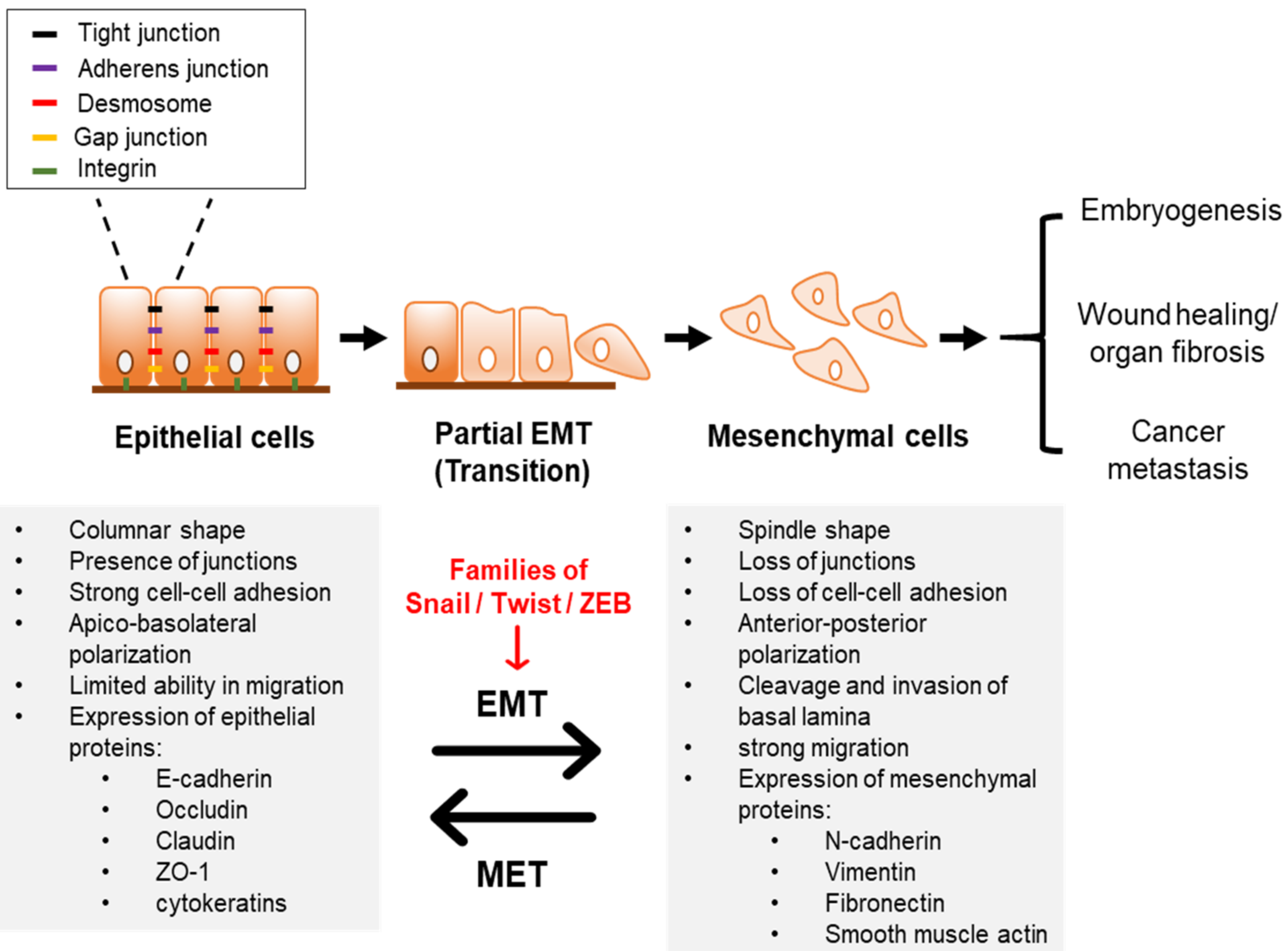
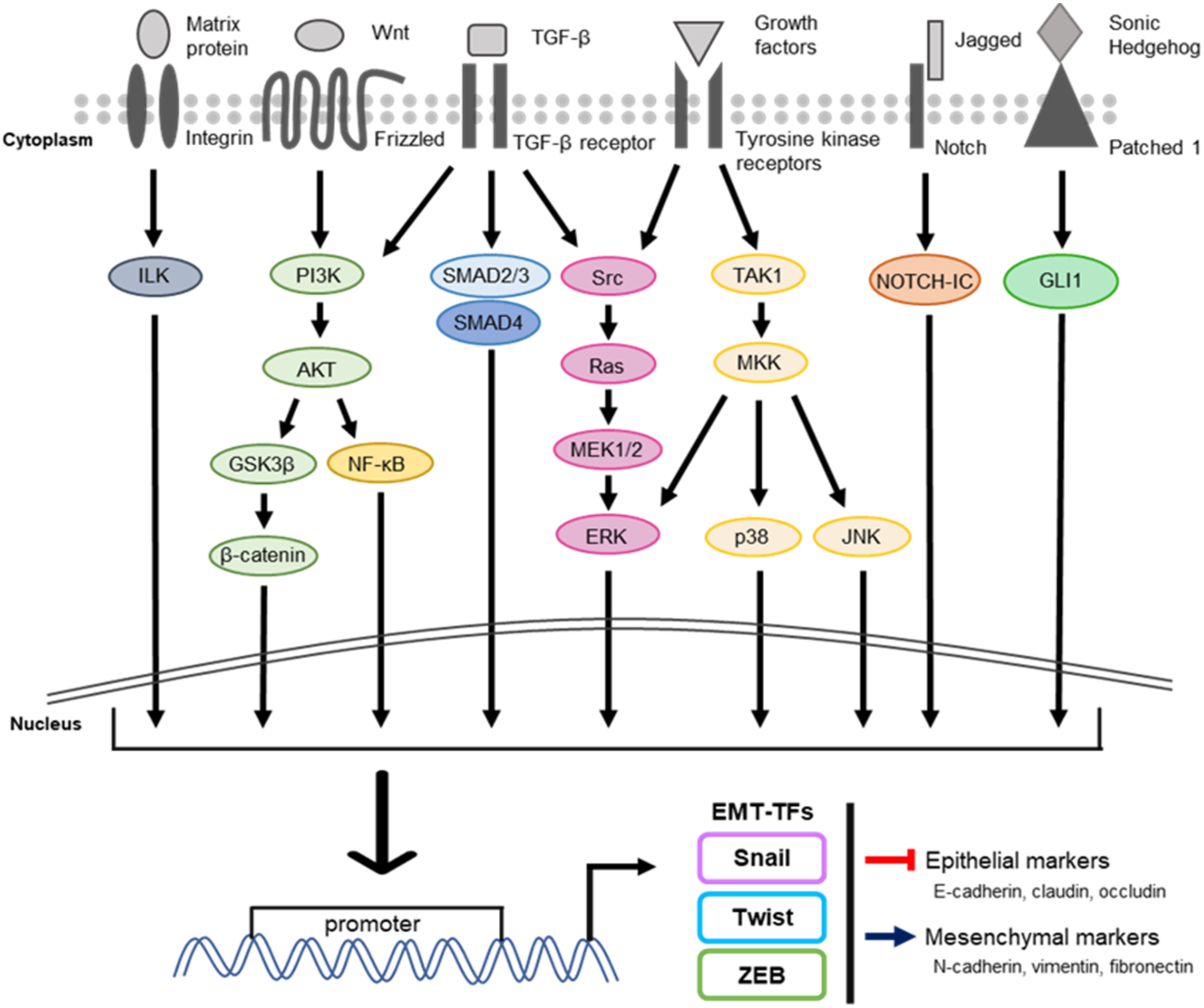
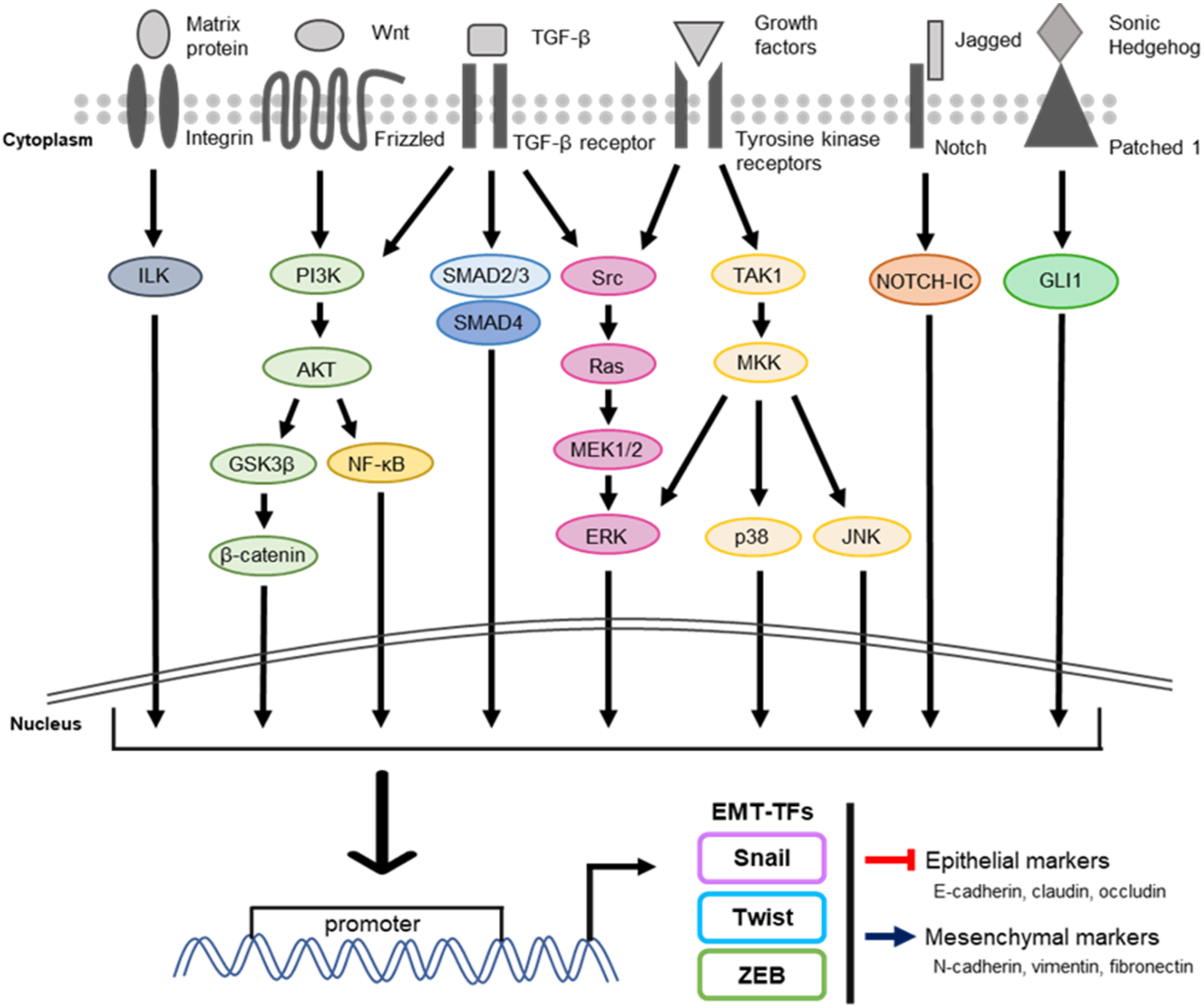
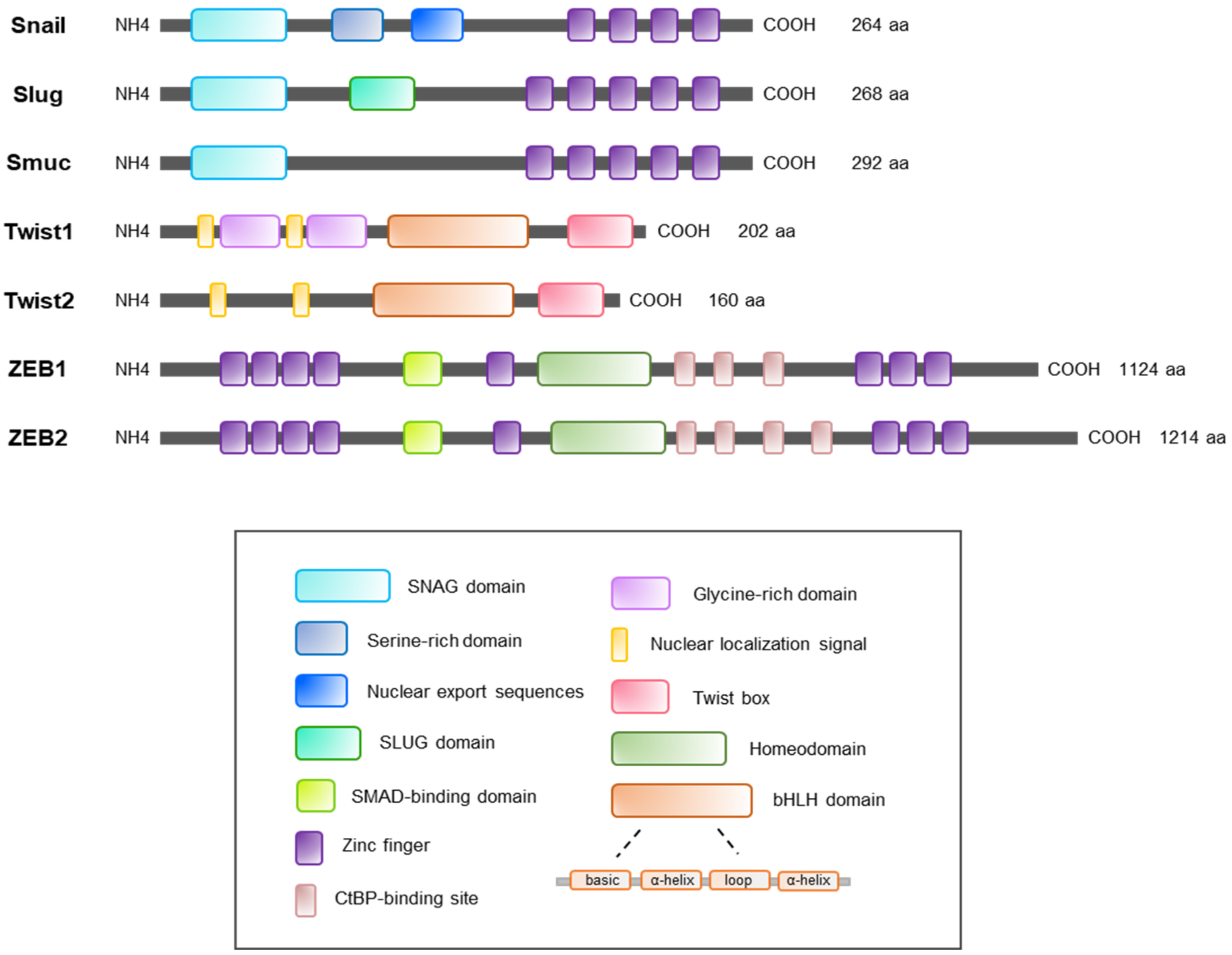
:1. Introduction
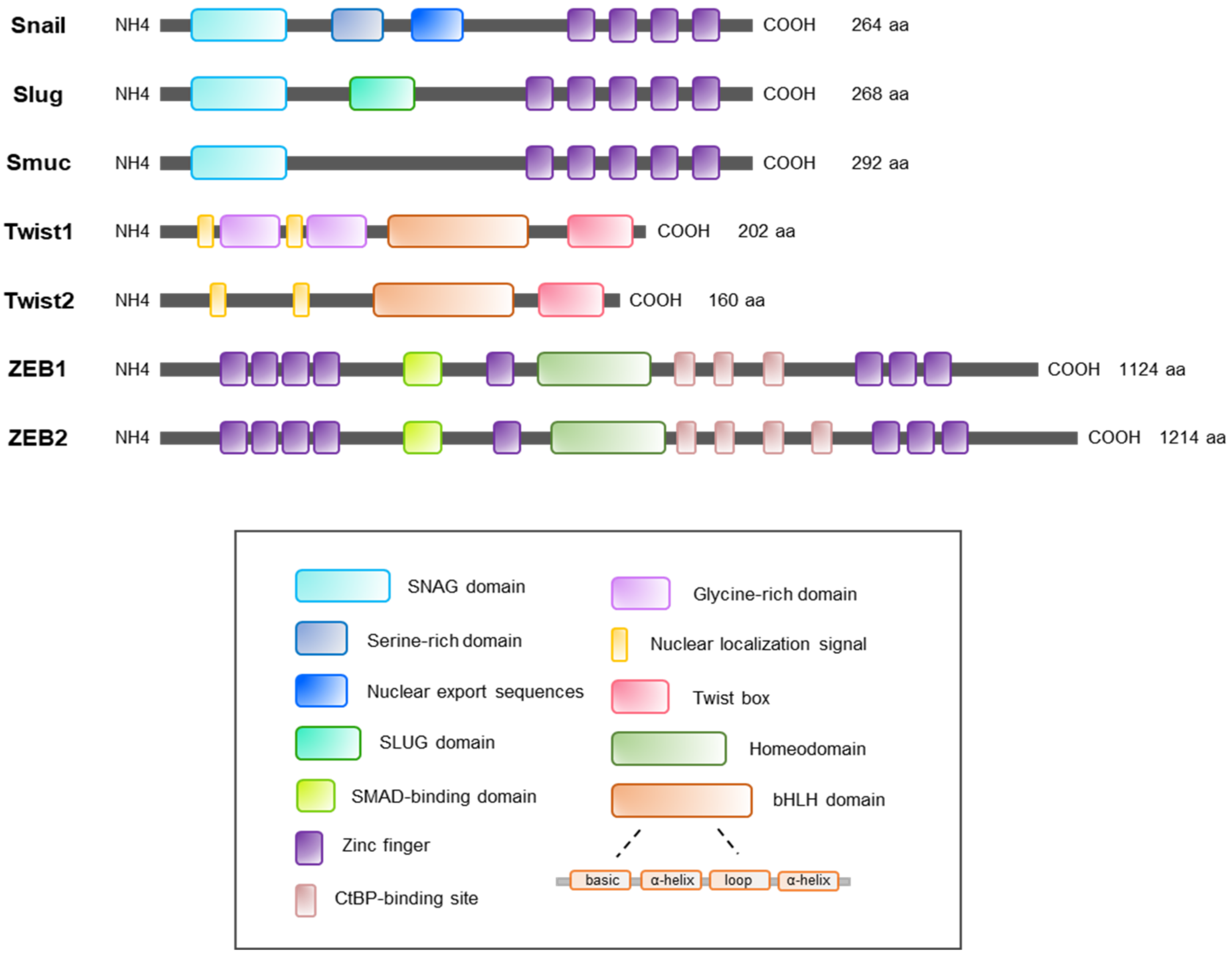
2. Regulation of the Snail Family
2.1. Snail
2.1.1. Phosphorylation of Snail
2.1.2. Ubiquitination of Snail
2.1.3. Acetylation of Snail
2.1.4. Glycosylation of Snail
2.2. Slug
2.2.1. Phosphorylation of Slug
2.2.2. Ubiquitination of Slug
2.2.3. Sumoylation of Slug
2.2.4. Acetylation of Slug
2.3. Smuc
3. Regulation of the Twist Family
3.1. Twist1
3.1.1. Phosphorylation of Twist1
3.1.2. Ubiquitination of Twist1
3.1.3. Acetylation of Twist1
3.1.4. Methylation of Twist1
4. Regulation of the ZEB Family
4.1. ZEB1 and ZEB2
4.1.1. Phosphorylation of ZEB1 and ZEB2
4.1.2. Ubiquitination of ZEB1 and ZEB2
4.1.3. Sumoylation of ZEB1 and ZEB2
4.1.4. Acetylation of ZEB1 and ZEB2
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | Ataxia–telangiectasia mutated kinase |
| BLBCs | Basal-like breast cancer cells |
| β-TrCP | Beta-transducin repeat-containing protein |
| CBP | CREB-binding protein |
| CDK2 | Cyclin-dependent kinase 2 |
| CSN5 | COP9 signalosome subunit 5 |
| CtBP | C-terminal binding protein |
| CZF | C-terminal zinc finger |
| DUBs | Deubiquitinating enzymes |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| EMT-TFs | EMT-transcription factors |
| EndoMT | Endothelial-to-mesenchymal transition |
| ERK | Extracellular signal-regulated kinases |
| FBXL14 | F-Box and leucine-rich repeat protein 14 |
| FBXL5 | F-box and leucine-rich repeat protein 5 |
| FBXO11 | F-box only protein 11 |
| FBXO45 | F-box only protein 45 |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| GSK3β | Glycogen synthase kinase 3 |
| HAT | Histone acetyltransferase |
| HCC | Hepatocellular carcinoma |
| HDAC1/2 | Histone deacetylase 1/2 |
| HNSCC | Head and neck squamous cell carcinoma |
| IGF-1 | Insulin-like growth factor-1 |
| IL-6 | Interleukin 6 |
| Lats2 | Large tumor suppressor kinase 2 |
| MAPK | Mitogen-activated protein kinase |
| MDM2 | Mouse double minute 2 homolog |
| MET | Mesenchymal–epithelial transition |
| MMPs | Matrix metalloproteinases |
| NF-κB | Nuclear factor-kappa B |
| NSCLC | Non-small cell lung cancer |
| NuRD | Nucleosome remodeling and deacetylation |
| NZF | N-terminal zinc finger |
| O-GlcNAc | O-linked β-N-acetylglucosamine |
| Pc2 | Polycomb protein 2 |
| PCAF | p300/CBP-associated factor |
| PI3K | Phosphoinositide 3-kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PKCα | Protein kinase C alpha |
| PKD1 | Protein kinase D1 |
| PRMTs | Protein arginine methyl transferases |
| PTM | Post-translational modification |
| SBD | SMAD-binding domain |
| SCP | Small C-terminal domain phosphatase |
| SRD | Serine-rich domain |
| TGF-β | Transforming growth factor beta |
| Tip60 | Tat-interacting protein of 60 kDa |
| USP51 | Ubiquitin specific peptidase 51 |
| ZnF | Zinc finger |
References
- Li, L.; Li, W. Epithelial-mesenchymal transition in human cancer: Comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol. Ther. 2015, 150, 33–46. [Google Scholar] [CrossRef]
- Škovierová, H.; Okajčeková, T.; Strnádel, J.; Vidomanová, E.; Halašová, E. Molecular regulation of epithelial-to-mesenchymal transition in tumorigenesis (Review). Int. J. Mol. Med. 2018, 41, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Garg, M. Epithelial-mesenchymal transition-activating transcription factors-multifunctional regulators in cancer. World J. Stem Cells 2013, 5, 188. [Google Scholar] [CrossRef]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef]
- Navas, T.; Kinders, R.J.J.; Lawrence, S.M.M.; Ferry-Galow, K.V.V.; Borgel, S.; Hollingshead, M.G.G.; Srivastava, A.K.K.; Alcoser, S.Y.Y.; Makhlouf, H.R.R.; Chuaqui, R.; et al. Clinical evolution of epithelial–mesenchymal transition in human carcinomas. Cancer Res. 2020, 80, 304–318. [Google Scholar] [CrossRef]
- Francou, A.; Anderson, K.V. The Epithelial-to-Mesenchymal Transition in Development and Cancer. Annu. Rev. Cancer Biol. 2020, 4, 197–220. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Liang, X.; Zheng, M.; Tang, Y. Cellular Phenotype Plasticity in Cancer Dormancy and Metastasis. Front. Oncol. 2018, 8, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Onuchic, J.N.; Levine, H.; Ben-Jacob, E. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 1–13. [Google Scholar] [CrossRef]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and ENDMT: Emerging roles in age-related macular degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K. Contribution of epithelial-mesenchymal transitions to organogenesis and cancer metastasis. Curr. Opin. Cell Biol. 2018, 55, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.; Chauhan, P.; Saha, B.; Kubatzky, K.F. Conceptual Evolution of Cell Signaling. Int. J. Mol. Sci. 2019, 20, 3292. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yun, F.; Shi, L.; Li, Z.-H.; Luo, N.-R.; Jia, Y.-F. Roles of Signaling Pathways in the Epithelial-Mesenchymal Transition in Cancer. Asian Pacific J. Cancer Prev. 2015, 16, 6201–6206. [Google Scholar] [CrossRef] [Green Version]
- Perona, R. Cell signalling: Growth factors and tyrosine kinase receptors. Clin. Transl. Oncol. 2006, 8, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Kang, H.E.; Kim, N.H.; Yook, J.I. Therapeutic implications of cancer epithelial-mesenchymal transition (EMT). Arch. Pharm. Res. 2019, 42, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal. Cell. Pathol. 2019, 2019, 9423907. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 1–20. [Google Scholar] [CrossRef]
- Song, K.-A.; Faber, A.C. Epithelial-to-mesenchymal transition and drug resistance: Transitioning away from death. J. Thorac. Dis. 2019, 11, E82–E85. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, N.; Wang, Y.; Xu, M.; Liu, N.; Pang, X.; Cao, J.; Ma, N.; Pang, H.; Liu, L.; et al. Zinc finger E-box binding homeobox 1 promotes invasion and bone metastasis of small cell lung cancer in vitro and in vivo. Cancer Sci. 2012, 103, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Rai, B.; Qi, F.; Liu, T.; Wang, J.; Wang, X.; Ma, B. Influence of the Twist gene on the invasion and metastasis of colon cancer. Oncol. Rep. 2018, 39, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.C.; Lebleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Peterson, S.E.; Loring, J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 24, 143–160. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Miao, B.; Bi, J.; Wang, Z.; Li, Y. Prioritizing functional phosphorylation sites based on multiple feature integration. Sci. Rep. 2016, 6, 24735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Li, Y.; Tuerhanjiang, A.; Wang, W.; Wu, Z.; Yuan, M.; Maitituoheti, M.; Wang, S. Twist2 contributes to cisplatin-resistance of ovarian cancer through the AKT/GSK-3β signaling pathway. Oncol. Lett. 2014, 7, 1102–1108. [Google Scholar] [CrossRef] [Green Version]
- Balcik-Ercin, P.; Cetin, M.; Yalim-Camci, I.; Uygur, T.; Yagci, T. Hepatocellular Carcinoma Cells with Downregulated ZEB2 Become Resistant to Resveratrol by Concomitant Induction of ABCG2 Expression. Mol. Biol. 2020, 54, 75–81. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.E.; Yin, M.; Paznekas, W.A.; Schertzer, M.; Wood, S.; Jabs, E.W. HumanSLUGGene Organization, Expression, and Chromosome Map Location on 8q. Genomics 1998, 51, 468–471. [Google Scholar] [CrossRef]
- Savagner, P.; Yamada, K.M.; Thiery, J.P. The Zinc-Finger Protein Slug Causes Desmosome Dissociation, an Initial and Necessary Step for Growth Factor–induced Epithelial–Mesenchymal Transition. J. Cell Biol. 1997, 137, 1403–1419. [Google Scholar] [CrossRef]
- Twigg, S.R.F.; Wilkie, A.O.M. Characterisation of the human snail (SNAI1) gene and exclusion as a major disease gene in craniosynostosis. Hum. Genet. 1999, 105, 320–326. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, Y.M.; Yang, C.H.; Cho, S.K.; Lee, J.W.; Cho, M. Functional regulation of Slug/Snail2 is dependent on GSK-3β-mediated phosphorylation. FEBS J. 2012, 279, 2929–2939. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Identification and characterization of human SNAIL3 (SNAI3) gene in silico. Int. J. Mol. Med. 2003, 11, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Hung, M.-C.C. Wnt, Hedgehog, and Snail: Sister Pathways That Control by GSK-3beta and beta-Trcp in the Regulation of Metastasis. Cell Cycle 2005, 4, 772–776. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ni, T.K.; Wronski, A.; Glass, B.; Skibinski, A.; Beck, A.; Kuperwasser, C. The SIRT2 Deacetylase Stabilizes Slug to Control Malignancy of Basal-like Breast Cancer. Cell Rep. 2016, 17, 1302–1317. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Rodriguez-Aznar, E.; Yabuta, N.; Owen, R.J.; Mingot, J.M.; Nojima, H.; Nieto, M.A.; Longmore, G.D. Lats2 kinase potentiates Snail1 activity by promoting nuclear retention upon phosphorylation. EMBO J. 2012, 31, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.; Sato, M.; Kim, R.-J.J.; Kwak, M.-K.K.; Naka, K.; Gim, J.; Kadota, M.; Tang, B.; Flanders, K.C.; Kim, T.-A.A.; et al. Definition of Smad3 Phosphorylation Events That Affect Malignant and Metastatic Behaviors in Breast Cancer Cells. Cancer Res. 2014, 74, 6139–6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Guo, X.; Qian, X.; Wang, H.; Yang, C.; Brinkman, K.L.; Serrano-Gonzalez, M.; Jope, R.S.; Zhou, B.; Engler, D.A.; et al. Activation of the ATM-Snail pathway promotes breast cancer metastasis. J. Mol. Cell Biol. 2012, 4, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Zhang, C.; Hassan, S.; Biswas, M.H.U.; Balaji, K.C. Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res. 2010, 70, 7810–7819. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Pan, X.W.; Huang, H.; Gao, Y.; Yang, Q.W.; Wang, L.H.; Cui, X.G.; Xu, D.F. Epithelial-mesenchymal transition induced by GRO-α-CXCR2 promotes bladder cancer recurrence after intravesical chemotherapy. Oncotarget 2017, 8, 45274–45285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Corsa, C.A.; Ponik, S.M.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.W.; Keely, P.J.; Longmore, G.D. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rayala, S.; Nguyen, D.; Vadlamudi, R.K.; Chen, S.; Kumar, R. Pak1 phosphorylation of Snail, a master regulator of epithelial-to- mesenchyme transition, modulates Snail’s subcellular localization and functions. Cancer Res. 2005, 65, 3179–3184. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.-C.C. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Shen, M.; Zha, Y.L.; Li, W.; Wei, Y.; Blanco, M.A.; Ren, G.; Zhou, T.; Storz, P.; Wang, H.Y.; et al. PKD1 Phosphorylation-Dependent Degradation of SNAIL by SCF-FBXO11 Regulates Epithelial-Mesenchymal Transition and Metastasis. Cancer Cell 2014, 26, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Viñas-Castells, R.; Beltran, M.; Valls, G.; Gómez, I.; García, J.M.; Montserrat-Sentís, B.; Baulida, J.; Bonilla, F.; De Herreros, A.G.; Díaz, V.M. The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J. Biol. Chem. 2010, 285, 3794–3805. [Google Scholar] [CrossRef] [Green Version]
- Vinas-Castells, R.; Frias, A.; Robles-Lanuza, E.; Zhang, K.; Longmore, G.D.; Garcia de Herreros, A.; Diaz, V.M. Nuclear ubiquitination by FBXL5 modulates Snail1 DNA binding and stability. Nucleic Acids Res. 2014, 42, 1079–1094. [Google Scholar] [CrossRef]
- Wu, Y.; Evers, B.M.; Zhou, B.P.; Mark Evers, B.; Zhou, B.P. Small C-terminal Domain Phosphatase Enhances Snail Activity through Dephosphorylation. J. Biol. Chem. 2009, 284, 640–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Liu, J.; Chen, F.; Feng, X.-H. C-terminal domain small phosphatase-like 2 promotes epithelial-to-mesenchymal transition via Snail dephosphorylation and stabilization. Open Biol. 2018, 8, 170274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, D.S.-S.; Wang, H.-J.; Tai, S.-K.; Chou, C.-H.; Hsieh, C.-H.; Chiu, P.-H.; Chen, N.-J.; Yang, M.-H. Acetylation of Snail Modulates the Cytokinome of Cancer Cells to Enhance the Recruitment of Macrophages. Cancer Cell 2014, 26, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Kim, H.S.; Kim, N.H.; Ji, S.; Cha, S.Y.; Kang, J.G.; Ota, I.; Shimada, K.; Konishi, N.; Nam, H.W.; et al. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J. 2010, 29, 3787–3796. [Google Scholar] [CrossRef] [Green Version]
- Virtakoivu, R.; Mai, A.; Mattila, E.; De Franceschi, N.; Imanishi, S.Y.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E.; et al. Vimentin–ERK Signaling Uncouples Slug Gene Regulatory Function. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-L.; Huang, H.-C.; Kao, S.-H.; Hsu, Y.-C.; Wang, Y.-T.; Li, K.-C.; Chen, Y.-J.; Yu, S.-L.; Wang, S.-P.; Hsiao, T.-H.; et al. Slug is temporally regulated by cyclin E in cell cycle and controls genome stability. Oncogene 2015, 34, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Park, M.H.; Oh, E.H.; Soung, N.K.; Lee, S.J.; Jung, J.K.; Lee, O.J.; Yun, S.J.; Kim, W.J.; Shin, E.Y.; et al. The p21-activated kinase 4-Slug transcription factor axis promotes epithelial−mesenchymal transition and worsens prognosis in prostate cancer. Oncogene 2018, 37, 5147–5159. [Google Scholar] [CrossRef]
- Vernon, A.E. Slug stability is dynamically regulated during neural crest development by the F-box protein Ppa. Development 2006, 133, 3359–3370. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.Q.Z.-Q.; Li, X.Y.X.-Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.-H.; Wang, W.-L.; Chen, C.-Y.; Chang, Y.-L.; Wu, Y.-Y.; Wang, Y.-T.; Wang, S.-P.; Nesvizhskii, A.I.; Chen, Y.-J.; Hong, T.-M.; et al. GSK3β controls epithelial–mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene 2014, 33, 3172–3182. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.K.; Kim, C.K.; Hwang, K.R.; Park, H.Y.; Koh, J.; Chung, D.H.; Lee, C.W.; Ha, G.H. Pellino-1 promotes lung carcinogenesis via the stabilization of Slug and Snail through K63-mediated polyubiquitination. Cell Death Differ. 2017, 24, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates E-cadherin repression via p19Arf in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xin, Y.; Xu, W.; Tian, X.; Wei, X.; Zhang, H. CBP-mediated Slug acetylation stabilizes Slug and promotes EMT and migration of breast cancer cells. Sci. China Life Sci. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef]
- Mingot, J.M.; Vega, S.; Maestro, B.; Sanz, J.M.; Nieto, M.A. Characterization of snail nuclear import pathways as representatives of C2H2 zinc finger transcription factors. J. Cell Sci. 2009, 122, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Domínguez, D.; Montserrat-Sentís, B.; Virgós-Soler, A.; Guaita, S.; Grueso, J.; Porta, M.; Puig, I.; Baulida, J.; Francí, C.; García de Herreros, A. Phosphorylation Regulates the Subcellular Location and Activity of the Snail Transcriptional Repressor. Mol. Cell. Biol. 2003, 23, 5078–5089. [Google Scholar] [CrossRef] [Green Version]
- Manzanares, M.; Locascio, A.; Nieto, M.A. The increasing complexity of the Snail gene superfamily in metazoan evolution. Trends Genet. 2001, 17, 178–181. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/Histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Cobaleda, C.; Pérez-Caro, M.; Vicente-Dueñas, C.; Sánchez-García, I. Function of the Zinc-Finger Transcription Factor SNAI2 in Cancer and Development. Annu. Rev. Genet. 2007, 41, 41–61. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Pasini, D.; Díaz, V.M.; Francí, C.; Gutierrez, A.; Dave, N.; Escrivà, M.; Hernandez-Muñoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb Complex 2 Is Required for E-cadherin Repression by the Snail1 Transcription Factor. Mol. Cell. Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, H.; Scott, I.C.; Cross, J.C. The transition to endoreduplication in trophoblast giant cells is regulated by the mSNA zinc finger transcription factor. Dev. Biol. 1998, 199, 150–163. [Google Scholar] [CrossRef] [Green Version]
- Grimes, H.L.; Chan, T.O.; Zweidler-McKay, P.A.; Tong, B.; Tsichlis, P.N. The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol. Cell. Biol. 1996, 16, 6263–6272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Ortiz, P.; Villarejo, A.; MacPherson, M.; Santos, V.; Montes, A.; Souchelnytskyi, S.; Portillo, F.; Cano, A. Characterization of the SNAG and SLUG domains of Snail2 in the repression of E-cadherin and EMT induction: Modulation by serine 4 phosphorylation. PLoS ONE 2012, 7, e36132. [Google Scholar] [CrossRef] [Green Version]
- Sefton, M.; Sánchez, S.; Nieto, M.A. Conserved and divergent roles for members of the Snail family of transcription factors in the chick and mouse embryo. Development 1998, 125, 3111–3121. [Google Scholar] [CrossRef] [PubMed]
- Gras, B.; Jacqueroud, L.; Wierinckx, A.; Lamblot, C.; Fauvet, F.F.; Lachuer, J.; Puisieux, A.; Ansieau, S.S. Snail Family Members Unequally Trigger EMT and Thereby Differ in Their Ability to Promote the Neoplastic Transformation of Mammary Epithelial Cells. PLoS ONE 2014, 9, e92254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jürgens, G.; Wieschaus, E.; Nüsslein-Volhard, C.; Kluding, H. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster-II. Zygotic loci on the third chromosome. Wilhelm Roux’s Arch. Dev. Biol. 1984, 193, 283–295. [Google Scholar] [CrossRef]
- Grau, Y.; Carteret, C.; Simpson, P. Mutations and Chromosomal Rearrangements Affecting the Expression of Snail, a Gene Involved in Embryonic Patterning in DROSOPHILA MELANOGASTER. Genetics 1984, 108, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2016, 129, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, ZEB and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Kataoka, H.; Murayama, T.; Yokode, M.; Mori, S.; Sano, H.; Ozaki, H.; Yokota, Y.; Nishikawa, S.I.; Kita, T. A novel Snail-related transcription factor Smuc regulates basic helix-loop-helix transcription factor activities via specific E-box motifs. Nucleic Acids Res. 2000, 28, 626–633. [Google Scholar] [CrossRef]
- Guo, S.; Yan, X.; Shi, F.; Ma, K.; Chen, Z.-J.; Zhang, C. Expression and distribution of the zinc finger protein, SNAI3, in mouse ovaries and pre-implantation embryos. J. Reprod. Dev. 2018, 64, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Lee, S.H.; Kim, H.S.; Kim, N.H.; Piao, S.; Park, S.H.; Jung, Y.S.; Yook, J.I.; Park, B.J.; Ha, N.C. Role of CK1 in GSK3Β-mediated phosphorylation and degradation of Snail. Oncogene 2010, 29, 3124–3133. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.-H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of β-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Díaz, V.; Viñas-Castells, R.; García de Herreros, A. Regulation of the protein stability of EMT transcription factors. Cell Adh. Migr. 2014, 8, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Rane, C.K.; Minden, A. P21 activated kinases: Structure, regulation, and functions. Small GTPases 2014, 5, e28003. [Google Scholar] [CrossRef]
- Kumar, R.; Gururaj, A.E.; Barnes, C.J. P21-Activated Kinases in Cancer. Nat. Rev. Cancer 2006, 6, 459–471. [Google Scholar] [CrossRef]
- Boohaker, R.J.; Cui, X.; Stackhouse, M.; Xu, B. ATM-mediated Snail Serine 100 phosphorylation regulates cellular radiosensitivity. Radiother. Oncol. 2013, 108, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Bastea, L.I.; Döppler, H.; Balogun, B.; Storz, P. Protein kinase D1 maintains the epithelial phenotype by inducing a DNA-bound, inactive SNAI1 transcriptional repressor complex. PLoS ONE 2012, 7, e30459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacPherson, M.R.; Molina, P.; Souchelnytskyi, S.; Wernstedt, C.; Martin-Pérez, J.; Portillo, F.; Cano, A. Phosphorylation of serine 11 and serine 92 as new positive regulators of human Snail1 function: Potential involvement of casein kinase-2 and the cAMP-activated kinase protein kinase A. Mol. Biol. Cell 2010, 21, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef]
- Fang, X.; Zhou, W.; Wu, Q.; Huang, Z.; Shi, Y.; Yang, K.; Chen, C.; Xie, Q.; Mack, S.C.; Wang, X.; et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J. Exp. Med. 2017, 214, 245–267. [Google Scholar] [CrossRef] [Green Version]
- Schauvliege, R.; Janssens, S.; Beyaert, R. Pellino Proteins: Novel Players in TLR and IL-1R Signalling. J. Cell. Mol. Med. 2007, 11, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bae, S.; An, S.; Park, J.K.; Kim, E.M.; Hwang, S.; Kim, W.; Um, H. Cooperative actions of p21 WAF 1 and p53 induce Slug protein degradation and suppress cell invasion. EMBO Rep. 2014, 15, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Yang, J.; Murphy, R.F.; Agrawal, D.K. Regulation of the p14ARF-Mdm2-p53 pathway: An overview in breast cancer. Exp. Mol. Pathol. 2006, 81, 115–122. [Google Scholar] [CrossRef]
- Ichimura, K.; Bolin, M.B.; Goike, H.M.; Schmidt, E.E.; Moshref, A.; Collins, V.P. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res. 2000, 60, 417–424. [Google Scholar]
- Abida, W.M.; Gu, W. p53-Dependent and p53-Independent Activation of Autophagy by ARF. Cancer Res. 2008, 68, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J. Autophagy by ARF: A Short Story. Mol. Cell 2006, 22, 436–437. [Google Scholar] [CrossRef]
- Zhuge, X.; Kataoka, H.; Tanaka, M.; Murayama, T.; Kawamoto, T.; Sano, H.; Togi, K.; Yamauchi, R.; Ueda, Y.; Xu, Y.; et al. Expression of the novel Snai-related zinc-finger transcription factor gene Smuc during mouse development. Int. J. Mol. Med. 2005, 15, 945–948. [Google Scholar] [CrossRef]
- Bradley, C.K.; Norton, C.R.; Chen, Y.; Han, X.; Booth, C.J.; Yoon, J.K.; Krebs, L.T.; Gridley, T. The Snail Family Gene Snai3 Is Not Essential for Embryogenesis in Mice. PLoS ONE 2013, 8, e65344. [Google Scholar] [CrossRef] [Green Version]
- Franco, H.L.; Casasnovas, J.; Rodríguez-Medina, J.R.; Cadilla, C.L. Redundant or separate entities?-Roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011, 39, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Merindol, N.; Riquet, A.; Szablewski, V.; Eliaou, J.F.; Puisieux, A.; Bonnefoy, N. The emerging role of Twist proteins in hematopoietic cells and hematological malignancies. Blood Cancer J. 2014, 4, e206. [Google Scholar] [CrossRef]
- Zhu, Q.-Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumor Biol. 2016, 37, 185–197. [Google Scholar] [CrossRef]
- Castanon, I.; Baylies, M.K. A Twist in fate: Evolutionary comparison of Twist structure and function. Gene 2002, 287, 11–22. [Google Scholar] [CrossRef]
- Barnes, R.M.; Firulli, A.B. A twist of insight-The role of Twist-family bHLH factors in development. Int. J. Dev. Biol. 2009, 53, 909–924. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, Y.; Gonzalez-Mendez, R.R.; Cadilla, C.L. Evolution of the Twist Subfamily Vertebrate Proteins: Discovery of a Signature Motif and Origin of the Twist1 Glycine-Rich Motifs in the Amino-Terminus Disordered Domain. PLoS ONE 2016, 11, e0161029. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Pei, H.; Li, Y.; Liu, M.; Chen, Y. Targeting Twist expression with small molecules. Medchemcomm 2017, 8, 268–275. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Lowe, G.; Flanagan, S.; Kuchler, K.; Glackin, C.A. Human dermo-1 has attributes similar to twist in early bone development. Bone 2000, 27, 591–602. [Google Scholar] [CrossRef]
- Chang, A.T.; Liu, Y.; Ayyanathan, K.; Benner, C.; Jiang, Y.; Prokop, J.W.; Paz, H.; Wang, D.; Li, H.R.; Fu, X.D.; et al. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes Dev. 2015, 29, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.Q.; Li, J.H.; Wen, J.F.; Zhou, Y.H.; Hu, Y.B.; Zhou, J.H. Effect and mechanism of the Twist gene on invasion and metastasis of gastric carcinoma cells. World J. Gastroenterol. 2008, 14, 2487–2493. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Hamada, S.; Kimura, K.; Kanno, A.; Hirota, M.; Umino, J.; Fujibuchi, W.; Masamune, A.; Tanaka, N.; Miura, K.; et al. Up-Regulation of MSX2 Enhances the Malignant Phenotype and Is Associated with Twist 1 Expression in Human Pancreatic Cancer Cells. Am. J. Pathol. 2008, 172, 926–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Qin, L.; Sun, T.; Wu, H.; He, T.; Yang, Z.; Mo, Q.; Liao, L.; Xu, J. Twist1 promotes breast cancer invasion and metastasis by silencing Foxa1 expression. Oncogene 2017, 36, 1157–1166. [Google Scholar] [CrossRef] [Green Version]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by Twist Proteins as a Collateral Effect of Tumor-Promoting Inactivation of Premature Senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, W.; Wang, W.; Yang, R.; Wang, T.; Su, T.; Weng, D.; Tao, T.; Li, W.; Ma, D.; et al. Correlation of TWIST2 up-regulation and epithelial-mesenchymal transition during tumorigenesis and progression of cervical carcinoma. Gynecol. Oncol. 2012, 124, 112–118. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, T.; Che, N.; Sun, D.; Zhao, N.; Dong, X.; Gu, Q.; Yao, Z.; Sun, B. Promotion of hepatocellular carcinoma metastasis through matrix metalloproteinase activation by epithelial-mesenchymal transition regulator Twist1. J. Cell. Mol. Med. 2011, 15, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of twist1 by MAPKs stabilizes twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Shin, D.M. Multiple biological functions of Twist1 in various cancers. Oncotarget 2017, 8, 20380–20393. [Google Scholar] [CrossRef] [Green Version]
- Vichalkovski, A.; Gresko, E.; Hess, D.; Restuccia, D.F.; Hemmings, B.A. PKB/AKT phosphorylation of the transcription factor Twist-1 at Ser42 inhibits p53 activity in response to DNA damage. Oncogene 2010, 29, 3554–3565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.W.; Xie, T.X.; Sano, D.; Myers, J.N. IL-6 stabilizes twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PLoS ONE 2011, 6, e19412. [Google Scholar] [CrossRef]
- Tedja, R.; Roberts, C.M.; Alvero, A.B.; Cardenas, C.; Yang-Hartwich, Y.; Spadinger, S.; Pitruzzello, M.; Yin, G.; Glackin, C.A.; Gil Mor, X. Protein kinase C–mediated phosphorylation of Twist1 at Ser-144 prevents Twist1 ubiquitination and stabilizes it. J. Biol. Chem. 2019, 294, 5082–5093. [Google Scholar] [CrossRef]
- Wang, J.; Nikhil, K.; Viccaro, K.; Chang, L.; Jacobsen, M.; Sandusky, G.; Shah, K. The Aurora-A-Twist1 axis promotes highly aggressive phenotypes in pancreatic carcinoma. J. Cell Sci. 2017, 130, 1078–1093. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.W.; Wong, G.; Earle, C.; Krueger, K.; Spevak, C.C. Hyaluronan-CD44 interaction promotes c-Src-mediated twist signaling, microRNA-10b expression, and RhoA/RhoC up-regulation, leading to Rho-kinase-associated cytoskeleton activation and breast tumor cell invasion. J. Biol. Chem. 2010, 285, 36721–36735. [Google Scholar] [CrossRef] [Green Version]
- Li, C.W.; Xia, W.; Lim, S.O.; Hsu, J.M.J.L.; Huo, L.; Wu, Y.; Li, L.Y.; Lai, C.C.; Chang, S.S.; Hsu, Y.H.; et al. AKT1 inhibits epithelial-to-mesenchymal transition in breast cancer through phosphorylation-dependent twist1 degradation. Cancer Res. 2016, 76, 1451–1462. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Ogura, K.; Wang, Z.; Inuzuka, H. Degradation of the transcription factor twist, an oncoprotein that promotes cancer metastasis. Discov. Med. 2013, 15, 7–15. [Google Scholar] [PubMed]
- Sun, T.; Fu, J.; Shen, T.; Lin, X.; Liao, L.; Feng, X.H.; Xu, J. The small C-terminal domain phosphatase 1 inhibits cancer cell migration and invasion by dephosphorylating Ser(P)68-Twist1 to accelerate Twist1 protein degradation. J. Biol. Chem. 2016, 291, 11518–11528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, R.; Nordin, K.; LaBonne, C. The F-box protein Ppa is a common regulator of core EMT factors Twist, Snail, Slug, and Sip1. J. Cell Biol. 2011, 194, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Shiota, M.; Yokomizo, A.; Tada, Y.; Uchiumi, T.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Yamamoto, K.; Seki, N.; Naito, S. P300/CBP-associated factor regulates Y-box binding protein-1 expression and promotes cancer cell growth, cancer invasion and drug resistance. Cancer Sci. 2010, 101, 1797–1806. [Google Scholar] [CrossRef]
- Wang, L.-T.; Wang, S.-N.; Chiou, S.-S.; Liu, K.-Y.; Chai, C.-Y.; Chiang, C.-M.; Huang, S.-K.; Yokoyama, K.K.; Hsu, S.-H. TIP60-dependent acetylation of the SPZ1-TWIST complex promotes epithelial–mesenchymal transition and metastasis in liver cancer. Oncogene 2019, 38, 518–532. [Google Scholar] [CrossRef]
- Shi, J.; Cao, J.; Zhou, B. Twist-BRD4 Complex: Potential Drug Target for Basal-like Breast Cancer. Curr. Pharm. Des. 2015, 21, 1256–1261. [Google Scholar] [CrossRef] [Green Version]
- Avasarala, S.; Van Scoyk, M.; Rathinam, M.K.K.; Zerayesus, S.; Zhao, X.; Zhang, W.; Pergande, M.R.; Borgia, J.A.; DeGregori, J.; Port, J.D.; et al. PRMT1 Is a novel regulator of epithelial-mesenchymal-transition in non-small cell lung cancer. J. Biol. Chem. 2015, 290, 13479–13489. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Crowley, S.D. Twist1: A Double-Edged Sword in Kidney Diseases. Kidney Dis. 2020, 6, 247–257. [Google Scholar] [CrossRef]
- Xue, G.; Restuccia, D.F.; Lan, Q.; Hynx, D.; Dirnhofer, S.; Hess, D.; Rüegg, C.; Hemmings, B.A. Akt/PKB-Mediated Phosphorylation of Twist1 Promotes Tumor Metastasis via Mediating Cross-Talk between PI3K/Akt and TGF-β Signaling Axes. Cancer Discov. 2012, 2, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhou, B.P. Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 2011, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.; Zhang, P.; You, J. Post-translational modifications of EMT transcriptional factors in cancer metastasis. Open Life Sci. 2016, 11, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Luo, Y.; Gu, X.; Zhan, W.; Wang, X. Twist1 Promotes Gastric Cancer Cell Proliferation through Up-Regulation of FoxM1. PLoS ONE 2013, 8, e77625. [Google Scholar] [CrossRef] [Green Version]
- Hamamori, Y.; Sartorelli, V.; Ogryzko, V.; Puri, P.L.; Wu, H.-Y.; Wang, J.Y.J.; Nakatani, Y.; Kedes, L. Regulation of Histone Acetyltransferases p300 and PCAF by the bHLH Protein Twist and Adenoviral Oncoprotein E1A. Cell 1999, 96, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Bikkavilli, R.K.; Avasarala, S.; Vanscoyk, M.; Sechler, M.; Kelley, N.; Malbon, C.C.; Winn, R.A. Dishevelled3 is a novel arginine methyl transferase substrate. Sci. Rep. 2012, 2, 805. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, R.M.; Morettin, A.; Côté, J. Role of PRMTs in cancer: Could minor isoforms be leaving a mark? World J. Biol. Chem. 2014, 5, 115. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Liu, Y.; De Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef]
- Remacle, J.E.; Kraft, H.; Lerchner, W.; Wuytens, G.; Collart, C.; Verschueren, K.; Smith, J.C.; Huylebroeck, D. New mode of DNA binding of multi-zinc finger transcription factors: δEF1 family members bind with two hands to two target sites. EMBO J. 1999, 18, 5073–5084. [Google Scholar] [CrossRef] [Green Version]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef]
- Gheldof, A.; Hulpiau, P.; van Roy, F.; De Craene, B.; Berx, G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell. Mol. Life Sci. 2012, 69, 2527–2541. [Google Scholar] [CrossRef]
- Wong, T.-S.S.; Gao, W.; Chan, J.Y.-W.W. Transcription regulation of E-cadherin by zinc finger E-box binding homeobox proteins in solid tumors. Biomed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; Van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; Van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Yu, J.; Zhang, M.; Qin, F.; Lan, X. ZEB1 promotes tumorigenesis and metastasis in hepatocellular carcinoma by regulating the expression of vimentin. Mol. Med. Rep. 2019, 19, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noël, A.; van Roy, F.; Berx, G.; Foidart, J.-M.; Gilles, C. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 2006, 25, 4975–4985. [Google Scholar] [CrossRef] [Green Version]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, À.; Beltran, M.; Peiró, S.; de Herreros, A.G. Functional Cooperation between Snail1 and Twist in the Regulation of ZEB1 Expression during Epithelial to Mesenchymal Transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef] [Green Version]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wei, Y.; Wang, L.i.L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorens, M.C.; Lorenzatti, G.; Cavallo, N.L.; Vaglienti, M.V.; Perrone, A.P.; Carenbauer, A.L.; Darling, D.S.; Cabanillas, A.M. Phosphorylation Regulates Functions of ZEB1 Transcription Factor. J. Cell. Physiol. 2016, 231, 2205–2217. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhang, P.; Hu, X.; Kim, J.; Yao, F.; Xiao, Z.; Zeng, L.; Chang, L.; Sun, Y.; Ma, L. USP51 promotes deubiquitination and stabilization of ZEB1. Am. J. Cancer Res. 2017, 7, 2020–2031. [Google Scholar]
- Zhang, S.; Hong, Z.; Chai, Y.; Liu, Z.; Du, Y.; Li, Q.; Liu, Q. CSN5 promotes renal cell carcinoma metastasis and EMT by inhibiting ZEB1 degradation. Biochem. Biophys. Res. Commun. 2017, 488, 101–108. [Google Scholar] [CrossRef]
- Long, J.; Zuo, D.; Park, M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef] [Green Version]
- Postigo, A.A.; Depp, J.L.; Taylor, J.J.; Kroll, K.L. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003, 22, 2453–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlubek, F.; Löhberg, C.; Meiler, J.; Jung, A.; Kirchner, T.; Brabletz, T.; Lohberg, C.; Meiler, J.; Jung, A.; Kirchner, T.; et al. Tip60 is a cell-type-specific transcriptional regulator. J. Biochem. 2001, 129, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Krämer, O.H.; Saur, D. A ZEB1-HDAC pathway enters the epithelial to mesenchymal transition world in pancreatic cancer. Gut 2012, 61, 329–330. [Google Scholar] [CrossRef] [Green Version]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.-O.O.; Heidecke, C.-D.D.; Friess, H.; Büchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef]
- Li, N.; Babaei-Jadidi, R.; Lorenzi, F.; Spencer-Dene, B.; Clarke, P.; Domingo, E.; Tulchinsky, E.; Vries, R.G.J.; Kerr, D.; Pan, Y.; et al. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis 2019, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Zhu, C.; Zhao, X.; Chen, C.; Zhang, H.; Yuan, H.; Deng, R.; Dou, J.; Wang, Y.; Huang, J.; et al. Atypical ubiquitin E3 ligase complex Skp1-Pam-Fbxo45 controls the core epithelial-to-mesenchymal transition-inducing transcription factors. Oncotarget 2015, 6, 979–994. [Google Scholar] [CrossRef] [PubMed]
- Costantino, M.E.; Stearman, R.P.; Smith, G.E.; Darling, D.S. Cell-specific phosphorylation of Zfhep transcription factor. Biochem. Biophys. Res. Commun. 2002, 296, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Hakuno, F.; Takahashi, S.-I. 40 YEARS OF IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Turcu, F.E.; Wilkinson, K.D. Polyubiquitin binding and disassembly by deubiquitinating enzymes. Chem. Rev. 2009, 109, 1495–1508. [Google Scholar] [CrossRef] [Green Version]
- Schütz, A.K.; Hennes, T.; Jumpertz, S.; Fuchs, S.; Bernhagen, J. Role of CSN5/JAB1 in Wnt/β-catenin activation in colorectal cancer cells. FEBS Lett. 2012, 586, 1645–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postigo, A.A.; Dean, D.C. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. USA 1999, 96, 6683–6688. [Google Scholar] [CrossRef] [Green Version]
- Hoffmeister, H.; Fuchs, A.; Erdel, F.; Pinz, S.; Gröbner-Ferreira, R.; Bruckmann, A.; Deutzmann, R.; Schwartz, U.; Maldonado, R.; Huber, C.; et al. CHD3 and CHD4 form distinct NuRD complexes with different yet overlapping functionality. Nucleic Acids Res. 2017, 45, 10534–10554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruneel, K.; Verstappe, J.; Vandamme, N.; Berx, G. Intrinsic Balance between ZEB Family Members Is Important for Melanocyte Homeostasis and Melanoma Progression. Cancers 2020, 12, 2248. [Google Scholar] [CrossRef]
- Manshouri, R.; Coyaud, E.; Kundu, S.T.; Peng, D.H.; Stratton, S.A.; Alton, K.; Bajaj, R.; Fradette, J.J.; Minelli, R.; Peoples, M.D.; et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat. Commun. 2019, 10, 5125. [Google Scholar] [CrossRef]
- Wu, L.M.N.; Wang, J.; Conidi, A.; Zhao, C.; Wang, H.; Ford, Z.; Zhang, L.; Zweier, C.; Ayee, B.G.; Maurel, P.; et al. Zeb2 recruits HDAC–NuRD to inhibit Notch and controls Schwann cell differentiation and remyelination. Nat. Neurosci. 2016, 19, 1060–1072. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The roles of ZEB1 in tumorigenic progression and epigenetic modifications. Biomed. Pharmacother. 2019, 110, 400–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Transcription Factor | Function | Effects on EMT-TFs | Regulation Factor | Role or Mechanism | Reference |
|---|---|---|---|---|---|
| Snail | Phosphorylation | Stabilization | Lats2 | Phosphorylation at Thr203 | [52] |
| PTK6 | Phosphorylation at Tyr342 | [53] | |||
| ATM | Phosphorylation at Ser100 | [54] | |||
| Degradation | PKD1 | Phosphorylation at Ser11 | [55] | ||
| Nuclear accumulation | GROα | Phosphorylation at Ser246 | [56] | ||
| ERK | Phosphorylation at Ser82 and Ser104 | [57] | |||
| PAK1 | Phosphorylation at Ser246 | [58] | |||
| Ubiquitination | Degradation | β-TrCP | Ubiquitination at pSer96 and pSer100 | [59] | |
| FBXO11 | Ubiquitination at pSer11 | [60] | |||
| FBXL14 | Ubiquitination at Lys98, Lys137, and Lys146 | [61] | |||
| FBXL5 | Ubiquitination at Lys85, Lys146, and Lys234 | [62] | |||
| Dephosphorylation | Stabilization | SCP | Dephosphorylation at Ser96 and Ser100 | [63,64] | |
| Acetylation | Increasing transcriptional activity | CBP | Acetylation at Lys126 and Lys187 | [65] | |
| Glycosylation | Stabilization | O-GlcNAc | O-GlcNAc at Ser112 | [66] | |
| Slug | Phosphorylation | Nuclear translocation | ERK-vimentin complex | Phosphorylation at Ser87 | [67] |
| Degradation | GSK3β | Phosphorylation at Ser92, Ser96, Ser100, and Ser104 | [48] | ||
| CDK2 | Phosphorylation at Ser54 and Ser104 | [68] | |||
| Stabilization | PAK4 | Phosphorylation at Ser158 and Ser254 | [69] | ||
| Ubiquitination | Degradation | FBXL14 | Ubiquitination depends on Leu33, Tyr34, Val58, and Trp59 | [70] | |
| β-TrCP | GSK3β-mediated ubiquitination | [71] | |||
| CHIP | GSK3β-mediated ubiquitination | [72] | |||
| p53 | MDM2-mediated ubiquitination | [73] | |||
| Stabilization | pellino-1 | Ubiquitination at Lys63 | [74] | ||
| Sumoylation | Stabilization | p14Arf | Sumoylation at Lys192 | [75] | |
| Acetylation | Stabilization | Sirtuin 2 | Deacetylation at Lys116 | [51] | |
| CBP | Acetylation at Lys166 and Lys211 | [76] |
| Transcription Factor | Function | Effects on EMT-TFs | Regulation Factor | Role or Mechanism | Reference |
|---|---|---|---|---|---|
| Twist1 | Phosphorylation | Stabilization | MAPK | Phosphorylation at Ser68 | [133] |
| Akt2 | Phosphorylation at Ser42 | [134,135] | |||
| CK2α | Phosphorylation at Ser18 and Ser20 | [136] | |||
| PKCα | Phosphorylation at Ser144 | [137] | |||
| Aurora A | Phosphorylation at Ser123, Thr148, and Ser184 | [138] | |||
| Nuclear translocation | CD44 | c-Src-dependent phosphorylation at tyrosine | [139] | ||
| Degradation | Akt1 | Phosphorylation at Ser42, Thr121, and Ser123 | [140] | ||
| IKKβ | Phosphorylation at Thr125 and Ser127 | [141] | |||
| Dephosphorylation | Degradation | SCP1 | Dephosphorylation at Ser68 | [142] | |
| Ubiquitination | Degradation | β-TrCP | Ubiquitination at Thr121 and Ser123 | [140] | |
| FBXL14 | C-terminal Twist box-dependent ubiquitination | [143] | |||
| Acetylation | Nuclear translocation | PCAF | Acetylation at Lys73, Lys76, and Lys77 | [144] | |
| Nuclear translocation | Tip60 | Acetylation at Lys73 and Lys76 | [145,146] | ||
| Methylation | PRMT1 | Methylation at Arg34 | [147] |
| Transcription Factor | Function | Effects on EMT-TFs | Regulation Factor | Role or Mechanism | Reference |
|---|---|---|---|---|---|
| ZEB1 | Phosphorylation | Stabilization | ATM | Phosphorylation at Ser585 | [168,169] |
| Inhibition of transcriptional activity | PKC | Phosphorylation at Thr851, Ser852, and Ser853 | [170] | ||
| ERK | Phosphorylation at Thr867 | [170] | |||
| Deubiquitination | Stabilization | USP51 | Binding to N-terminal of ZEB1 | [171] | |
| CSN5 | Binding to ZEB1 | [172] | |||
| Sumoylation | Inhibition of transcriptional activity | Pc2 | Sumoylation at Lys347 and Lys774 | [173] | |
| Acetylation | Inhibition of transcriptional activity | p300 and PCAF | Acetylation at Lys741, Lys774, and Lys775 | [174] | |
| Tip60 | Binding to N-terminal of ZEB1 | [175] | |||
| Deacetylation | Increasing transcriptional activity | HDAC1/2 | Binding to ZEB1 | [176,177] | |
| ZEB2 | Phosphorylation | Degradation | GSK3β | Phosphorylation at Ser705 and Tyr802 | [178] |
| Ubiquitination | Degradation | FBXL14 | Binding to ZEB2 | [143] | |
| FBXO45 | Ubiquitination at Lys48 | [179] | |||
| FBXW7 | GSK3β-mediated ubiquitination | [178] | |||
| Sumoylation | Inhibition of transcriptional activity | Pc2 | Sumoylation at Lys391 and Lys866 | [173] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, E.; Seo, J.; Yoon, H.; Cho, S. The Post-Translational Regulation of Epithelial–Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis. Int. J. Mol. Sci. 2021, 22, 3591. https://doi.org/10.3390/ijms22073591
Kang E, Seo J, Yoon H, Cho S. The Post-Translational Regulation of Epithelial–Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis. International Journal of Molecular Sciences. 2021; 22(7):3591. https://doi.org/10.3390/ijms22073591
Chicago/Turabian StyleKang, Eunjeong, Jihye Seo, Haelim Yoon, and Sayeon Cho. 2021. "The Post-Translational Regulation of Epithelial–Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis" International Journal of Molecular Sciences 22, no. 7: 3591. https://doi.org/10.3390/ijms22073591