2.1. Does the Reduced and Denatured RNase Spontaneously Recover Its Native Structure and Activity?
In the pioneering report by Sela and co-workers [
8], these authors reported that totally reduced RNase obtained using thioglycolic acid as reducing agent and 8 M urea as denaturant, after removal of these reagents and oxidation at pH 7.0 or 8.0 by air bubbling at room temperature (25 °C), no more than 12–19% than the original activity can be restored. A more efficient but not complete recovery of activity was obtained (55% of the native activity) starting from a partially reduced enzyme with one intact native disulfide [
8]. In a meticulous study by White [
12], eight samples of RNase were reduced using a purified sample of thioglycolic acid. The re-oxidation process gave 85% as mean of reactivation. However, only 50% of the original soluble enzyme was recovered after lyophilization, indicating that relevant amounts of oxidized RNase acquired scarce solubility probably due to incorrect inter- or intra-molecular disulfides. Thus, taking into account this loss, an amount of native and active enzyme of 43% that was formed through this procedure is reasonable. Very similar results were obtained using β-ME as reducing agent [
12].
Only a few years later, Anfinsen described an almost complete recovery of activity (80–100%) starting from a full reduced RNase with β-ME in 8 M urea, removal this reagent with a Sephadex G-25 column, and subsequent exposition to air (20 h) at low RNase concentrations (~25 µM) at pH 8.0–8.5 [
9].
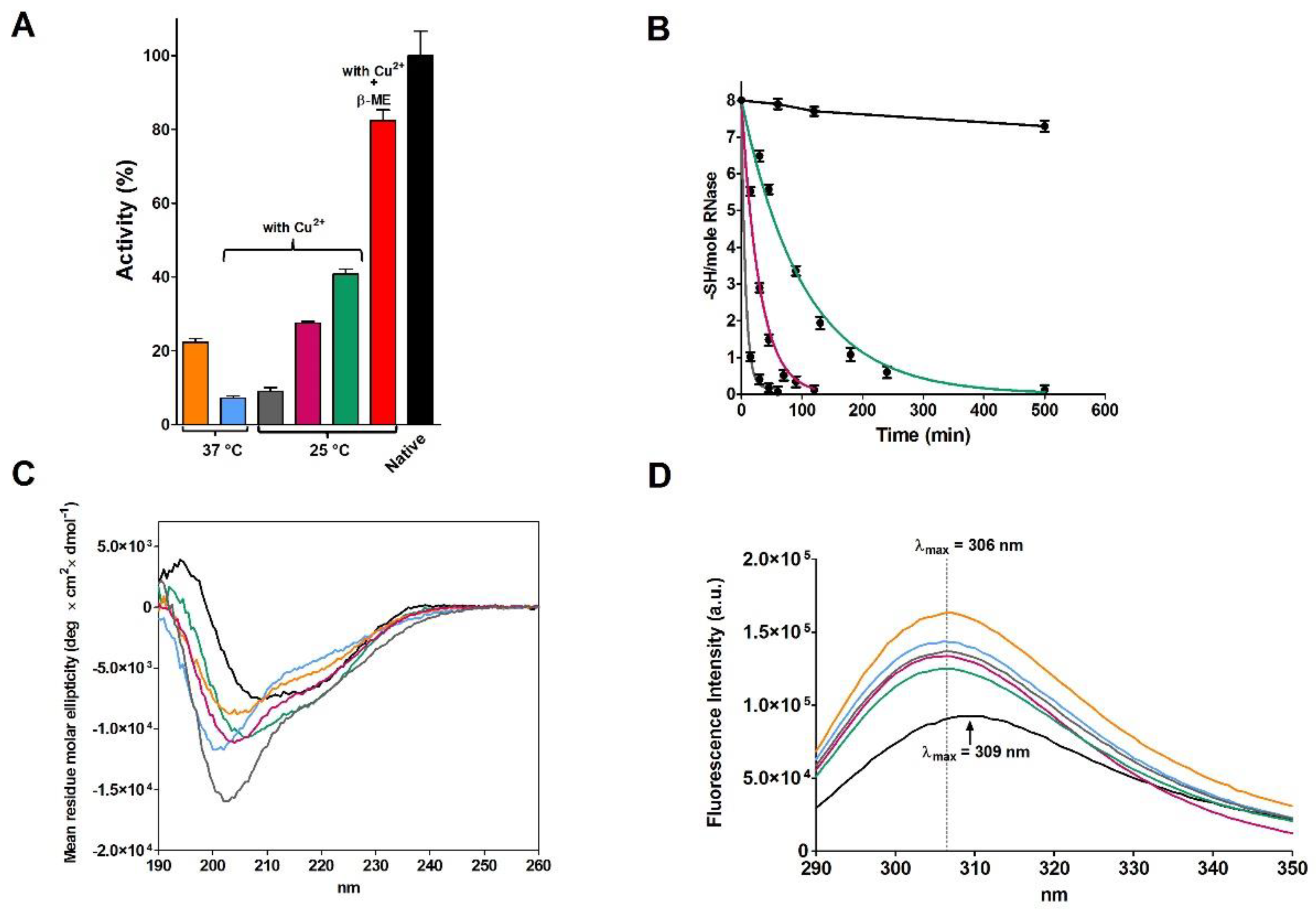
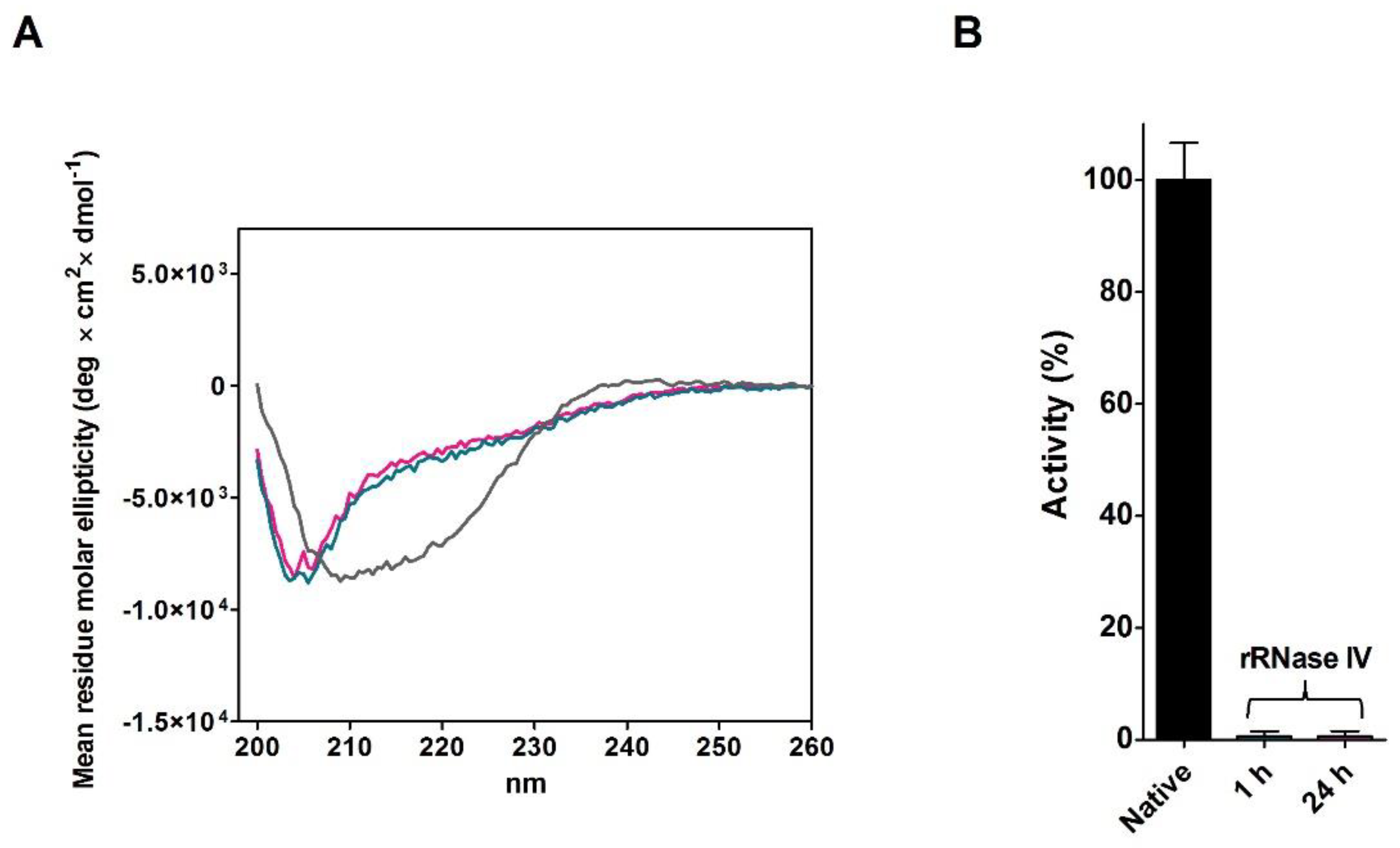
Our attempts to reproduce these results were disappointing in both kinetics of disulfide formation and activity recovery under various temperature and protein concentrations. More precisely, starting from the reduced and denatured RNase I (rRNase I and other rRNase samples, i.e., rRNase II, III and IV, differ mainly in the chromatographic purification step, see Materials and Methods), incubation at pH 8.5 with very low protein concentration (14 µM) did not give a complete oxidation before two days had elapsed (
Table 1), and no more than 20–30% of the original activity (
Figure 1A and
Table 1). We believe that the faster oxidation observed by Anfinsen and Haber (20 h) [
9] was possibly due to an amount of spurious metals, a not uncommon flaw in buffer preparations in the mid-20th century and, in traces, even present in our experiments (
Figure 1A). In fact, EDTA strongly inhibits the re-oxidation and the presence of trace of Cu
2+ and other metals greatly enhances the process as also observed by other authors (
Figure 1B) [
10,
13]. The presence of 10 µM Cu
2+ gave very low recovery of activity compared to the 0.3 µM Cu
2+ possibly due to a fast and tumultuous disulfide formation (mostly incorrect).
For sake of simplicity in the present work, we will continue to use the term auto-oxidation or spontaneous oxidation (used by Anfinsen and many other authors and textbooks) instead of the more correct “metal catalyzed oxidation”.
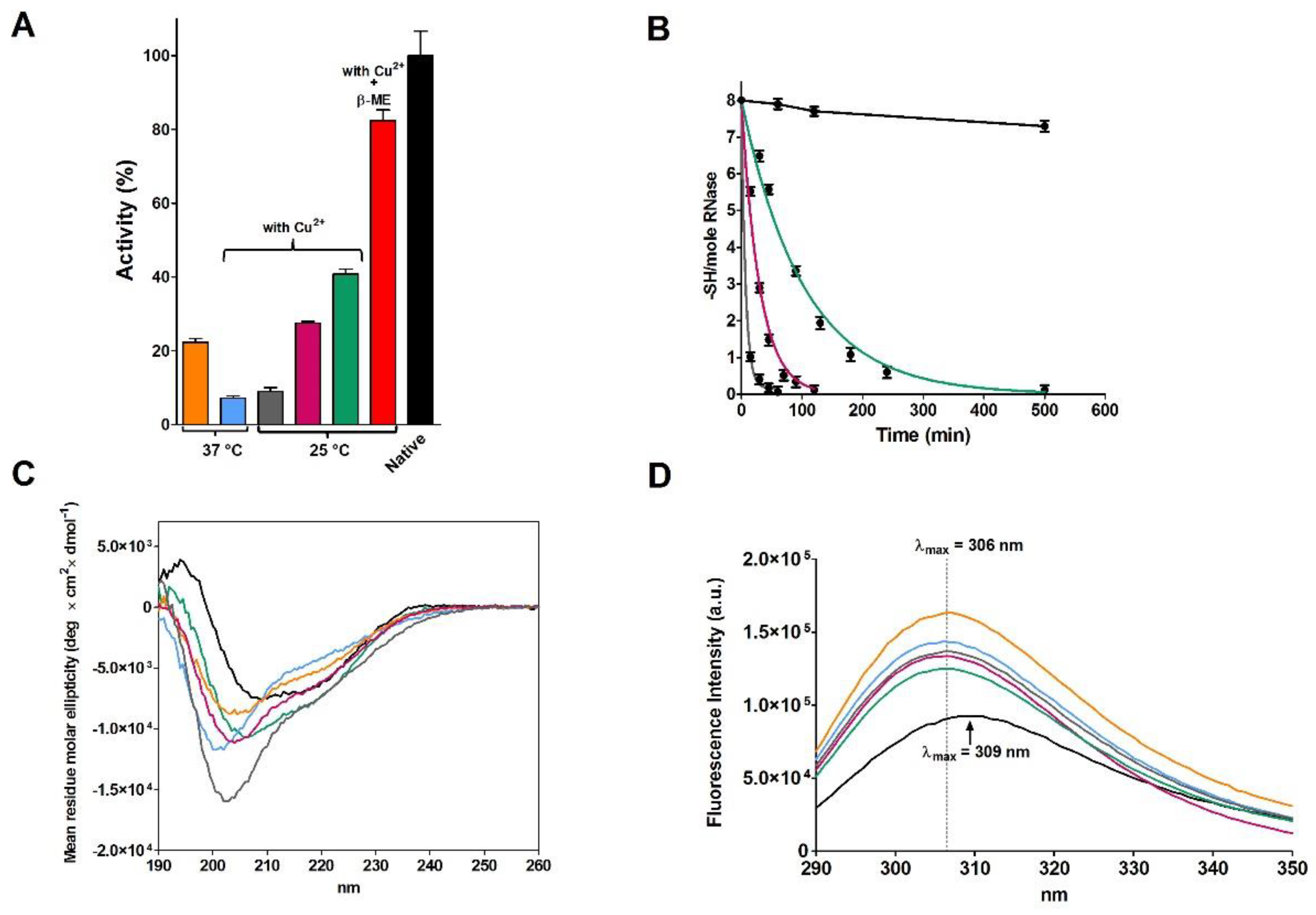
The CD spectrum of the partially active and fully re-oxidized rRNase I at pH 8.5 with or without Cu
2+ did not overlap the one of the native enzyme, indicating lower amounts of beta sheets and turns (
Figure 1C and
Table 2). The intrinsic fluorescence of the six tyrosines of rRNase I gave further insights. In fact, the spectrum of the re-oxidized protein does not overlap the one of the native enzyme, despite all disulfides were formed. As it will be discussed below, this is a signal that tyrosines, in the re-oxidized rRNase I, reside in an increased non-polar environment compared to the native enzyme (
Figure 1D) [
14]. This indicates different tertiary structures formed as a consequence of non-native disulfides. Interestingly, the presence of sub-stoichiometric amount of β-ME (11 μM against 14 μM of rRNase I) allows a relevant recovery of activity (82%) in the range of that found by Anfinsen (80–100%) (
Figure 1A and
Table 1).
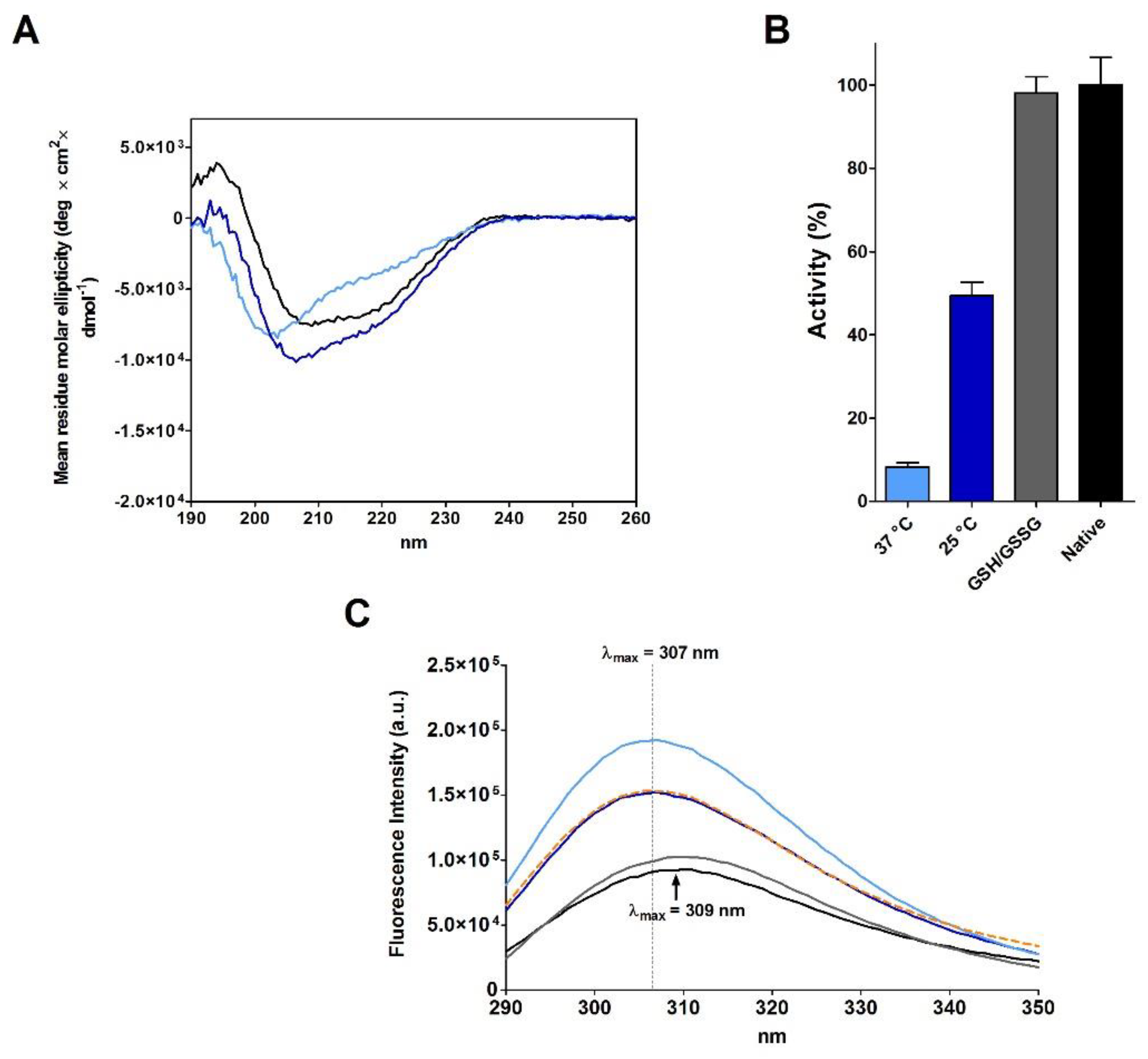
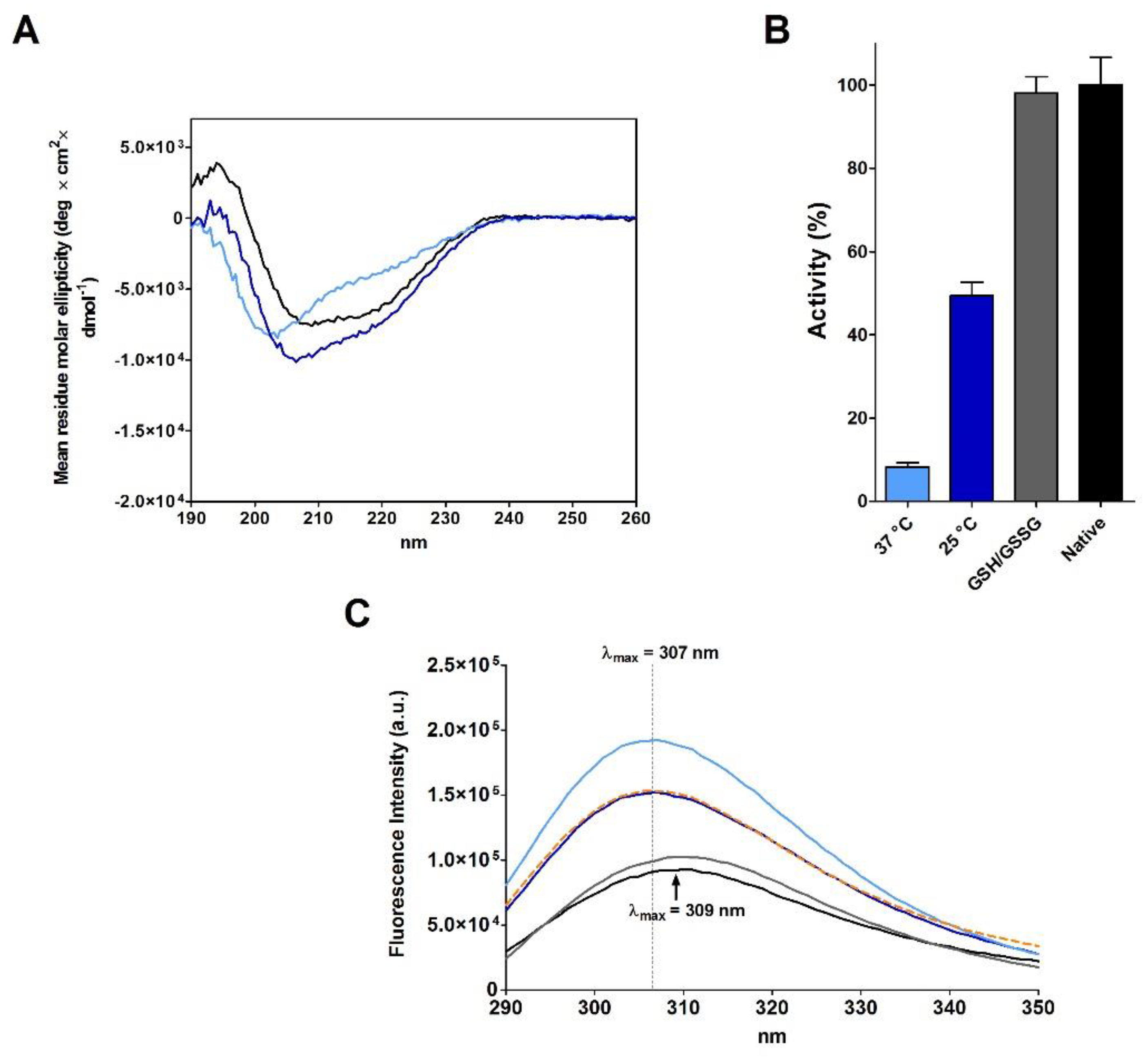
As Anfinsen, after the reduction step, removed β-ME and urea using a Sephadex G-25 column equilibrated with acetic acid (pH 3.5), we also replicated this procedure obtaining a new reduced RNase (rRNase II, see Materials and Methods). The full oxidation of this reduced protein is again slow (4.5 day at 25 °C) (
Table 1) and CD analyses suggest a secondary structure only approaching that found in the native enzyme, showing a lower amount of beta structures (
Figure 2A and
Table 3). The complete oxidized enzyme also displays 49% of the native RNase activity, which represents the maximum recovery reached in all our experiments without a classical reshuffling mixture (GSH/GSSG) or without a sub-stoichiometric amount of β-ME (
Figure 2B and
Table 1). The intrinsic fluorescence spectra also indicate that oxidized protein acquired a tertiary structure different compared to the native RNase with the tyrosines inserted in a more hydrophobic environment (
Figure 2C). This represents a further indication that incorrect disulfides oppose a complete re-formation of native secondary and tertiary structures of this enzyme.
Furthermore, the replica of a famous experiment of re-oxidation of rRNase II at extreme low concentrations (1.8 µM), capable of recovering 100% of the original activity in 1 h, as described by Anfinsen [
7], gave disappointing results. The full re-oxidation of rRNase II requires three days (
Table 1), which must be compared to one hour described by Anfinsen [
7]. Moreover, the recovery of activity is only of 23% (
Table 1).
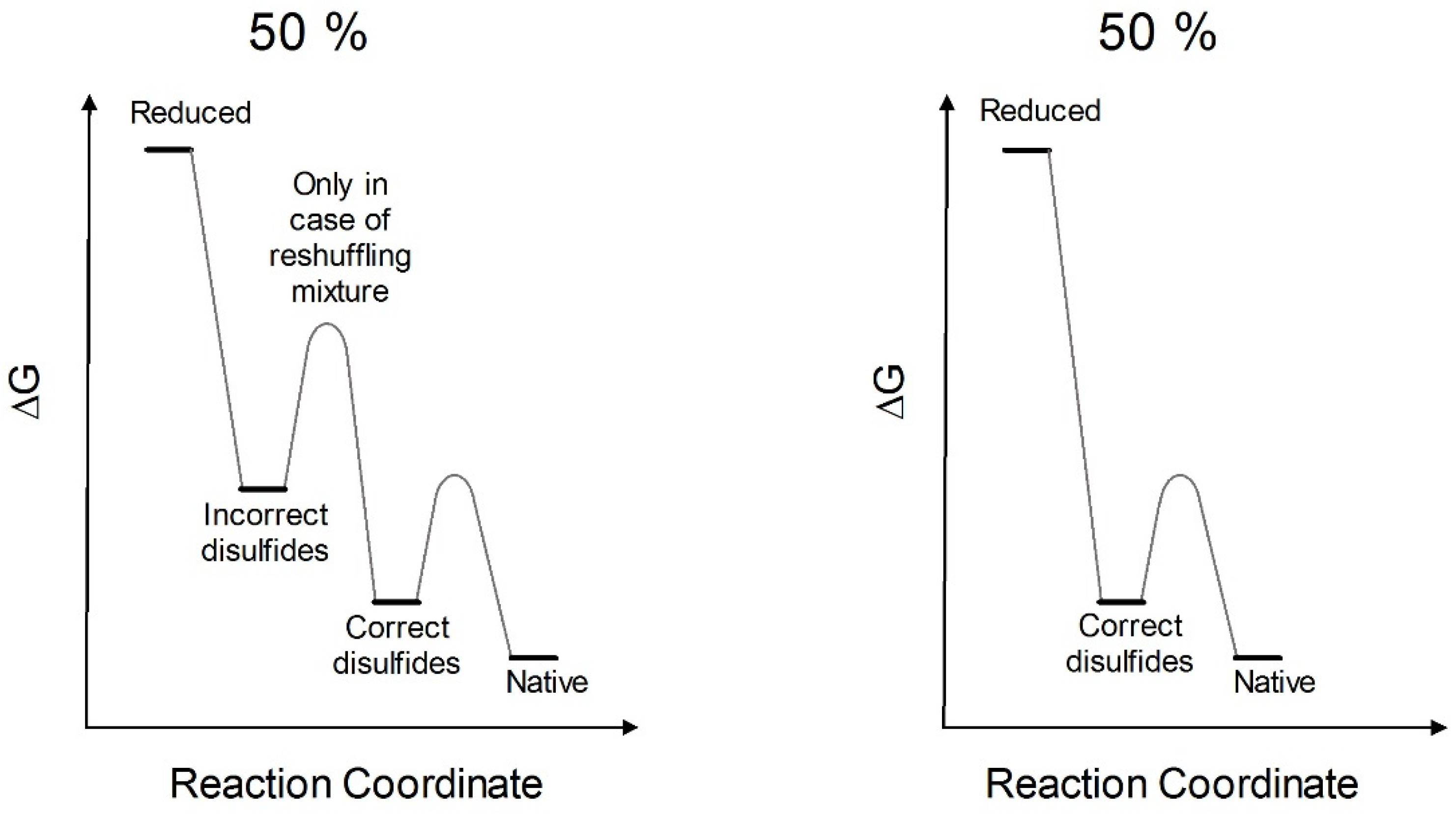
The results claimed by Anfinsen for a spontaneous complete recovery of activity generated the belief that the reduced RNase spontaneously forms all its native structure and that proper disulfides can be formed later as stabilizers. This is what is described in many textbooks [
1,
2,
3]. However, this is disputed by evidence that insufficient recovery of the original structure and activity can be obtained, likely due to the formation of incorrect disulfides. In fact, more recently, a quasi-stochastic mechanism for its oxidative folding has been proposed [
15,
16]. In addition, more recent studies agree with our results, observing that incomplete native structure can be recovered using a particular oxidizing agent [
17]. Actually, we obtained a full recovery of the native activity and an emission spectrum close to the native one only after incubation with a reshuffling solution of GSH/GSSG (
Figure 2B,C). This agrees with later studies which observed that the use of a mixture of reduced/oxidized DTT may contribute to restore the native conformation [
18,
19]. As above reported, also traces of β-ME (
Table 1) can produce very high recovery of the original activity.
2.3. RNase Reduction in the Absence of Denaturing Agent
Anfinsen used 8 M urea and a relevant amount of β-ME (about 0.6 M) to reduce all the four disulfides in RNase [
9]. The presence of urea was necessary because, in its absence, only a partially reduced RNase can be obtained, even after long incubation times at pH 8.5 [
12]. In this way, however, nobody could distinguish between the effect due to urea and that due to the disulfide breakdown on activity and structure. The use of a stronger reducing agent such as DTT allows us to explore what happens to the enzyme activity and structure when the disulfides are progressively broken in the absence of a denaturing solution. DTT is a reagent of particular interest as its oxidized form is greatly stabilized being a cyclic disulfide, and it cannot form mixed disulfides when it interacts with protein cysteines.
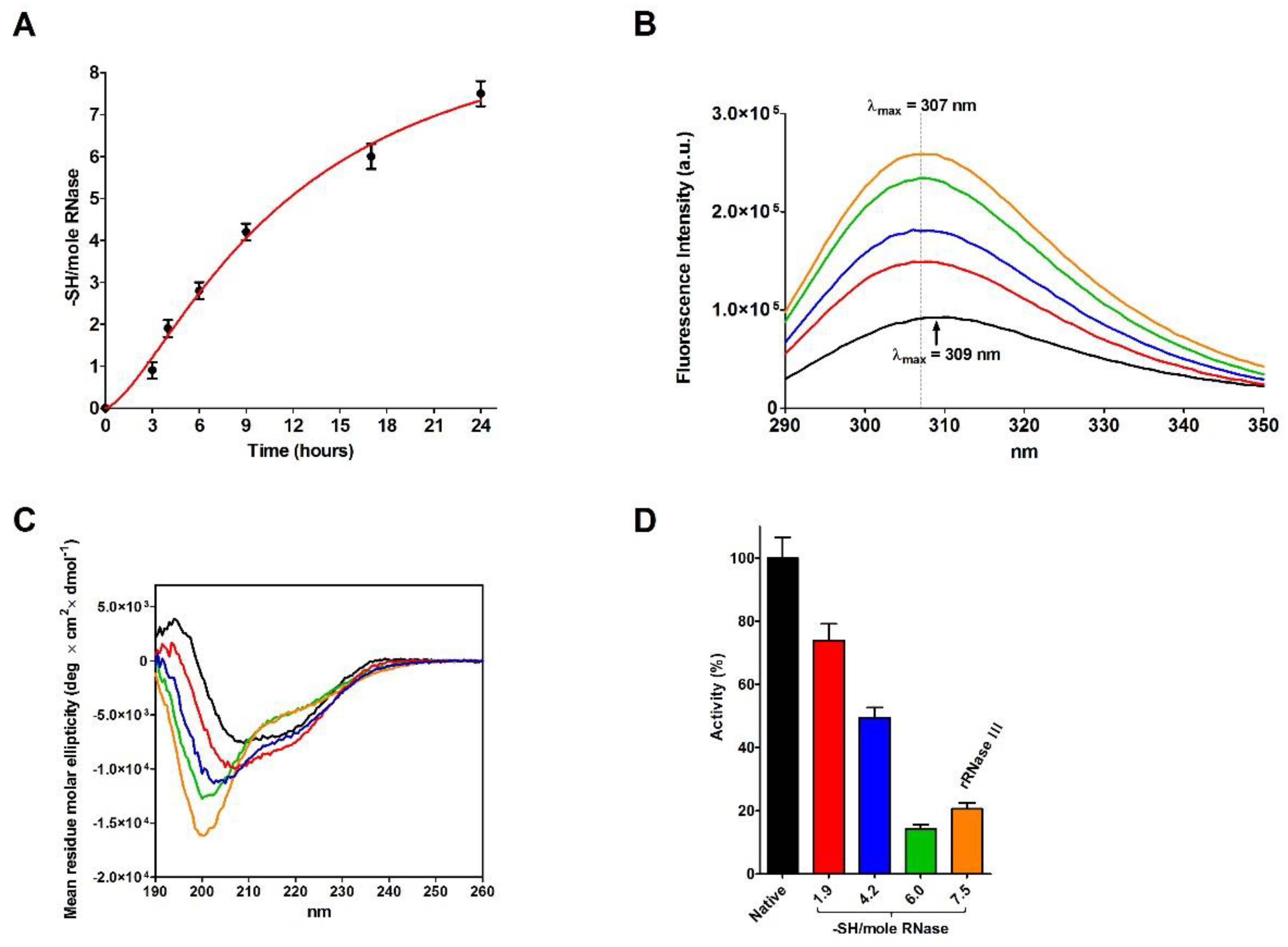
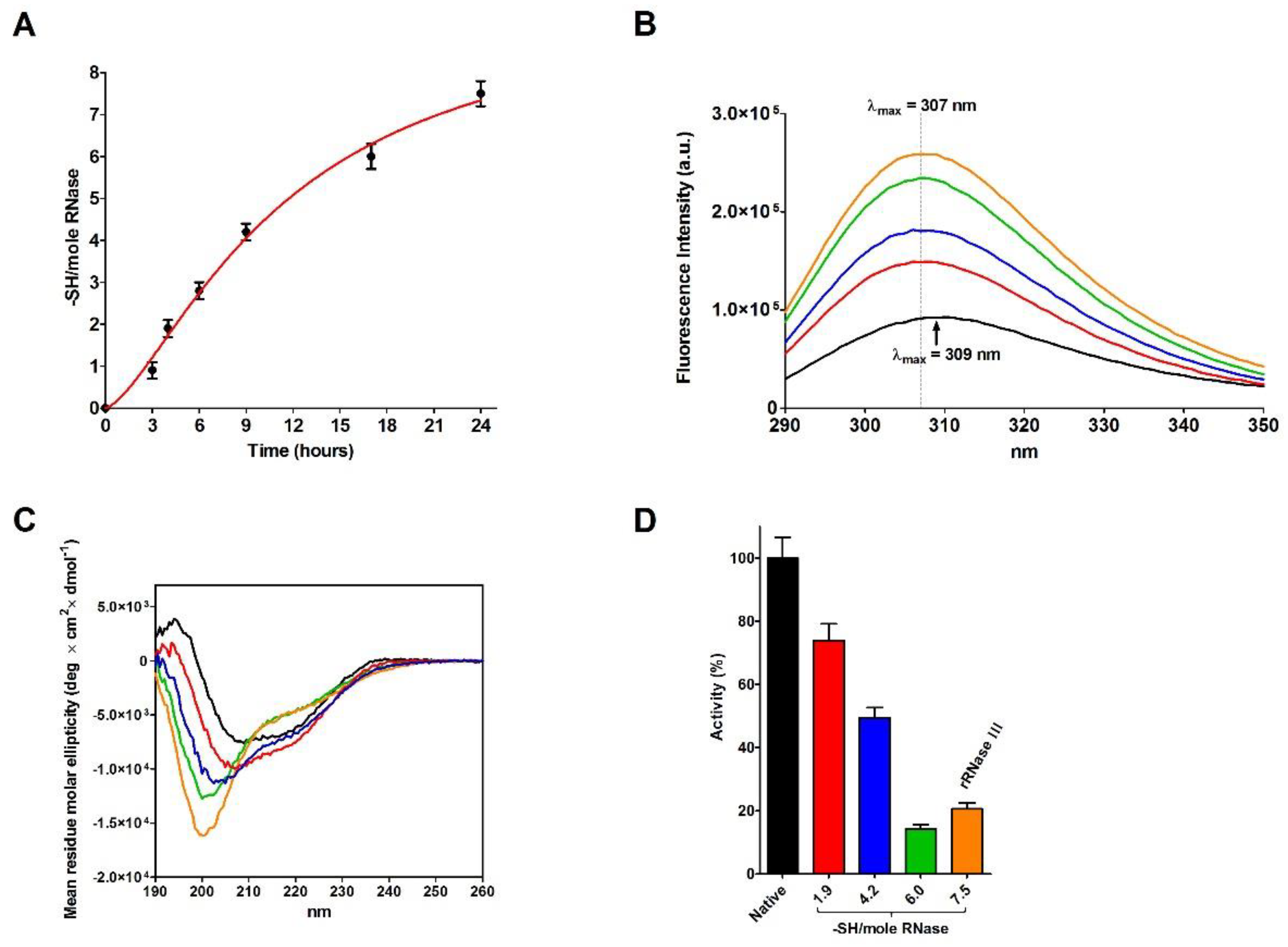
The incubation of native RNase with DTT causes a progressive and complete reduction of the four disulfides even in the absence of urea (
Figure 4A). During this process, a relevant increase in the emission fluorescence of tyrosines has been found probably due to a shift in these residues toward a more hydrophobic environment and then to a change in the tertiary structure (
Figure 4B) [
20].
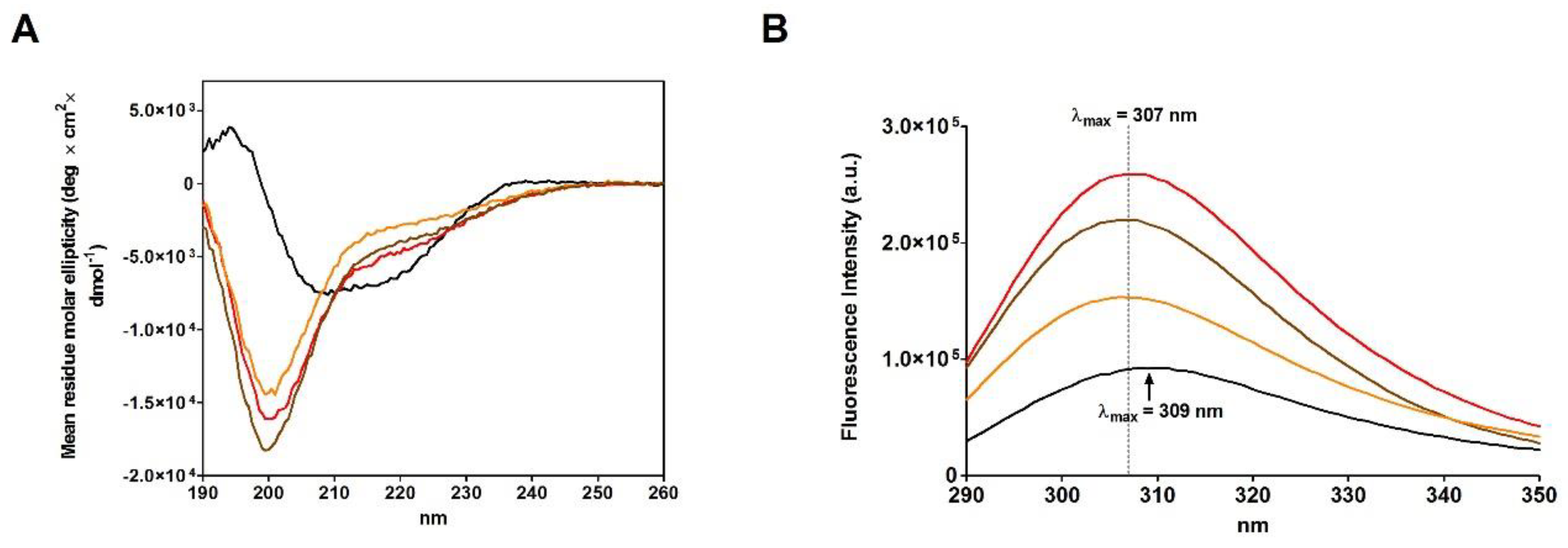
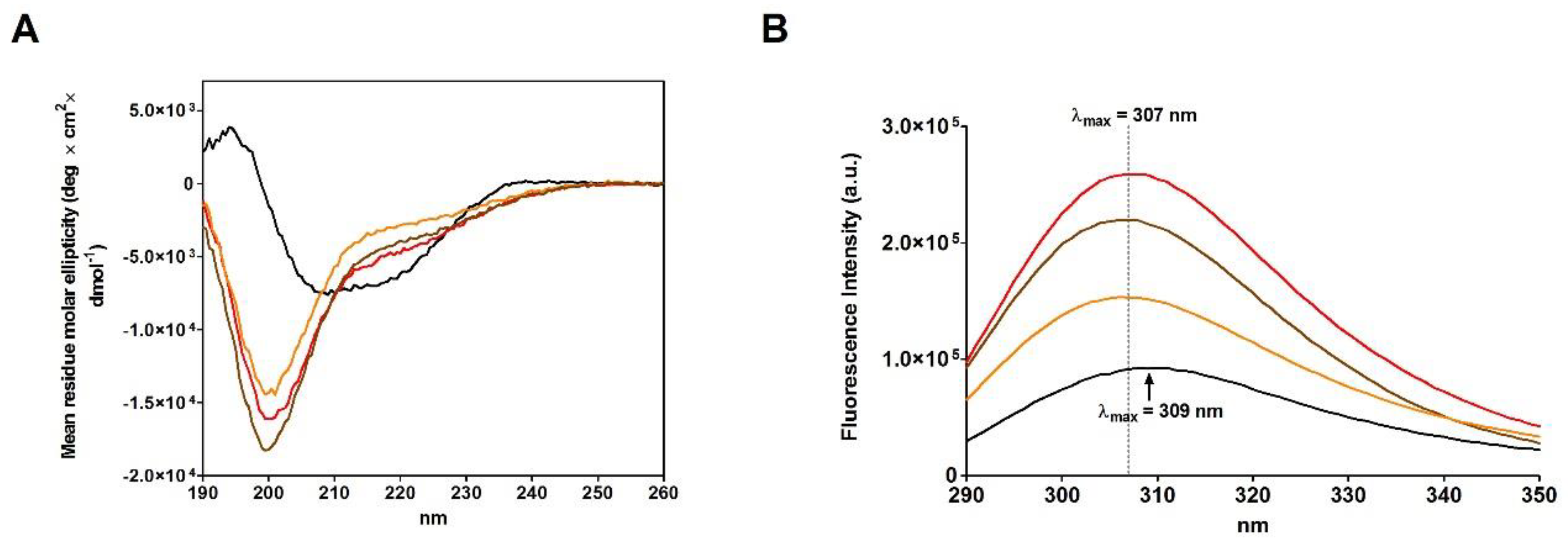
A comparison of the CD spectra of the rRNase I and II, obtained in the presence of urea, with rRNase III, obtained in absence of urea (see Materials and Methods), suggests very similar reduced structures showing a relevant lack of secondary structures more evident for beta-sheets compared to the native form (
Figure 3A and
Table 4). Therefore, the presence of urea is not essential for the modification of the secondary structure, only determined by breakage of disulfides (
Figure 3A and
Figure 4C).
Surprisingly, the reduction of only one disulfide is able to generate some not negligible structural changes by lowering the amount of beta structures and reducing the activity to 74% (
Figure 4C,D and
Table 5). Other prominent structural and activity perturbations occur when other disulfides are broken step by step (
Figure 4C,D and
Table 5).
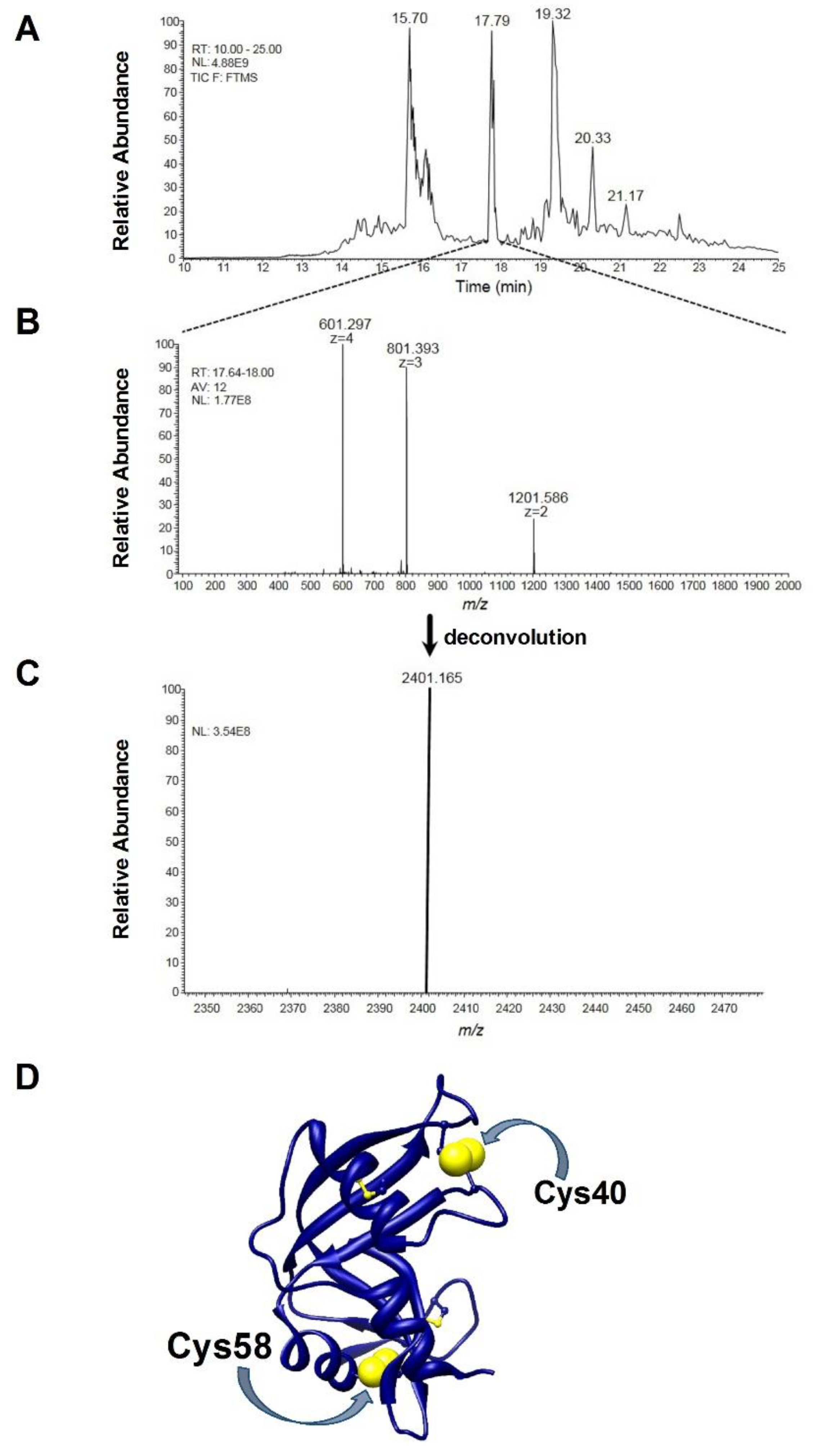
A further indication that the breaking of just one disulfide is responsible for important structural changes comes from mass spectrometry data. When the reduction process with DTT was stopped when only one disulfide was broken, the partially reduced protein was incubated with bromopyruvate, a reagent that alkylates reduced cysteines in a few seconds. Digestion and ESI mass analysis of this modified protein revealed a quantitatively relevant amount of one peptide with an experimental [M + H]
+1 monisot. = 2401.166
m/z, corresponding to the fragment 40–61 of RNase (CKPVNTFVHESLADVQAVCSQK) with a non-native disulfide bridge, i.e., Cys40-Cys58 (theoretical [M + H]
+1 monisot. = 2401.164
m/z) (
Figure 5A–C). Both these cysteines are very distant in the native protein and linked with Cys95 and Cys110, respectively (
Figure 5D).
This finding is a strong indication that one of the two free cysteines, originated by DTT reduction of a single disulfide, interchanges with another distant disulfide and this becomes possible only if relevant secondary and tertiary structural changes occurred.
Only a few experimental studies about the partial reduction of RNase have been performed, but they support our results. In particular, the study of Li and co-workers [
21] identified the Cys40-Cys95 as one of the two disulfides first broken upon selective reduction. Even the theoretical study of Krupa and co-workers identified Cys40-Cys95 as the more susceptible disulfide to be the first cleaved [
22].
2.5. The Effect of Urea on the Activity of the Native RNase
In 1955, Anfinsen described a curious behavior of RNase. Apparently, prolonged incubation of this enzyme in 8 M urea at 5 °C did not produce any loss of activity [
23]. This result was not disproved or modified later but only corrected in its interpretation. In fact, in 1989, Anfinsen, commenting his own work, says that it represents “
a beautiful example of how an entirely acceptable conclusion can be reached that is entirely wrong because of the paucity of knowledge at that particular time” [
24] (pp. 197–198). This refers to his previous (wrong) conclusion that an ordered shape of a protein is not strictly needed for its catalytic function if the structure of the active site is intact. In fact, further studies revealed that some apparent increment of activity occurs at high urea concentration probably due to an increased solubility of the reaction product or to a denaturation of the RNA thereby making it more available to the digestion by the enzyme [
25]. In reality, the RNase activity is slightly lowered at similar urea concentration [
25].
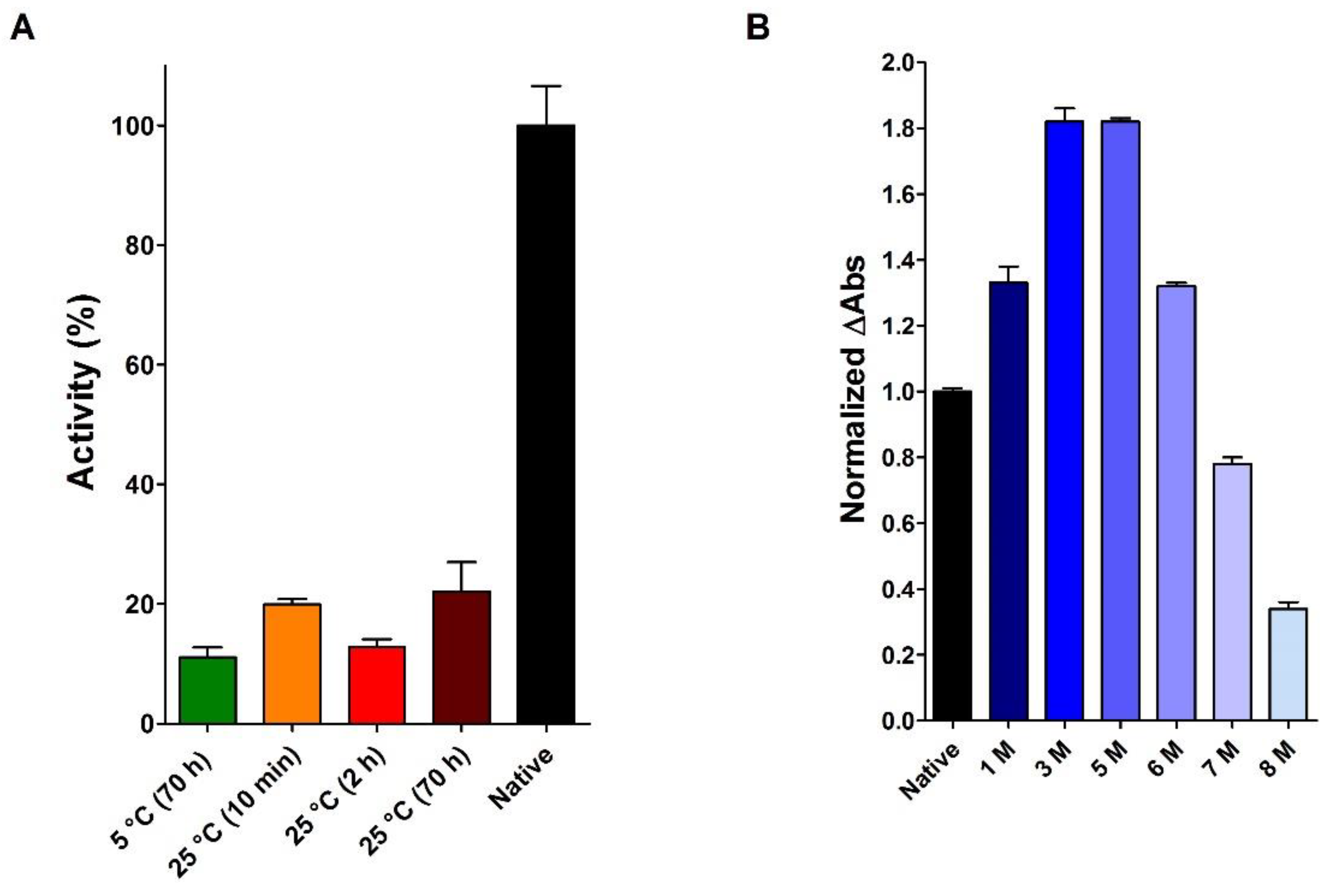
Thus, the full preservation of the RNase activity in 8 M urea, as reported by Anfinsen, is certainly a curious result, and my co-workers and I have been interested in replicating this experiment. However, as shown in
Figure 7A the presence of 8 M urea strongly inhibits RNase either after short or long time of incubation. Our experiments also confirmed an apparent increase in activity at lower urea concentrations (up to 6 M) and a slight decrease in activity at 7 M urea (
Figure 7B). Thus, the lack of inhibition due to 8 M urea found by Anfinsen and co-workers [
23] was possibly due to some overestimation of the urea concentration occurred in their activity measurements.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}