
Integrative Analyses of Transcriptomes and Metabolomes Reveal Associated Genes and Metabolites with Flowering Regulation in Common Vetch (Vicia sativa L.)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
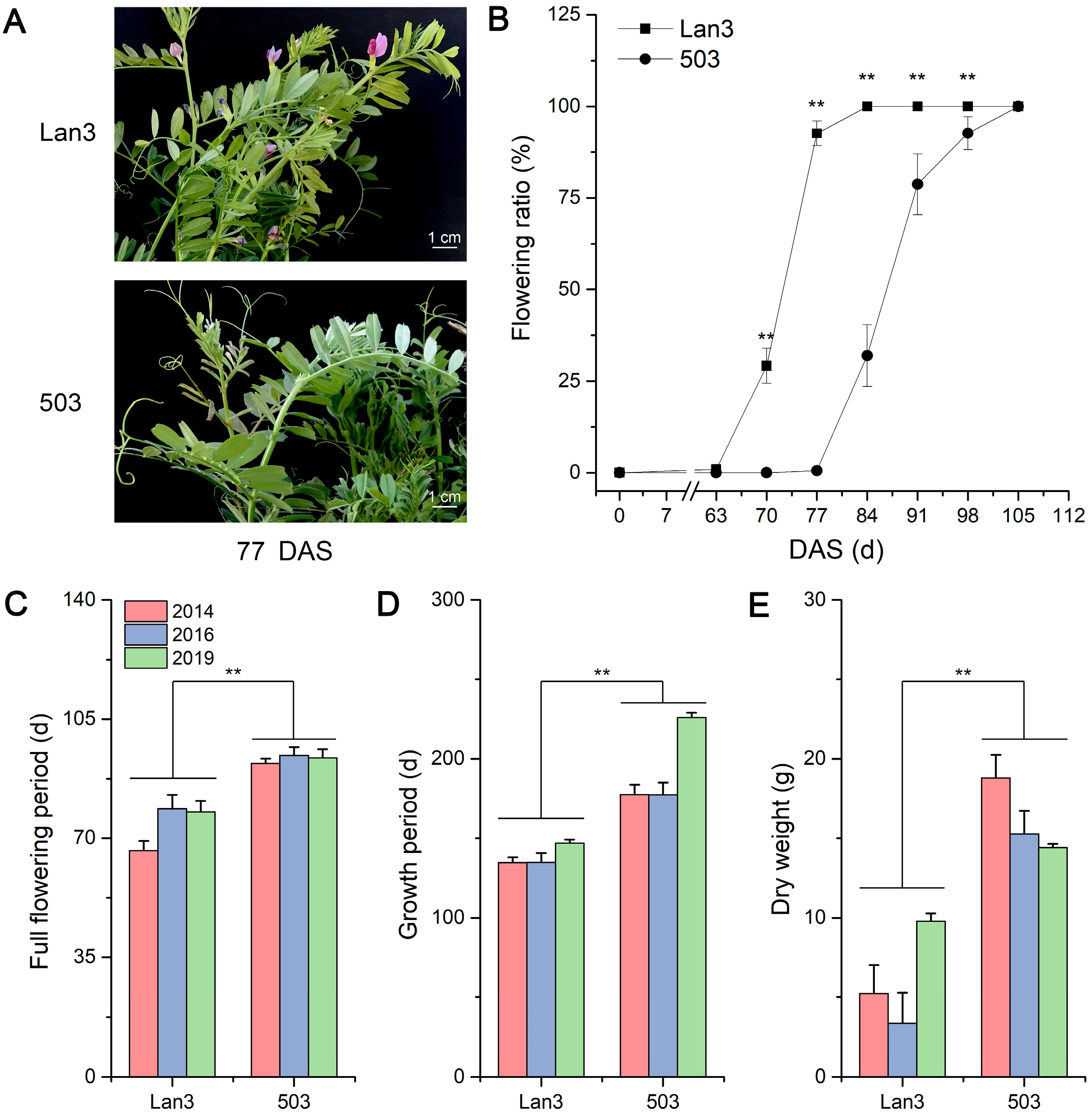
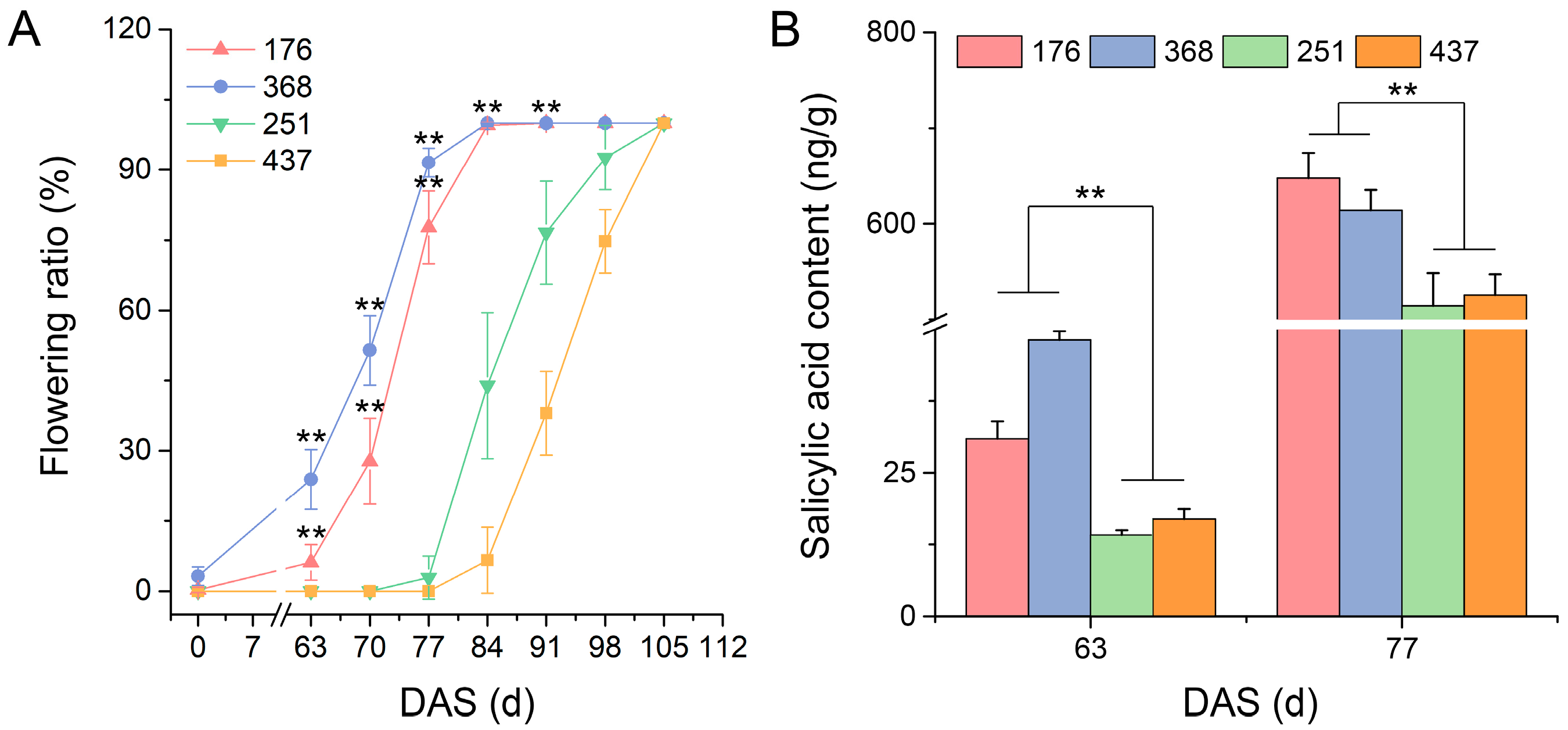
2.1. Flowering Dynamics and the Dry Weight of V. sativa
2.2. De Novo Assembly of Transcriptome and the Functional Annotation of Unigenes
2.3. qRT-PCR Verification
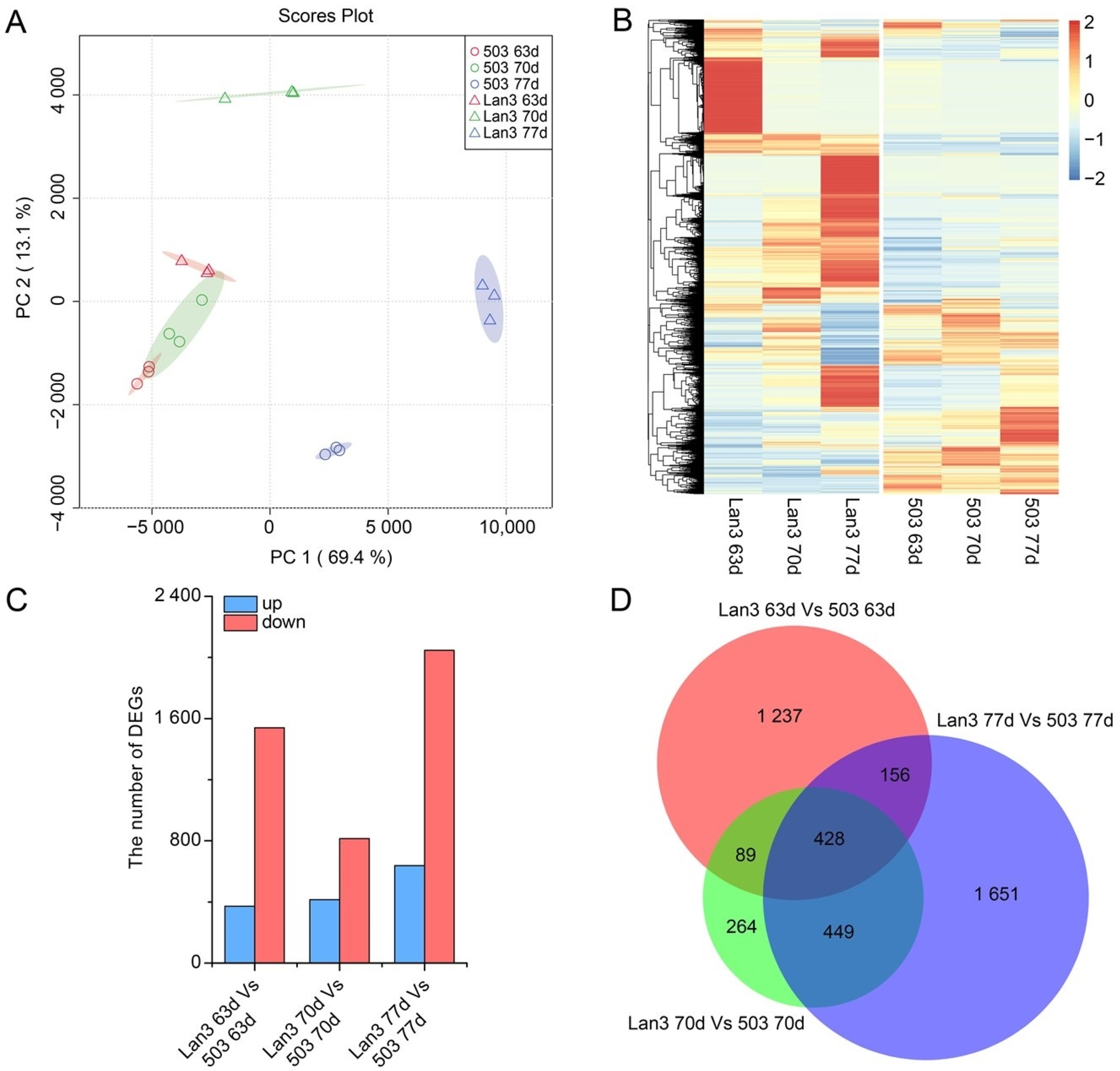
2.4. Identification and Expression Pattern Analysis of DEGs
2.5. GO Functional Analysis
2.6. Identification of TFs
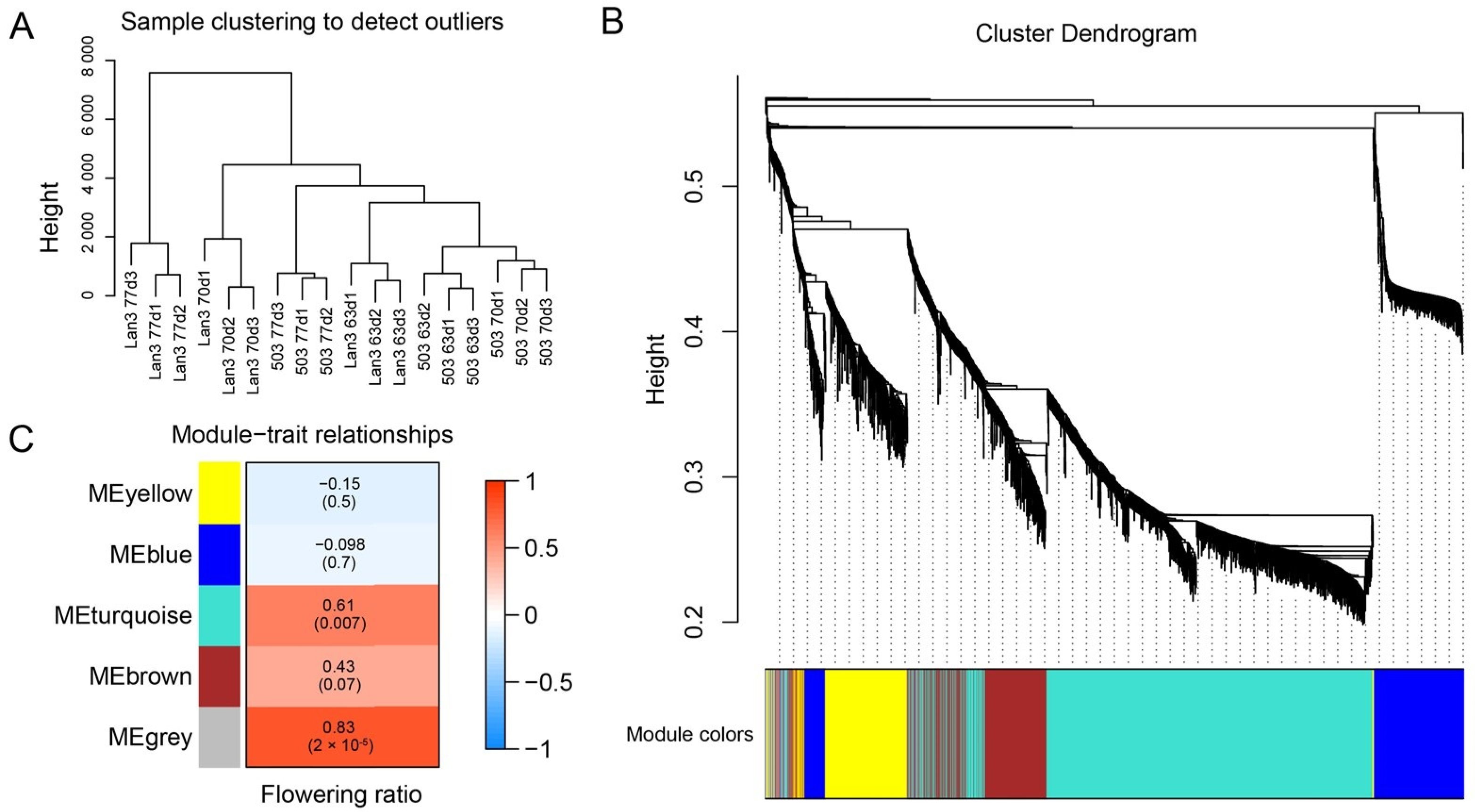
2.7. WGCNA of Flowering Regulation Genes
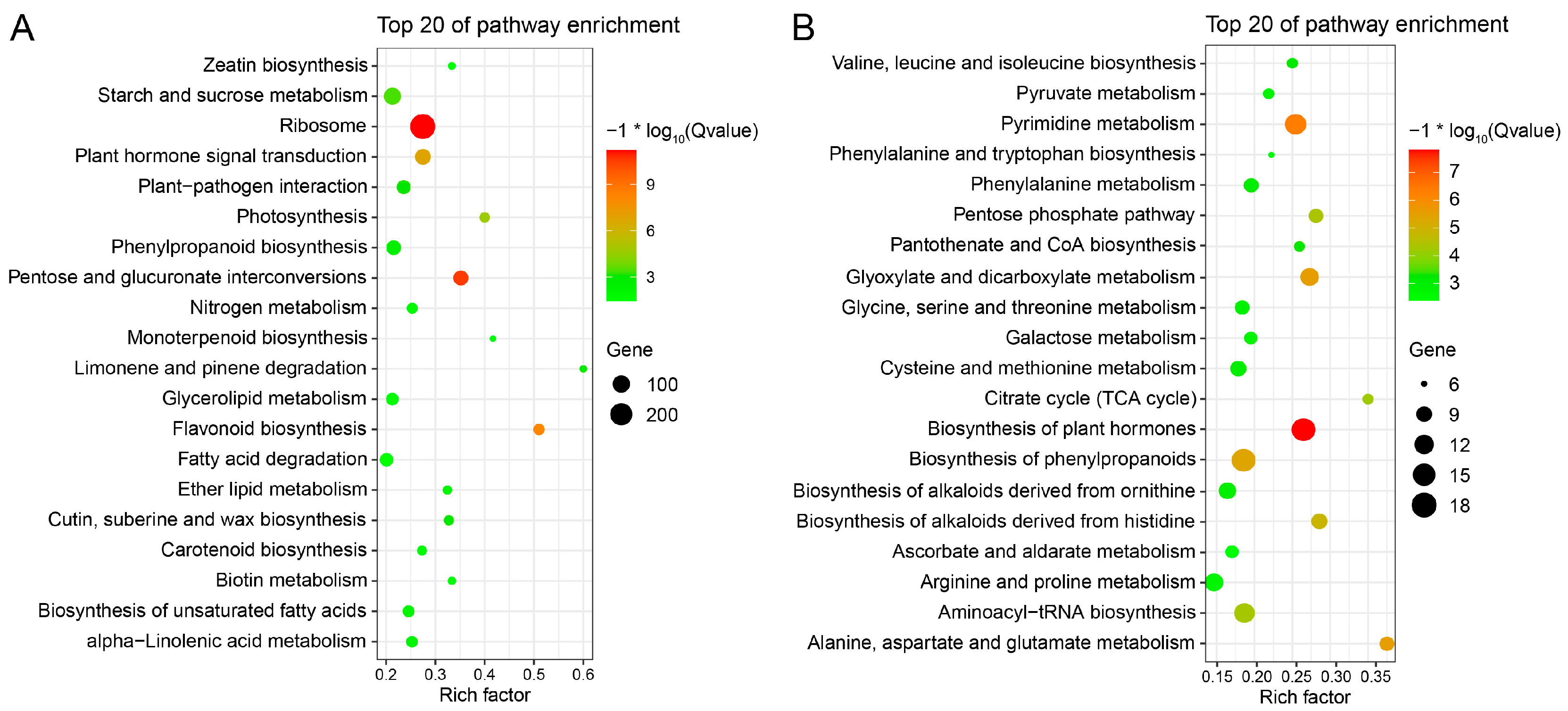
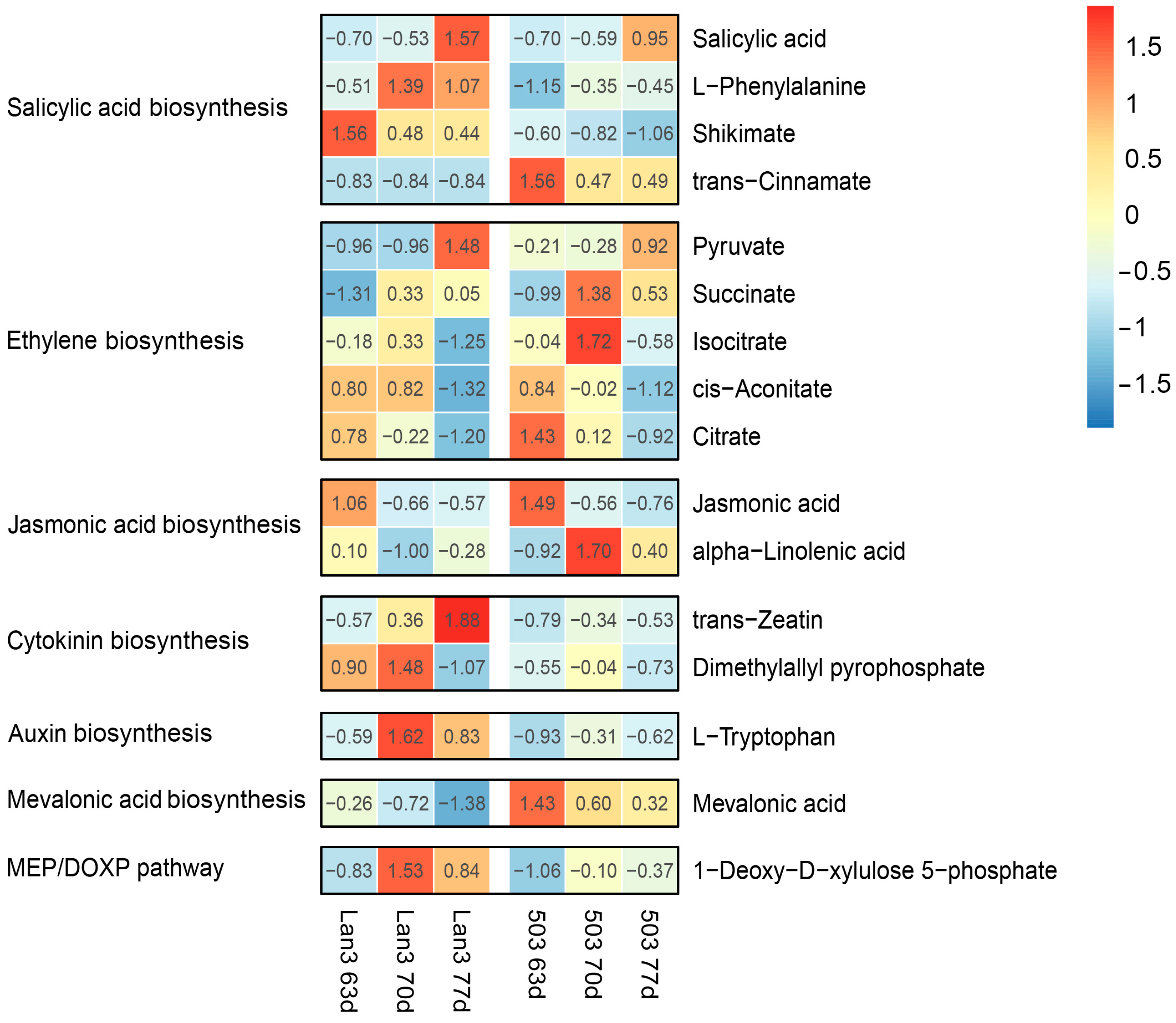
2.8. Metabolic Characteristics of the Early and Late Flowering Genotype
2.9. Integrative Analysis of DEGs and Differential Metabolites
2.10. Variation of Salicylic Acid Content in Various V. sativa Accessions
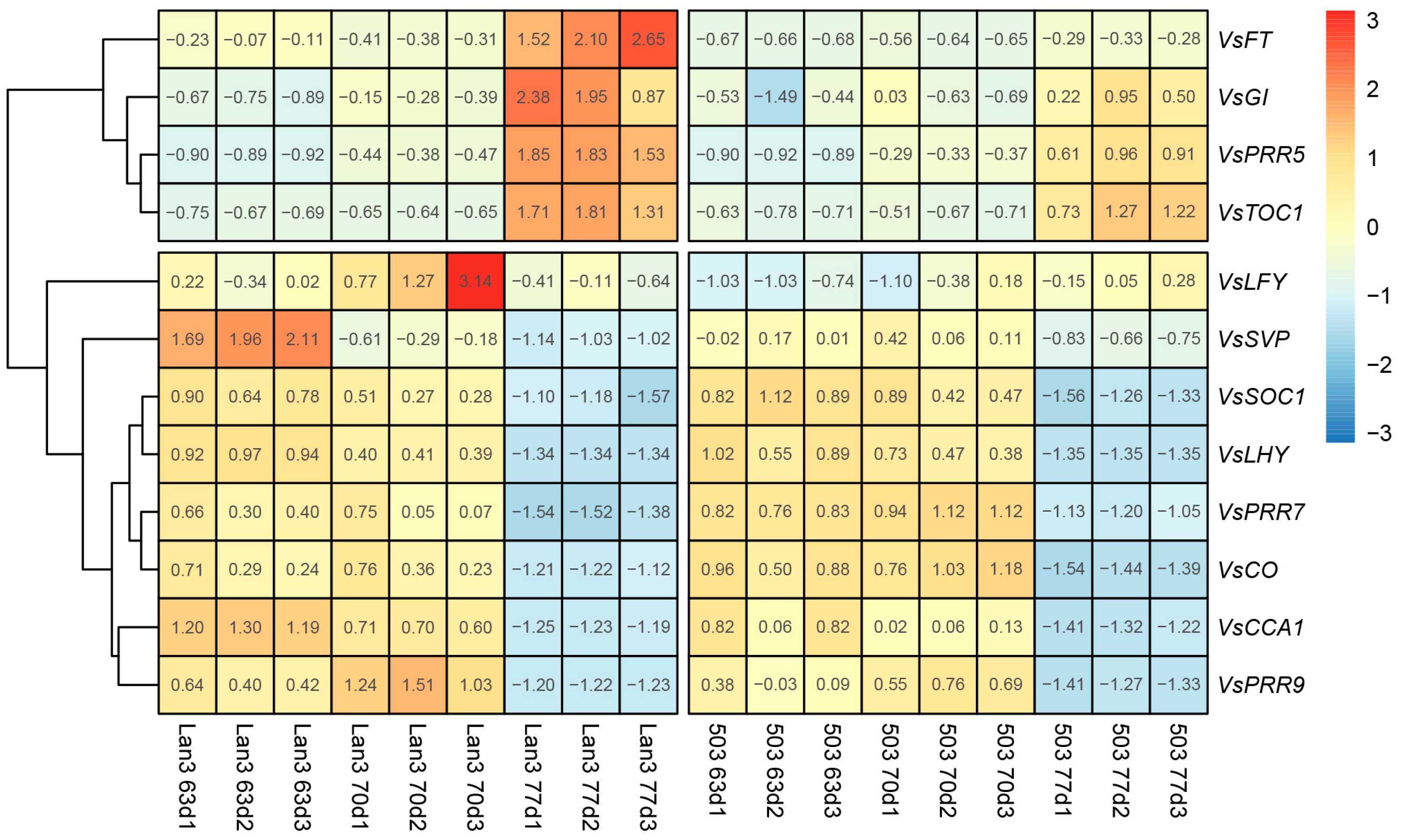
2.11. Flowering-Related Genes in V. sativa
3. Discussion
3.1. Flowering Time Affects the Yield of V. sativa
3.2. TFs Involved in the Flowering Process in V. sativa
3.3. Role of Plant Hormones in the Flowering Process in V. sativa
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. RNA Extraction and Library Construction for Transcriptome Analysis
4.3. Sequence Filtering, De Novo Assembly, and Functional Annotation
4.4. Quantitative Real-Time PCR Analysis
4.5. Differential Expression of Unigenes
4.6. DEG Analysis
4.7. Untargeted LC-MS
4.8. Determination of the Salicylic Acid Content
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, D.; Liu, B.; Ge, L.; Weng, Y.; Cao, X.; Liu, F.; Mao, P.; Ma, X. Identification and characterization of regulatory pathways involved in early flowering in the new leaves of alfalfa (Medicago sativa L.) by transcriptome analysis. BMC Plant Biol. 2021, 21, 8. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, A.; Jo, S.H.; Park, H.J.; Jung, W.Y.; Kim, H.S.; Lee, H.J.; Jeong, S.G.; Kim, Y.S.; Cho, H.S. Nitrogen signaling genes and SOC1 determine the flowering time in a reciprocal negative feedback loop in Chinese cabbage (Brassica rapa L.) based on CRISPR/Cas9-mediated mutagenesis of multiple BrSOC1 homologs. Int. J. Mol. Sci. 2021, 22, 4631. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Liu, Z.; Wang, Y.; Liu, W. Comparative transcriptomic analysis provides insights into the coordinated mechanisms of leaves and roots response to cold stress in common vetch. Ind. Crops Prod. 2020, 158, 112949. [Google Scholar] [CrossRef]
- Vicia sativa. Available online: https://plants.ces.ncsu.edu/plants/vicia-sativa/ (accessed on 11 May 2022).
- Li, R.; Zhang, Z.; Tang, W.; Huang, Y.; Nan, Z. Effect of row configuration on yield and radiation use of common vetch-oat strip intercropping on the Qinghai-Tibetan plateau. Eur. J. Agron. 2021, 128, 126290. [Google Scholar] [CrossRef]
- Mikić, A.; Mihailović, V.; Ćupina, B.; Milić, D.; Katić, S.; Karagić, Đ.; Pataki, I.; D’Ottavio, P.; Kraljević-Balalić, M. Forage yield components and classification of common vetch (Vicia sativa L.) cultivars of diverse geographic origin. Grass Forage Sci. 2014, 69, 315–322. [Google Scholar] [CrossRef]
- Cakmakci, S.; Aydinoglu, B.; Karaca, M.; Bilgen, M. Heritability of yield components in common vetch (Vicia sativa L.). Acta Agric. Scand. Sect. B-Soil Plant Sci. 2006, 56, 54–59. [Google Scholar]
- Larbi, A.; El-Moneim, A.M.A.; Nakkoul, H.; Jammal, B.; Hassan, S. Intra-species variations in yield and quality determinants in Vicia species: 3. Common vetch (Vicia sativa ssp. sativa L.). Anim. Feed. Sci. Technol. 2011, 164, 241–251. [Google Scholar] [CrossRef]
- Sánchez-Gutiérrez, R.A.; Figueroa-Gonzáles, J.J.; Rivera Vázquez, J.S.; Reveles-Hernández, M.; Gutierrez-Bañuelos, H.; Espinoza-Canales, A. Yield and nutritional value of common vetch (Vicia sativa L.) during fall-winter in Zacatecas, Mexico. Rev. Mex. Cienc. Pecu. 2020, 11, 294–303. [Google Scholar] [CrossRef]
- Fırıncıoğlu, H.K.; Ünal, S.; Erbektaş, E.; Doğruyol, L. Relationships between seed yield and yield components in common vetch (Vicia sativa ssp. sativa) populations sown in spring and autumn in central Turkey. Field Crops Res. 2010, 116, 30–37. [Google Scholar] [CrossRef]
- Hu, X.; Li, T.; Wang, J.; Wang, Y.; Baskin, C.C.; Baskin, J.M. Seed dormancy in four Tibetan Plateau Vicia species and characterization of physiological changes in response of seeds to environmental factors. Seed Sci. Res. 2013, 23, 133–140. [Google Scholar] [CrossRef]
- Park, I.W.; Ramirez-Parada, T.; Mazer, S.J. Advancing frost dates have reduced frost risk among most North American angiosperms since 1980. Glob. Chang. Biol. 2021, 27, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Pardee, G.L.; Inouye, D.W.; Irwin, R.E. Direct and indirect effects of episodic frost on plant growth and reproduction in subalpine wildflowers. Glob. Chang. Biol. 2018, 24, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Gruber, K.; Ladinig, U.; Buchner, O.; Neuner, G. Winter frosts reduce flower bud survival in high-mountain plants. Plants 2021, 10, 1507. [Google Scholar] [CrossRef]
- Neuner, G.; Erler, A.; Ladinig, U.; Hacker, J.; Wagner, J. Frost resistance of reproductive tissues during various stages of development in high mountain plants. Physiol. Plant. 2013, 147, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Kaya, O.; Kose, C. Cell death point in flower organs of some apricot (Prunus armeniaca L.) cultivars at subzero temperatures. Sci. Hortic. 2019, 249, 299–305. [Google Scholar] [CrossRef]
- Rowland, L.J.; Ogden, E.L.; Takeda, F.; Glenn, D.M.; Ehlenfeldt, M.K.; Vinyard, B.T. Variation among highbush blueberry cultivars for frost tolerance of open flowers. HortScience 2013, 48, 692–695. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, L.; Nan, Z.; Wang, Y. Comparative transcriptional profiling provides insights into the evolution and development of the zygomorphic flower of Vicia sativa (Papilionoideae). PLoS ONE 2013, 8, e57338. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Duan, W.; Huang, Z.; Liu, G.; Wu, P.; Liu, T.; Li, Y.; Hou, X. Comprehensive analysis of the flowering genes in Chinese cabbage and examination of evolutionary pattern of CO-like genes in plant kingdom. Sci. Rep. 2015, 5, 14631. [Google Scholar] [CrossRef]
- Zhang, X.; Meng, L.; Liu, B.; Hu, Y.; Cheng, F.; Liang, J.; Aarts, M.G.; Wang, X.; Wu, J. A transposon insertion in FLOWERING LOCUS T is associated with delayed flowering in Brassica rapa. Plant Sci. 2015, 241, 211–220. [Google Scholar] [CrossRef]
- Yang, X.; Liu, D.; Yan, Z.; Liu, C.; Feng, G. Regulation of flowering under short photoperiods based on transcriptomic and metabolomic analysis in Phaseolus vulgaris L. Mol. Genet. Genom. 2021, 296, 379–390. [Google Scholar] [CrossRef]
- Kong, F.; Nan, H.; Cao, D.; Li, Y.; Wu, F.; Wang, J.; Lu, S.; Yuan, X.; Cober, E.R.; Abe, J.; et al. A new dominant gene E9 conditions early flowering and maturity in soybean. Crop Sci. 2014, 54, 2529–2535. [Google Scholar] [CrossRef]
- Catt, S.C.; Paull, J.G. Effects of ambient temperature and photoperiod on flowering time in faba bean (Vicia faba L.). Crop Pasture Sci. 2017, 68, 893–901. [Google Scholar] [CrossRef]
- Iannucci, A.; Terribile, M.R.; Martiniello, P. Effects of temperature and photoperiod on flowering time of forage legumes in a Mediterranean environment. Field Crops Res. 2008, 106, 156–162. [Google Scholar] [CrossRef]
- Santner, A.; Calderon-Villalobos, L.I.; Estelle, M. Plant hormones are versatile chemical regulators of plant growth. Nat. Chem. Biol. 2009, 5, 301–307. [Google Scholar] [CrossRef]
- Wang, Y.H.; Irving, H.R. Developing a model of plant hormone interactions. Plant Signal. Behav. 2011, 6, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Campos-Rivero, G.; Osorio-Montalvo, P.; Sanchez-Borges, R.; Us-Camas, R.; Duarte-Ake, F.; De-la-Pena, C. Plant hormone signaling in flowering: An epigenetic point of view. J. Plant Physiol. 2017, 214, 16–27. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Pan, J.; Lou, D.; Hu, Y.; Yu, D. The bHLH transcription factors MYC2, MYC3, and MYC4 are required for jasmonate-mediated inhibition of flowering in Arabidopsis. Mol. Plant 2017, 10, 1461–1464. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Ye, T.; Lu, Y.; Chen, X.; Wu, Y. The inhibitory effect of ABA on floral transition is mediated by ABI5 in Arabidopsis. J. Exp. Bot. 2013, 64, 675–684. [Google Scholar] [CrossRef]
- Cheng, S.; Chen, P.; Su, Z.; Ma, L.; Hao, P.; Zhang, J.; Ma, Q.; Liu, G.; Liu, J.; Wang, H.; et al. High-resolution temporal dynamic transcriptome landscape reveals a GhCAL-mediated flowering regulatory pathway in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2021, 19, 153–166. [Google Scholar] [CrossRef]
- Hong, L.; Niu, F.; Lin, Y.; Wang, S.; Chen, L.; Jiang, L. MYB117 is a negative regulator of flowering time in Arabidopsis. Plant Signal. Behav. 2021, 16, e1901448. [Google Scholar] [CrossRef]
- Andres, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Yu, H. Florigen trafficking integrates photoperiod and temperature signals in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Doyle, M.R.; Sung, S.; Amasino, R.M. Vernalization: Winter and the timing of flowering in plants. Annu. Rev. Cell Dev. Biol. 2009, 25, 277–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.H.; Shim, J.S.; Kinmonth-Schultz, H.A.; Imaizumi, T. Photoperiodic flowering: Time measurement mechanisms in leaves. Annu. Rev. Plant Biol. 2015, 66, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Waseem, M.; Li, N.; Su, D.; Chen, J.; Li, Z. Overexpression of a basic helix-loop-helix transcription factor gene, SlbHLH22, promotes early flowering and accelerates fruit ripening in tomato (Solanum lycopersicum L.). Planta 2019, 250, 173–185. [Google Scholar] [CrossRef]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.; Yanofsky, M.F. Three ways to learn the ABCs. Curr. Opin. Plant Biol. 2000, 3, 47–52. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, L.; Song, J.; Jiang, L.; Liu, S. Isolation and characterization of a MADS-box gene in cucumber (Cucumis sativus L.) that affects flowering time and leaf morphology in transgenic Arabidopsis. Biotechnol. Biotechnol. Equip. 2019, 33, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Su, Q.; Chen, L.; Cai, Y.; Chen, Y.; Yuan, S.; Li, M.; Zhang, J.; Sun, S.; Han, T.; Hou, W. Functional redundancy of FLOWERING LOCUS T 3b in soybean flowering time regulation. Int. J. Mol. Sci. 2022, 23, 2497. [Google Scholar] [CrossRef]
- Fudge, J.B.; Lee, R.H.; Laurie, R.E.; Mysore, K.S.; Wen, J.; Weller, J.L.; Macknight, R.C. Medicago truncatula SOC1 genes are up-regulated by environmental cues that promote flowering. Front. Plant Sci. 2018, 9, 496. [Google Scholar] [CrossRef]
- Lorenzo, C.D.; Garcia-Gagliardi, P.; Antonietti, M.S.; Sanchez-Lamas, M.; Mancini, E.; Dezar, C.A.; Vazquez, M.; Watson, G.; Yanovsky, M.J.; Cerdan, P.D. Improvement of alfalfa forage quality and management through the down-regulation of MsFTa1. Plant Biotechnol. J. 2020, 18, 944–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gu, Y.; Gao, H.; Qiu, L.; Chang, R.; Chen, S.; He, C. Molecular and geographic evolutionary support for the essential role of GIGANTEAa in soybean domestication of flowering time. BMC Evol. Biol. 2016, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yan, H.; Zhang, D.; Yu, D. Ectopic expression of a soybean SVP-like gene in tobacco causes abnormal floral organs and shortens the vegetative phase. Plant Growth Regul. 2016, 80, 345–353. [Google Scholar] [CrossRef]
- Zhang, T.; Chao, Y.; Kang, J.; Ding, W.; Yang, Q. Molecular cloning and characterization of a gene regulating flowering time from alfalfa (Medicago sativa L.). Mol. Biol. Rep. 2013, 40, 4597–4603. [Google Scholar] [CrossRef]
- Jaudal, M.; Monash, J.; Zhang, L.; Wen, J.; Mysore, K.S.; Macknight, R.; Putterill, J. Overexpression of Medicago SVP genes causes floral defects and delayed flowering in Arabidopsis but only affects floral development in Medicago. J. Exp. Bot. 2014, 65, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Hu, T.; Zhang, W.; Zhou, L.; Wang, Y.; Jiang, Z. Transcriptome profiling for floral development in reblooming cultivar ‘High Noon’ of Paeonia suffruticosa. Sci. Data 2019, 6, 217. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhang, Y.; Zhen, X.; Guo, D.; Guo, C.; Shu, Y. Transcriptome sequencing and expression profiling of genes involved in daylily (Hemerocallis citrina Borani) flower development. Biotechnol. Biotechnol. Equip. 2020, 34, 542–548. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics-the link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- KEGG Database. Available online: https://www.genome.jp/kegg/ (accessed on 13 May 2022).
- Tadege, M.; Chen, F.; Murray, J.; Wen, J.; Ratet, P.; Udvardi, M.K.; Dixon, R.A.; Mysore, K.S. Control of vegetative to reproductive phase transition improves biomass yield and simultaneously reduces lignin content in Medicago truncatula. BioEnergy Res. 2014, 8, 857–867. [Google Scholar] [CrossRef]
- Casler, M.D. Selection for flowering time as a mechanism to increase biomass yield of upland switchgrass. BioEnergy Res. 2019, 13, 100–108. [Google Scholar] [CrossRef]
- Mallikarjuna, B.P.; Viswanatha, K.P.; Samineni, S.; Gaur, P.M. Association of flowering time with phenological and productivity traits in chickpea. Euphytica 2019, 215, 77. [Google Scholar] [CrossRef]
- Nakamichi, N.; Takao, S.; Kudo, T.; Kiba, T.; Wang, Y.; Kinoshita, T.; Sakakibara, H. Improvement of Arabidopsis biomass and cold, drought and salinity stress tolerance by modified circadian clock-associated Pseudo-Response Regulators. Plant Cell Physiol. 2016, 57, 1085–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Jia, C.; Ma, W.; Cui, Y.; Jin, X.; Luo, D.; Min, X.; Liu, Z. MYB transcription factors in alfalfa (Medicago sativa): Genome-wide identification and expression analysis under abiotic stresses. PeerJ 2019, 7, e7714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liu, G.; Jia, J.; Zhao, G.; Xia, C.; Zhang, L.; Li, F.; Zhang, Q.; Dong, C.; Gao, S.; et al. The wheat MYB-related transcription factor TaMYB72 promotes flowering in rice. J. Integr. Plant Biol. 2016, 58, 701–704. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Guan, Y.; Liu, Y.; Zhang, Z.; Jaffar, M.A.; Song, A.; Chen, S.; Jiang, J.; Chen, F. Regulation of flowering time in chrysanthemum by the R2R3 MYB transcription factor CmMYB2 is associated with changes in gibberellin metabolism. Hortic. Res. 2020, 7, 96. [Google Scholar] [CrossRef]
- Aslam, M.; Jakada, B.H.; Fakher, B.; Greaves, J.G.; Niu, X.; Su, Z.; Cheng, Y.; Cao, S.; Wang, X.; Qin, Y. Genome-wide study of pineapple (Ananas comosus L.) bHLH transcription factors indicates that cryptochrome-interacting bHLH2 (AcCIB2) participates in flowering time regulation and abiotic stress response. BMC Genom. 2020, 21, 735. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, D.; Lin, X.; Ding, M.; Tong, Z. Genome-wide identification of MADS-box family genes in moso bamboo (Phyllostachys edulis) and a functional analysis of PeMADS5 in flowering. BMC Plant Biol. 2018, 18, 176. [Google Scholar] [CrossRef]
- Li, Y.H.; Zhang, W.; Li, Y. Transcriptomic analysis of flower blooming in Jasminum sambac through de novo RNA sequencing. Molecules 2015, 20, 10734–10747. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, Y.; Zhang, Y.; Zhao, T.; Jiang, J.; Li, J.; Xu, X.; Yang, H. Transcriptome analysis of flower development and mining of genes related to flowering time in tomato (Solanum lycopersicum). Int. J. Mol. Sci. 2021, 22, 8128. [Google Scholar] [CrossRef]
- Liang, Y.C.; Reid, M.S.; Jiang, C.Z. Controlling plant architecture by manipulation of gibberellic acid signalling in petunia. Hortic. Res. 2014, 1, 14061. [Google Scholar] [CrossRef] [Green Version]
- Martín-Mex, R.; Nexticapan-Garcéz, Á.; Villanueva-Couoh, E.; Uicab-Quijano, V.; Vergara-Yoisura, S.; Larqué-Saavedra, A. Salicylic acid stimulates flowering in micropopagated gloxinia plants. Rev. Fitotec. Mex. 2015, 38, 115–118. [Google Scholar] [CrossRef]
- Fu, L.; Tan, D.; Sun, X.; Ding, Z.; Zhang, J. Transcriptional analysis reveals potential genes and regulatory networks involved in salicylic acid-induced flowering in duckweed (Lemna gibba). Plant Physiol. Biochem. 2020, 155, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.K.A. Effect of ascorbic and salicylic acids on growth and flowering of Gazania cv. Frosty Kiss Mixed. Ornam. Hortic. 2020, 26, 537–544. [Google Scholar] [CrossRef]
- Murmu, J.; Bush, M.J.; DeLong, C.; Li, S.; Xu, M.; Khan, M.; Malcolmson, C.; Fobert, P.R.; Zachgo, S.; Hepworth, S.R. Arabidopsis basic leucine-zipper transcription factors TGA9 and TGA10 interact with floral glutaredoxins ROXY1 and ROXY2 and are redundantly required for anther development. Plant Physiol. 2010, 154, 1492–1504. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Xu, J.; Yuan, C.; Hu, Y.; Liu, Q.; Chen, Q.; Zhang, P.; Shi, N.; Qin, C. Characterization of genes associated with TGA7 during the floral transition. BMC Plant Biol. 2021, 21, 367. [Google Scholar] [CrossRef]
- Van Loon, L.C.; Rep, M.; Pieterse, C.M. Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 2006, 44, 135–162. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- AgriGO 2.0. Available online: http://systemsbiology.cau.edu.cn/agriGOv2/ (accessed on 1 February 2022).
- KOBAS 3.0. Available online: http://kobas.cbi.pku.edu.cn/ (accessed on 16 February 2022).
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- PlntfDB Database. Available online: http://plntfdb.bio.uni-potsdam.de/v3.0/ (accessed on 16 October 2021).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Zhang, L.; Niu, D.; Nan, S.; Miao, X.; Hu, X.; Li, C.; Fu, H. Transcriptome and co-expression network analysis reveal molecular mechanisms of mucilage formation during seed development in Artemisia sphaerocephala. Carbohyd. Polym. 2021, 251, 117044. [Google Scholar] [CrossRef]
- MetaboAnalyst 5.0. Available online: https://www.metaboanalyst.ca/MetaboAnalyst/ModuleView.xhtml (accessed on 10 May 2022).
- MBRole 2.0. Available online: http://csbg.cnb.csic.es/mbrole2/analysis.php (accessed on 20 March 2022).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Cui, Y.; Dong, R.; Luo, D.; Fang, L.; Nan, Z.; Liu, Z. Integrative Analyses of Transcriptomes and Metabolomes Reveal Associated Genes and Metabolites with Flowering Regulation in Common Vetch (Vicia sativa L.). Int. J. Mol. Sci. 2022, 23, 6818. https://doi.org/10.3390/ijms23126818
Zhou Q, Cui Y, Dong R, Luo D, Fang L, Nan Z, Liu Z. Integrative Analyses of Transcriptomes and Metabolomes Reveal Associated Genes and Metabolites with Flowering Regulation in Common Vetch (Vicia sativa L.). International Journal of Molecular Sciences. 2022; 23(12):6818. https://doi.org/10.3390/ijms23126818
Chicago/Turabian StyleZhou, Qiang, Yue Cui, Rui Dong, Dong Luo, Longfa Fang, Zhibiao Nan, and Zhipeng Liu. 2022. "Integrative Analyses of Transcriptomes and Metabolomes Reveal Associated Genes and Metabolites with Flowering Regulation in Common Vetch (Vicia sativa L.)" International Journal of Molecular Sciences 23, no. 12: 6818. https://doi.org/10.3390/ijms23126818