
Renin-Angiotensin System in Huntington′s Disease: Evidence from Animal Models and Human Patients

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Motor and Behavioral Tests in Mice
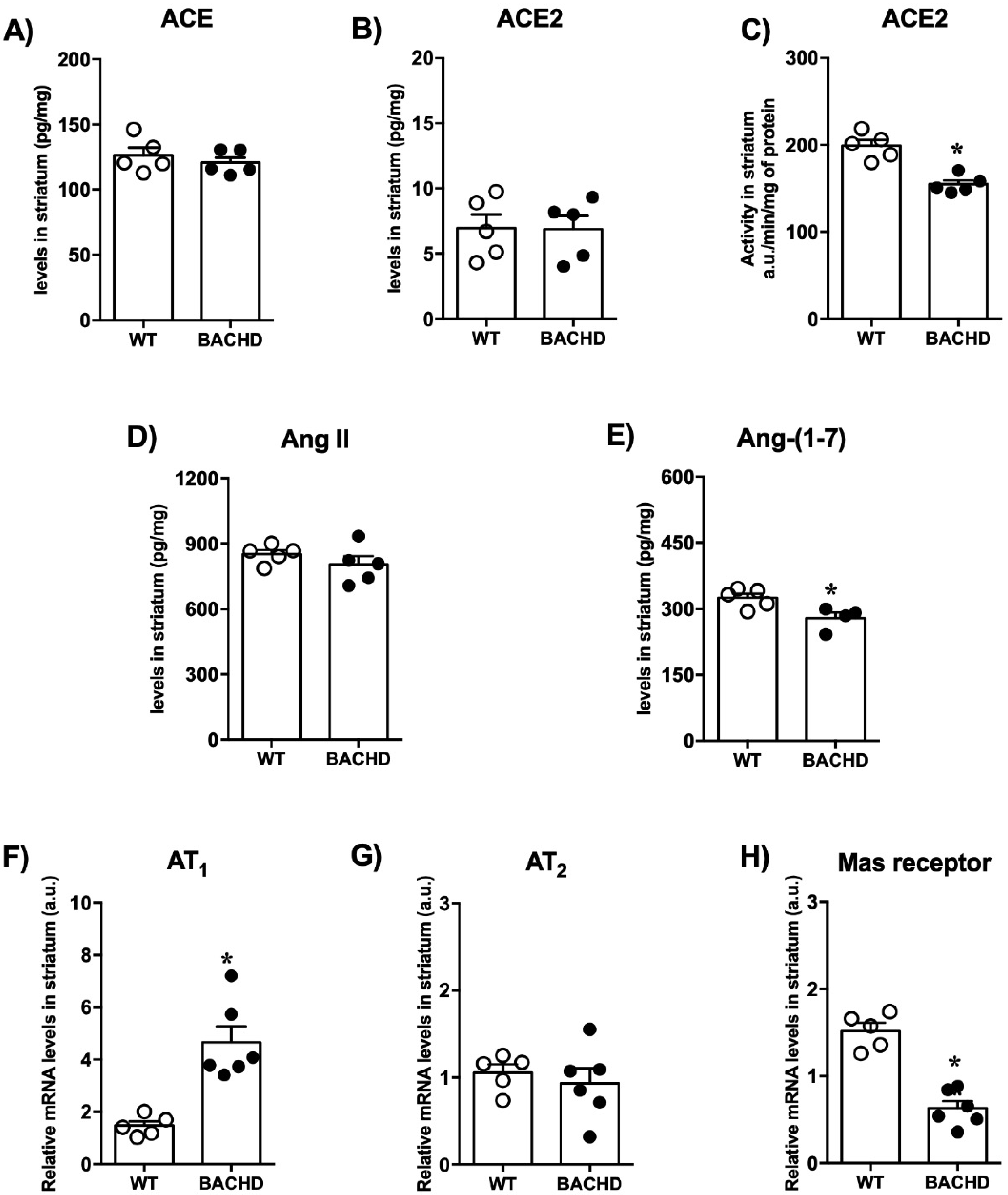
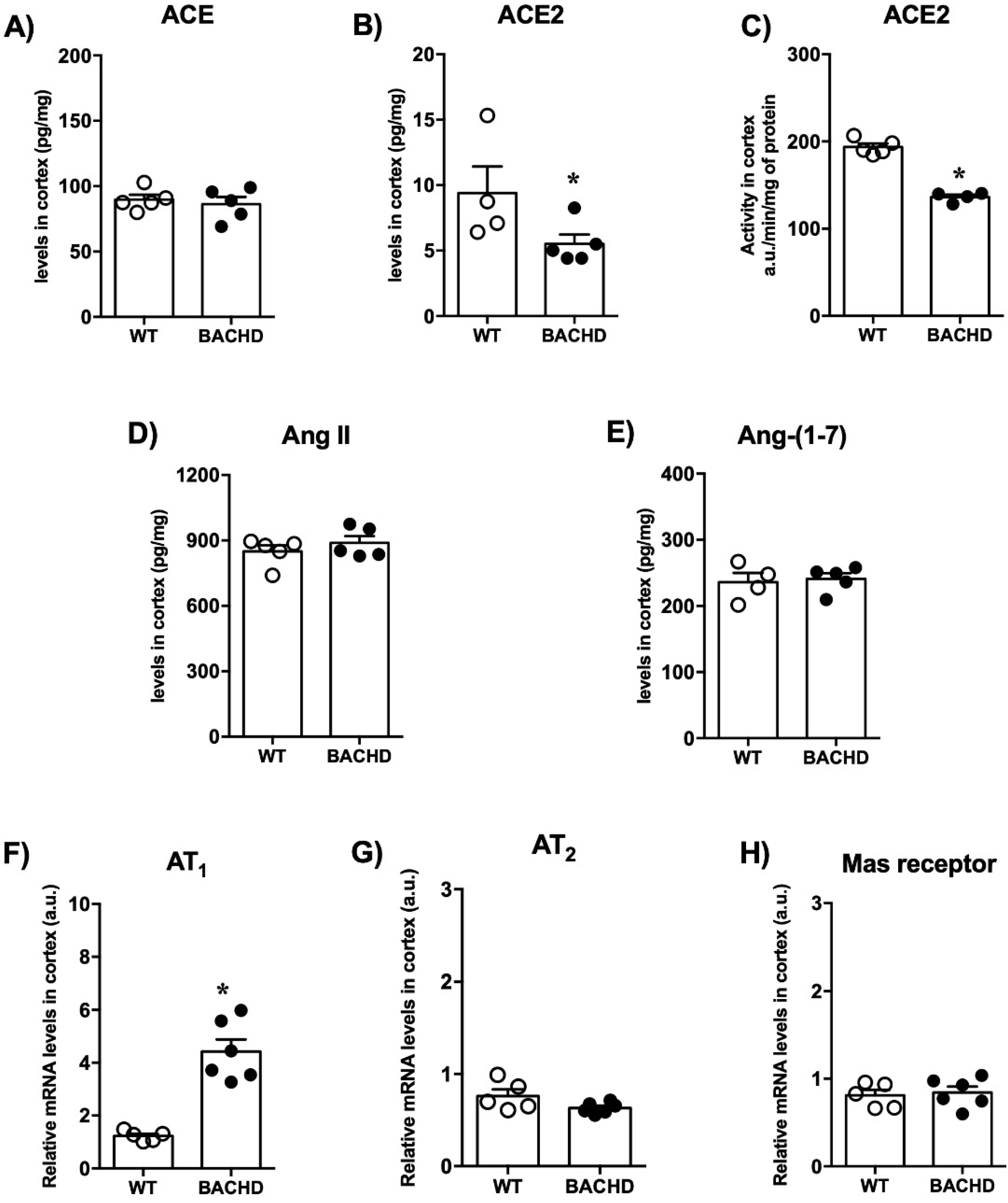
2.2. Renin-Angiotensin System Components
3. Discussion
4. Materials and Methods
4.1. Animal Model and Ethical Procedures
4.2. Open Field Test
4.3. Rotarod
4.4. Wire Hang Test
4.5. Measurement of RAS Components
4.6. Measurement of ACE2 Activity
4.7. Measurement of RAS Receptors’ mRNA Expression
4.8. Assessment of Plasma Levels of Proteins Related to the RAS in Human Samples
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Bano, D.; Zanetti, F.; Mende, Y.; Nicotera, P. Neurodegenerative processes in Huntington’s disease. Cell Death Dis. 2011, 2, e22. [Google Scholar] [CrossRef] [PubMed]
- Walker, F. Huntinton’s disease_Lancet 2007. Lancet Neurol. 2017, 369, 218–228. [Google Scholar] [CrossRef]
- Vonsattel, J.-P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P. Neuropathological Classification of Huntinqton’s Disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s Disease: From Molecular Pathogenesis to Clinical Treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Li, S.-H.; Li, X.-J. Huntingtin–protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef]
- van der Burg, J.; Björkqvist, M.; Brundin, P. Beyond the brain: Widespread pathology in Huntington’s disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef]
- Papoutsi, M.; Labuschagne, I.; Tabrizi, S.J.; Stout, J.C. The cognitive burden in Huntington’s disease: Pathology, phenotype, and mechanisms of compensation. Mov. Disord. 2014, 29, 673–683. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Baake, V.; Reijntjes, R.; Dumas, E. Cognitive decline in Huntington’s disease expansion gene carrier. Cortex 2017, 95, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, J.L.; Sigmund, C.D. Minireview: Overview of the renin-angiotensin system--an endocrine and paracrine system. Endocrinology 2003, 144, 2179–2183. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Mehr, A.P.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Fyhrquist, F.; Saijonmaa, O. Renin-angiotensin system revisited. J. Intern. Med. 2008, 264, 224–236. [Google Scholar] [CrossRef]
- Fountain, J.; Lappin, S. Physiology, Renin Angiotensin System; StatPearls Publishing: Treasure Island, FL, USA, 2017. [Google Scholar] [PubMed]
- Van Thiel, B.S.; Góes Martini, A.; Te Riet, L.; Severs, D.; Uijl, E.; Garrelds, I.M.; Leijten, F.P.J.; Van Der Pluijm, I.; Essers, J.; Qadri, F.; et al. Brain Renin-Angiotensin System Does It Exist? Hypertension 2017, 69, 1136–1144. [Google Scholar] [CrossRef]
- Von Bohlen Und Halbach, O.; Albrecht, D. The CNS renin-angiotensin system. Cell Tissue Res. 2006, 326, 599–616. [Google Scholar] [CrossRef]
- Ribeiro, V.T.; Cordeiro, T.M.E.; da Silva Filha, R.; Perez, L.G.; Caramelli, P.; Teixeira, A.L.; de Souza, L.C.; Simões e Silva, A.C. Circulating Angiotensin-(1–7) Is Reduced in Alzheimer’s Disease Patients and Correlates With White Matter Abnormalities: Results From a Pilot Study. Front. Neurosci. 2021, 15, 636754. [Google Scholar] [CrossRef]
- Kangussu, L.; Almeida-Santos, A.; Bader, M.; Alenina, N.; Fontes, M.A.P.; Santos, R.A.S.; Aguiar, D.C.; Campagnole-Santos, M.J. Angiotensin-(1–7) attenuates the anxiety and depression-like behaviors in transgenic rats with low brain angiotensinogen. Behav. Brain Res. 2013, 257, 25–30. [Google Scholar] [CrossRef]
- Almeida-Santos, A.; Kangussu, L. Reduced anxiety-like behavior in transgenic rats with chronically overproduction of angiotensin-(1–7): Role of the Mas receptor. Behav. Brain Res. 2017, 331, 193–198. [Google Scholar]
- Kehoe, P.G.; Wong, S.; Al Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-β and tau pathology. Alzheimers Res. Ther. 2016, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Zhu, D.; Xie, W. Central angiotensin II-induced Alzheimer-like tau phosphorylation in normal rat brains. FEBS Lett. 2012, 586, 3737–3745. [Google Scholar] [CrossRef] [Green Version]
- Mertens, B.; Vanderheyden, P.; Michotte, Y.; Sarre, S. The role of the central renin-angiotensin system in Parkinson’s disease. J. Renin Angiotensin Aldosterone Syst. 2010, 11, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, F.M.; Furr-Stimming, E.; Teixeira, A.L. Neuroimmunology of Huntington’s disease: Revisiting evidence from human studies. Mediat. Inflamm. 2016, 2016, 8653132. [Google Scholar]
- Kawajiri, M.; Mogi, M.; Higaki, N.; Matsuoka, T.; Ohyagi, Y.; Tsukuda, K.; Kohara, K.; Horiuchi, M.; Miki, T.; Kira, J.I. Angiotensin-converting enzyme (ACE) and ACE2 levels in the cerebrospinal fluid of patients with multiple sclerosis. Mult. Scler. 2009, 15, 262–265. [Google Scholar] [CrossRef]
- Arregui, A.; Bennett, J.P.; Bird, E.D.; Yamamura, H.I.; Iversen, L.L.; Snyder, S.H. Huntington’s chorea: Selective depletion of activity of angiotensin converting enzyme in the corpus striatum. Ann. Neurol. 1977, 2, 294–298. [Google Scholar] [CrossRef]
- De Mello, W.C.; Gerena, Y.; Ayala-Peña, S. Angiotensins and Huntington’s disease: A study on immortalized progenitor striatal cell lines. Front. Endocrinol. 2017, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Arregui, A.; Emson, P.C.; Spokes, E.G. Angiotensin-converting enzyme in substantia nigra: Reduction of activity in Huntington’s disease and after intrastriatal kainic acid in rats. Eur. J. Pharmacol. 1978, 52, 121–124. [Google Scholar] [CrossRef]
- Butterworth, J. Changes in Nine Enzyme Markers for Neurons, Glia, and Endothelial Cells in Agonal State and Huntington’s Disease Caudate Nucleus. J. Neurochem. 1986, 47, 583–587. [Google Scholar] [CrossRef]
- Schweisfurth, H.; Schiöberg-Schiegnitz, S.; Kuhn, W.; Parusel, B. Angiotensin I converting enzyme in cerebrospinal fluid of patients with neurological diseases. Klin. Wochenschr. 1987, 65, 955–958. [Google Scholar] [CrossRef]
- Ge, J.; Barnes, N.M. Alterations in angiotensin AT1 and AT2 receptor subtype levels in brain regions from patients with neurodegenerative disorders. Eur. J. Pharmacol. 1996, 297, 299–306. [Google Scholar] [CrossRef]
- Baake, V.; Reijntjes, R.H.A.M.; Dumas, E.M.; Thompson, J.C.; REGISTRY Investigators of the European Huntington’s Disease Network; Roos, R.A.C. Cognitive decline in Huntington’s disease expansion gene carriers. Cortex 2017, 95, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Heidecke, H.; Schröder, A.; Paul, F.; Wachter, R.; Hoffmann, R.; Ellrichmann, G.; Dragun, D.; Waschbisch, A.; Stegbauer, J.; et al. Increase of angiotensin II type 1 receptor auto-antibodies in Huntington’s disease. Mol. Neurodegener. 2014, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; André, V.M.; Wilburn, B.; Lu, X.-H.; Tao, J.; Yamazaki, I.; Li, S.-H.; Sun, Y.E.; et al. Full-Length Human Mutant Huntingtin with a Stable Polyglutamine Repeat Can Elicit Progressive and Selective Neuropathogenesis in BACHD Mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gironacci, M.M.; Vicario, A.; Cerezo, G.; Silva, M.G. The depressor axis of the renin–angiotensin system and brain disorders: A translational approach. Clin. Sci. 2018, 132, 1021–1038. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef]
- Menalled, L.; El-Khodor, B.F.; Patry, M.; Suárez-Fariñas, M.; Orenstein, S.J.; Zahasky, B.; Leahy, C.; Wheeler, V.; Yang, X.W.; MacDonald, M.; et al. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009, 35, 319–336. [Google Scholar] [CrossRef] [Green Version]
- Uekawa, K.; Hasegawa, Y.; Senju, S.; Nakagata, N.; Ma, M.; Nakagawa, T.; Koibuchi, N.; Kim-Mitsuyama, S. Intracerebroventricular Infusion of Angiotensin-(1–7) Ameliorates Cognitive Impairment and Memory Dysfunction in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, A.; Shetty, S.; Shirole, T.; Jagtap, A.G. Potential of protease inhibitor in 3-nitropropionic acid induced Huntington’s disease like symptoms: Mitochondrial dysfunction and neurodegeneration. Neurotoxicology 2014, 45, 139–148. [Google Scholar] [CrossRef]
- Kaur, P.; Muthuraman, A.; Kaur, M. The Implications of Angiotensin-Converting Enzymes and Their Modulators in Neurodegenerative Disorders: Current and Future Perspectives. ACS Chem. Neurosci. 2015, 6, 508–521. [Google Scholar] [CrossRef]
- Kamel, A.S.; Abdelkader, N.F.; El-Rahman, S.S.A.; Emara, M.; Zaki, H.F.; Khattab, M.M. Stimulation of ACE2/ANG(1–7)/Mas Axis by Diminazene Ameliorates Alzheimer’s Disease in the D-Galactose-Ovariectomized Rat Model: Role of PI3K/Akt Pathway. Mol. Neurobiol. 2018, 55, 8188–8202. [Google Scholar] [CrossRef]
- Rocha, N.P.; Toledo, A.; Corgosinho, L.T.; de Souza, L.C.; Guimarães, H.C.; Resende, E.P.; Braz, N.F.; Gomes, K.B.; e Silva, A.C.S.; Caramelli, P.; et al. Cerebrospinal Fluid Levels of Angiotensin-Converting Enzyme Are Associated with Amyloid-β42 Burden in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 1085–1090. [Google Scholar] [CrossRef]
- Rocha, N.P.; Scalzo, P.L.; Barbosa, I.G.; de Campos-Carli, S.M.; Tavares, L.D.; de Souza, M.S.; Christo, P.P.; Reis, H.J.; e Silva, A.C.S.; Teixeira, A.L. Peripheral levels of angiotensins are associated with depressive symptoms in Parkinson’s disease. J. Neurol. Sci. 2016, 368, 235–239. [Google Scholar] [CrossRef]
- Rocha, N.P.; Cleary, C.; Colpo, G.D.; Stimming, E.F.; Teixeira, A.L. Peripheral Levels of Renin-Angiotensin System Components Are Associated with Cognitive Performance in Huntington’s Disease. Front. Neurosci. 2020, 14, 594945. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, Y.-D.; Zhou, J.-S.; Zhu, X.-C.; Tian, Y.-Y.; Zhao, H.-D.; Lu, H.; Gao, Q.; Tan, L.; Yu, J.-T. Angiotensin-(1–7) is Reduced and Inversely Correlates with Tau Hyperphosphorylation in Animal Models of Alzheimer’s Disease. Mol. Neurobiol. 2015, 53, 2489–2497. [Google Scholar] [CrossRef]
- Chen, J.-L.; Zhang, D.-L.; Sun, Y.; Zhao, Y.-X.; Zhao, K.-X.; Pu, D.; Xiao, Q. Angiotensin-(1–7) administration attenuates Alzheimer’s disease-like neuropathology in rats with streptozotocin-induced diabetes via Mas receptor activation. Neuroscience 2017, 346, 267–277. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Kumar, V.B.; Armbrecht, H.J. The SAMP8 Mouse: A Model to Develop Therapeutic Interventions for Alzheimers Disease. Curr. Pharm. Des. 2012, 18, 1123–1130. [Google Scholar] [CrossRef]
- Duan, R.; Xue, X.; Zhang, Q.-Q.; Wang, S.-Y.; Gong, P.-Y.; E, Y.; Jiang, T.; Zhang, Y.-D. ACE2 activator diminazene aceturate ameliorates Alzheimer’s disease-like neuropathology and rescues cognitive impairment in SAMP8 mice. Aging 2020, 12, 14819–14829. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; De Aragão, B.C.; Andrade, J.N.; Magalhães-Gomes, M.P.S.; Foureaux, G.; Joviano-Santos, J.V.; Nogueira, J.C.; Ribeiro, F.; Tapia, J.C.; Guatimosim, C. Muscle atrophy is associated with cervical spinal motoneuron loss in BACHD mouse model for Huntington’s disease. Eur. J. Neurosci. 2016, 45, 785–796. [Google Scholar] [CrossRef]
- Leite, J.V.; Guimarães, F.S.; Moreira, F.A. Aripiprazole, an atypical antipsychotic, prevents the motor hyperactivity induced by psychotomimetics and psychostimulants in mice. Eur. J. Pharmacol. 2008, 578, 222–227. [Google Scholar] [CrossRef]
- Sousa, N.; Genes, O.A.; Wotjak, C.T. A hitchhiker’s guide to behavioral analysis in laboratory rodents. Genes Brain Behav. 2006, 5 (Suppl. S2), 5–24. [Google Scholar] [CrossRef]
- Viana, T.G.; Almeida-Santos, A.F.; Aguiar, D.C.; Moreira, F.A. Effects of Aripiprazole, an Atypical Antipsychotic, on the Motor Alterations Induced by Acute Ethanol Administration in Mice. Basic Clin. Pharmacol. Toxicol. 2012, 112, 319–324. [Google Scholar] [CrossRef]
- Tanaka, H.; Ma, J.; Tanaka, K.F.; Takao, K.; Komada, M.; Tanda, K.; Suzuki, A.; Ishibashi, T.; Baba, H.; Isa, T.; et al. Mice with Altered Myelin Proteolipid Protein Gene Expression Display Cognitive Deficits Accompanied by Abnormal Neuron–Glia Interactions and Decreased Conduction Velocities. J. Neurosci. 2009, 29, 8363–8371. [Google Scholar] [CrossRef]
- Huentelman, M.J.; Zubcevic, J.; Katovich, M.J.; Raizada, M.K. Cloning and characterization of a secreted form of angiotensin-converting enzyme 2. Regul. Pept. 2004, 122, 61–67. [Google Scholar] [CrossRef]
- Kangussu, L.M.; Guimaraes, P.S.; Nadu, A.P.; Melo, M.B.; Santos, R.A.; Campagnole-Santos, M.J. Activation of angiotensin-(1–7)/Mas axis in the brain lowers blood pressure and attenuates cardiac remodeling in hypertensive transgenic (mRen2)27 rats. Neuropharmacology 2015, 97, 58–66. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls (n = 16) | Manifest HD (n = 22) | p-Value | |
|---|---|---|---|
| Age in years | 48.12 ± 11.02 (47.5) | 49.88 ± 12.64 (49.4) | 0.661 a |
| Female sex, n (%) | 10 (62.5) | 14 (66.7) | 1.000 b |
| BMI (Kg/m2) | 28.51 ± 7.32 (25) | 27.62 ± 6.48 (25) | 0.650 c |
| CAG length (larger allele) | - | 44.63 ± 4.14 (43) | - |
| UHDRS TMS | - | 29.67 ± 12.91 (30) | - |
| TFC score | - | 8.43 ± 2.82 (8) | - |
| Independence Scale (%) | - | 84.52 ± 12.14 (85) | - |
| Ang II (pg/mL) | 1567.77 ± 325.19 (1608) | 1598.50 ± 282.83 (1618) | 0.761 a |
| ACE (pg/mL) | 447.21 ± 62.69 (463) | 435.49 ± 73.94 (426) | 0.614 a |
| Ang-(1-7) (pg/mL) | 22.80 ± 12.80 (21) | 39.78 ± 31.04 (33) | 0.048 a |
| ACE2 (pg/mL) | 7.27 ± 6.46 (5.7) | 6.05 ± 7.86 (4.7) | 0.189 c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kangussu, L.M.; Rocha, N.P.; Valadão, P.A.C.; Machado, T.C.G.; Soares, K.B.; Joviano-Santos, J.V.; Latham, L.B.; Colpo, G.D.; Almeida-Santos, A.F.; Furr Stimming, E.; et al. Renin-Angiotensin System in Huntington′s Disease: Evidence from Animal Models and Human Patients. Int. J. Mol. Sci. 2022, 23, 7686. https://doi.org/10.3390/ijms23147686
Kangussu LM, Rocha NP, Valadão PAC, Machado TCG, Soares KB, Joviano-Santos JV, Latham LB, Colpo GD, Almeida-Santos AF, Furr Stimming E, et al. Renin-Angiotensin System in Huntington′s Disease: Evidence from Animal Models and Human Patients. International Journal of Molecular Sciences. 2022; 23(14):7686. https://doi.org/10.3390/ijms23147686
Chicago/Turabian StyleKangussu, Lucas M., Natalia P. Rocha, Priscila A. C. Valadão, Thatiane C. G. Machado, Kívia B. Soares, Julliane V. Joviano-Santos, Leigh B. Latham, Gabriela D. Colpo, Ana Flávia Almeida-Santos, Erin Furr Stimming, and et al. 2022. "Renin-Angiotensin System in Huntington′s Disease: Evidence from Animal Models and Human Patients" International Journal of Molecular Sciences 23, no. 14: 7686. https://doi.org/10.3390/ijms23147686