
Codon Usage Provides Insights into the Adaptive Evolution of Mycoviruses in Their Associated Fungi Host

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Analysis of Mycoviral RP Genes Nucleotide Composition
2.2. Overall CUB among Mycoviral RP Genes
2.3. The Contribution of Mutation Pressure and Natural Selection in Forming Mycoviral CUB
2.4. The Relative Abundance of Dinucleotide among Mycoviral RP Genes
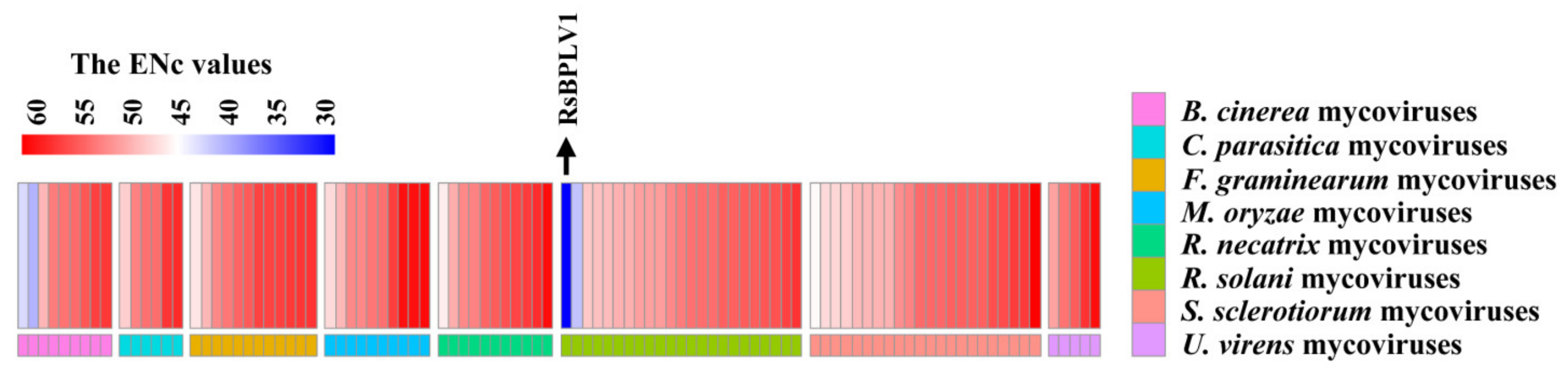
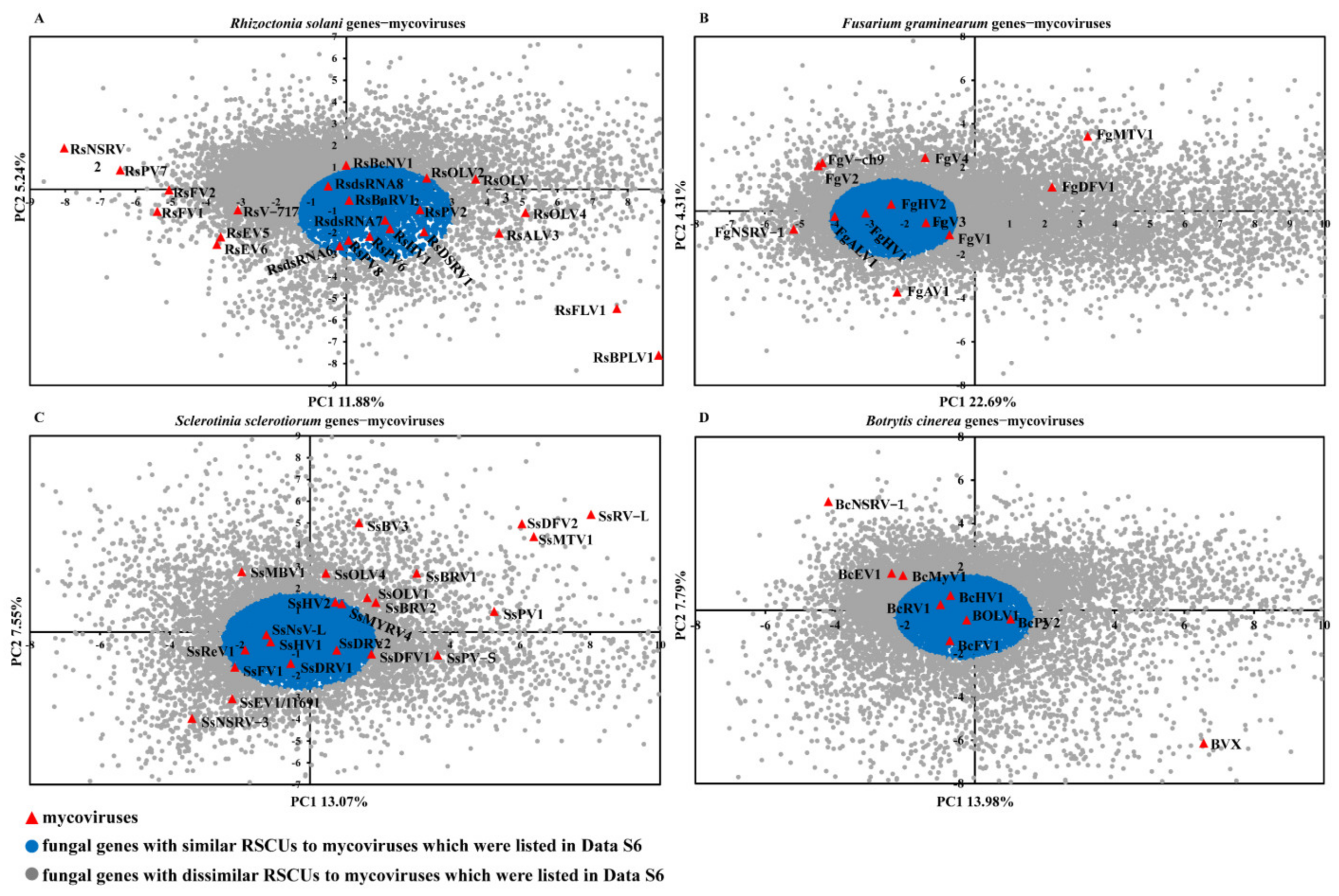
2.5. The Similarity of Codon Usage between Mycoviruses and Their Fungi Host
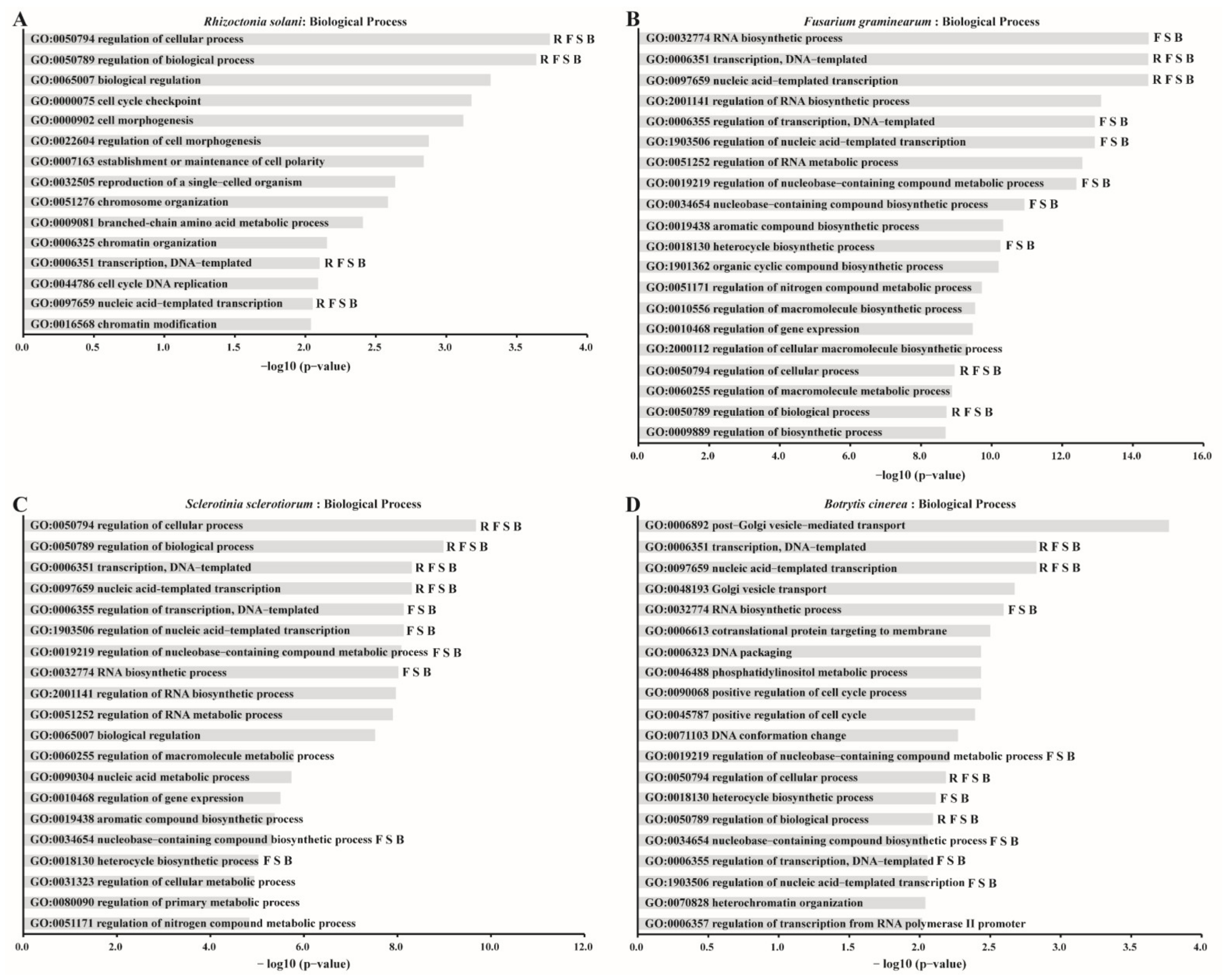
2.6. The Expression of Fungal Genes That Have Similar Codon Usage Pattern to Mycoviruses Response to Mycoviruses Infections
3. Discussion
4. Materials and Methods
4.1. Sequence Data Acquisition
4.2. The Effective Number of Codons Analysis
4.3. ENc-GC3s Plot Analysis
4.4. Parity Rule 2 (PR2) Analysis
4.5. Neutrality Plot (GC12 vs. GC3) Analysis
4.6. Relative Dinucleotide Abundance Analysis
4.7. Principal Component Analysis of RSCUs
4.8. Gene Ontology Enrichment Analysis
4.9. Correlation Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grantham, R.; Gautier, C.; Gouy, M.; Mercier, R.; Pave, A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980, 8, r49–r62. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoletzki, N.; Eyre-Walker, A. Synonymous codon usage in Escherichia coli: Selection for translational accuracy. Mol. Biol. Evol. 2007, 24, 374–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuller, T.; Waldman, Y.Y.; Kupiec, M.; Ruppin, E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. USA 2010, 107, 3645–3650. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Matassi, G. Codon usage and genome evolution. Curr. Opin. Genet. Dev. 1994, 4, 851–860. [Google Scholar] [CrossRef]
- Kane, J.F. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr. Opin. Biotechnol. 1995, 6, 494–500. [Google Scholar] [CrossRef]
- Gupta, S.K.; Bhattacharyya, T.K.; Ghosh, T.C. Synonymous codon usage in Lactococcus lactis: Mutational bias versus translational selection. J. Biomol. Struct. Dyn. 2004, 21, 527–536. [Google Scholar] [CrossRef]
- Naya, H.; Romero, H.; Carels, N.; Zavala, A.; Musto, H. Translational selection shapes codon usage in the GC-rich genome of Chlamydomonas reinhardtii. FEBS Lett. 2001, 501, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Emery, L.R.; Zeng, K. Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Stenico, M.; Peden, J.F.; Lloyd, A.T. Codon usage: Mutational bias, translational selection, or both? Biochem. Soc. Trans. 1993, 21, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Bibb, M.J.; Findlay, P.R.; Johnson, M.W. The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene 1984, 30, 157–166. [Google Scholar] [CrossRef]
- Gouy, M.; Gautier, C. Codon usage in bacteria: Correlation with gene expressivity. Nucleic Acids Res. 1982, 10, 7055–7074. [Google Scholar] [CrossRef]
- Eyre-Walker, A. Synonymous codon bias is related to gene length in Escherichia coli: Selection for translational accuracy? Mol. Biol. Evol. 1996, 13, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Dafu, D. The relationship between synonymous codon usage and protein structure. FEBS Lett. 1998, 434, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Duret, L. tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet. 2000, 16, 287–289. [Google Scholar] [CrossRef]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar]
- Adams, M.J.; Antoniw, J.F. Codon usage bias amongst plant viruses. Arch. Virol. 2004, 149, 113–135. [Google Scholar]
- Biswas, K.K.; Palchoudhury, S.; Chakraborty, P.; Bhattacharyya, U.K.; Ghosh, D.K.; Debnath, P.; Ramadugu, C.; Keremane, M.L.; Khetarpal, R.K.; Lee, R.F. Codon usage bias analysis of citrus tristeza virus: Higher codon adaptation to citrus reticulata host. Viruses 2019, 11, 331. [Google Scholar] [CrossRef] [Green Version]
- Franzo, G.; Tucciarone, C.M.; Cecchinato, M.; Drigo, M. Canine parvovirus type 2 (CPV-2) and Feline panleukopenia virus (FPV) codon bias analysis reveals a progressive adaptation to the new niche after the host jump. Mol. Phylogenet. Evol. 2017, 114, 82–92. [Google Scholar] [CrossRef]
- Jiang, Y.; Deng, F.; Wang, H.; Hu, Z. An extensive analysis on the global codon usage pattern of baculoviruses. Arch. Virol. 2008, 153, 2273–2282. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Pandit, A.; Sinha, S. Differential trends in the codon usage patterns in HIV-1 genes. PLoS ONE 2011, 6, e28889. [Google Scholar] [CrossRef]
- Bahir, I.; Fromer, M.; Prat, Y.; Linial, M. Viral adaptation to host: A proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009, 5, 311. [Google Scholar] [CrossRef]
- Burns, C.C.; Shaw, J.; Campagnoli, R.; Jorba, J.; Vincent, A.; Quay, J.; Kew, O. Modulation of poliovirus replicative fitness in HeLa cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 2006, 80, 3259–3272. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, e107. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The adaptation of codon usage of +ssRNA viruses to their hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef]
- Costafreda, M.I.; Perez-Rodriguez, F.J.; D’Andrea, L.; Guix, S.; Ribes, E.; Bosch, A.; Pinto, R.M. Hepatitis A virus adaptation to cellular shutoff is driven by dynamic adjustments of codon usage and results in the selection of populations with altered capsids. J. Virol. 2014, 88, 5029–5041. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.U.; Yao, X.; Li, X.; Chen, D.; Tao, S. Analysis of codon usage bias of Crimean-Congo hemorrhagic fever virus and its adaptation to hosts. Infect. Genet. Evol. 2018, 58, 1–16. [Google Scholar] [CrossRef]
- Ghabrial, S.A.; Caston, J.R.; Jiang, D.; Nibert, M.L.; Suzuki, N. 50-plus years of fungal viruses. Virology 2015, 479–480, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Kast, A.; Voges, R.; Schroth, M.; Schaffrath, R.; Klassen, R.; Meinhardt, F. Autoselection of cytoplasmic yeast virus like elements encoding toxin/antitoxin systems involves a nuclear barrier for immunity gene expression. PLoS Genet. 2015, 11, e1005005. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. The good viruses: Viral mutualistic symbioses. Nat. Rev. Microbiol. 2011, 9, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Je, M.; Kim, H.; Son, H.S. Analysis of the codon usage pattern of the RdRP gene of mycovirus infecting Aspergillus spp. Virol. J. 2019, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Je, M.; Kim, H.; Cho, M.; Son, H.S. Characterization of codon usage patterns and evolutionary relationships in Partitiviruses. In Proceedings of the 2019 3rd International Conference on Computational Biology and Bioinformatics (ICCBB’19), Nagoya, Japan, 17–19 October 2019; Association for Computing Machinery: New York, NY, USA, 2019; pp. 33–39. [Google Scholar]
- Simón, D.; Cristina, J.; Musto, H. Nucleotide composition and codon usage across viruses and their respective hosts. Front. Microbiol. 2021, 12, 646300. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Cheng, X.; Virk, N.; Chen, W.; Ji, S.; Ji, S.; Sun, Y.; Wu, X. CpG usage in RNA viruses: Data and hypotheses. PLoS ONE 2013, 8, e74109. [Google Scholar] [CrossRef] [Green Version]
- Lytras, S.; Hughes, J. Synonymous dinucleotide usage: A codon-aware metric for quantifying dinucleotide representation in viruses. Viruses 2020, 12, 462. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, S.; Zhang, B.; Wei, W. Analysis of synonymous codon usage bias of zika virus and its adaption to the hosts. PLoS ONE 2016, 11, e0166260. [Google Scholar] [CrossRef] [Green Version]
- Abdoulaye, A.H.; Foda, M.F.; Kotta-Loizou, I. Viruses infecting the plant pathogenic fungus Rhizoctonia solani. Viruses 2019, 11, 1113. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Bhattacharjee, P.; Wang, S.; Zhang, L.; Ahmed, I.; Guo, L. Mycoviruses in Fusarium species: An Update. Front. Cell Infect. Microbiol. 2019, 9, 257. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Fu, Y.; Li, G.; Ghabrial, S.A. Viruses of the plant pathogenic fungus Sclerotinia sclerotiorum. Adv. Virus Res. 2013, 86, 215–248. [Google Scholar]
- Pearson, M.N.; Bailey, A.M. Viruses of botrytis. Adv. Virus Res. 2013, 86, 249–272. [Google Scholar]
- Allen, T.D.; Nuss, D.L. Specific and common alterations in host gene transcript accumulation following infection of the chestnut blight fungus by mild and severe hypoviruses. J. Virol. 2004, 78, 4145–4155. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Cho, W.K.; Yu, J.; Son, M.; Choi, H.; Min, K.; Lee, Y.W.; Kim, K.H. A comparison of transcriptional patterns and mycological phenotypes following infection of Fusarium graminearum by four mycoviruses. PLoS ONE 2014, 9, e100989. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Li, P.; Qiu, D.; Guo, L. Transcriptome-based discovery of Fusarium graminearum stress responses to FgHV1 infection. Int. J. Mol. Sci. 2016, 17, 1922. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Bera, B.C.; Greenbaum, B.D.; Bhatia, S.; Sood, R.; Selvaraj, P.; Anand, T.; Tripathi, B.N.; Virmani, N. Revelation of influencing factors in overall codon usage bias of equine influenza viruses. PLoS ONE 2016, 11, e0154376. [Google Scholar] [CrossRef] [Green Version]
- Cristina, J.; Moreno, P.; Moratorio, G.; Musto, H. Genome-wide analysis of codon usage bias in Ebolavirus. Virus Res. 2015, 196, 87–93. [Google Scholar] [CrossRef]
- Kandeel, M.; Ibrahim, A.; Fayez, M.; Al-Nazawi, M. From SARS and MERS CoVs to SARS-CoV-2: Moving toward more biased codon usage in viral structural and nonstructural genes. J. Med. Virol. 2020, 92, 660–666. [Google Scholar] [CrossRef]
- Beutler, E.; Gelbart, T.; Han, J.H.; Koziol, J.A.; Beutler, B. Evolution of the genome and the genetic code: Selection at the dinucleotide level by methylation and polyribonucleotide cleavage. Proc. Natl. Acad. Sci. USA 1989, 86, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Breslauer, K.J.; Frank, R.; Blocker, H.; Marky, L.A. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 3746–3750. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, N.J.; Witteveldt, J.; Evans, D.J.; Simmonds, P. The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucleic Acids Res. 2014, 42, 4527–4545. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, G.; Bosch, A.; Pinto, R.M. Genome variability and capsid structural constraints of hepatitis a virus. J. Virol. 2003, 77, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; Nasrullah, I.; Tong, Y. Genome-wide analysis of codon usage and influencing factors in chikungunya viruses. PLoS ONE 2014, 9, e90905. [Google Scholar] [CrossRef] [Green Version]
- Jitobaom, K.; Phakaratsakul, S.; Sirihongthong, T.; Chotewutmontri, S.; Suriyaphol, P.; Suptawiwat, O.; Auewarakul, P. Codon usage similarity between viral and some host genes suggests a codon-specific translational regulation. Heliyon 2020, 6, e03915. [Google Scholar] [CrossRef]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.R. Dissimilation of synonymous codon usage bias in virus-host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef]
- Wu, S.; Cheng, J.; Fu, Y.; Chen, T.; Jiang, D.; Ghabrial, S.A.; Xie, J. Virus-mediated suppression of host non-self recognition facilitates horizontal transmission of heterologous viruses. PLoS Pathog. 2017, 13, e1006234. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Kondo, H.; Liu, L.; Guo, L.; Qiu, D. A novel virus in the family Hypoviridae from the plant pathogenic fungus Fusarium graminearum. Virus Res. 2013, 174, 69–77. [Google Scholar] [CrossRef]
- Kashif, M.; Jurvansuu, J.; Hyder, R.; Vainio, E.J.; Hantula, J. Phenotypic recovery of a Heterobasidion isolate infected by a debilitation-associated virus is related to altered host gene expression and reduced virus titer. Front. Microbiol. 2021, 12, 661554. [Google Scholar] [CrossRef]
- Lemus-Minor, C.G.; Cañizares, M.C.; García-Pedrajas, M.D.; Pérez-Artés, E. Fusarium oxysporum f. sp. dianthi virus 1 accumulation is correlated with changes in virulence and other phenotypic traits of its fungal host. Phytopathology 2018, 108, 957–963. [Google Scholar] [CrossRef]
- Uchida, K.; Sakuta, K.; Ito, A.; Takahashi, Y.; Katayama, Y.; Omatsu, T.; Mizutani, T.; Arie, T.; Komatsu, K.; Fukuhara, T.; et al. Two novel endornaviruses co-infecting a phytophthora pathogen of Asparagus officinalis modulate the developmental stages and fungicide sensitivities of the host oomycete. Front. Microbiol. 2021, 12, 633502. [Google Scholar] [CrossRef]
- Sharp, P.M.; Cowe, E. Synonymous codon usage in Saccharomyces cerevisiae. Yeast 1991, 7, 657–678. [Google Scholar] [CrossRef] [PubMed]
- Comeron, J.M.; Aguade, M. An evaluation of measures of synonymous codon usage bias. J. Mol. Evol. 1998, 47, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Karlin, S.; Doerfler, W.; Cardon, L.R. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 1994, 68, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycovirus | GO Term | Description |
|---|---|---|
| SsMYRV4 | GO:0006725 | cellular aromatic compound metabolic process |
| GO:0044260 | cellular macromolecule metabolic process | |
| GO:0046483 | heterocycle metabolic process | |
| GO:0090304 | nucleic acid metabolic process | |
| GO:0006139 | nucleobase-containing compound metabolic process | |
| GO:1901360 | organic cyclic compound metabolic process | |
| GO:0016070 | RNA metabolic process | |
| GO:0006396 | RNA processing | |
| FgV2 | GO:0019438 | aromatic compound biosynthetic process |
| GO:0018130 | heterocycle biosynthetic process | |
| GO:0097659 | nucleic acid-templated transcription | |
| GO:0034654 | nucleobase-containing compound biosynthetic process | |
| GO:1901362 | organic cyclic compound biosynthetic process | |
| GO:2000112 | regulation of cellular macromolecule biosynthetic process | |
| GO:0010556 | regulation of macromolecule biosynthetic process | |
| GO:1903506 | regulation of nucleic acid-templated transcription | |
| GO:0019219 | regulation of nucleobase-containing compound metabolic process | |
| GO:2001141 | regulation of RNA biosynthetic process | |
| GO:0051252 | regulation of RNA metabolic process | |
| GO:0006355 | regulation of transcription, DNA-templated | |
| GO:0032774 | RNA biosynthetic process | |
| GO:0006351 | transcription, DNA-templated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Lyu, X.; Cheng, J.; Fu, Y.; Lin, Y.; Abdoulaye, A.H.; Jiang, D.; Xie, J. Codon Usage Provides Insights into the Adaptive Evolution of Mycoviruses in Their Associated Fungi Host. Int. J. Mol. Sci. 2022, 23, 7441. https://doi.org/10.3390/ijms23137441
Wang Q, Lyu X, Cheng J, Fu Y, Lin Y, Abdoulaye AH, Jiang D, Xie J. Codon Usage Provides Insights into the Adaptive Evolution of Mycoviruses in Their Associated Fungi Host. International Journal of Molecular Sciences. 2022; 23(13):7441. https://doi.org/10.3390/ijms23137441
Chicago/Turabian StyleWang, Qianqian, Xueliang Lyu, Jiasen Cheng, Yanping Fu, Yang Lin, Assane Hamidou Abdoulaye, Daohong Jiang, and Jiatao Xie. 2022. "Codon Usage Provides Insights into the Adaptive Evolution of Mycoviruses in Their Associated Fungi Host" International Journal of Molecular Sciences 23, no. 13: 7441. https://doi.org/10.3390/ijms23137441