
In Silico Prediction of the Metabolic Resistance of Vitamin D Analogs against CYP3A4 Metabolizing Enzyme


Abstract
:1. Introduction
2. Results and Discussion
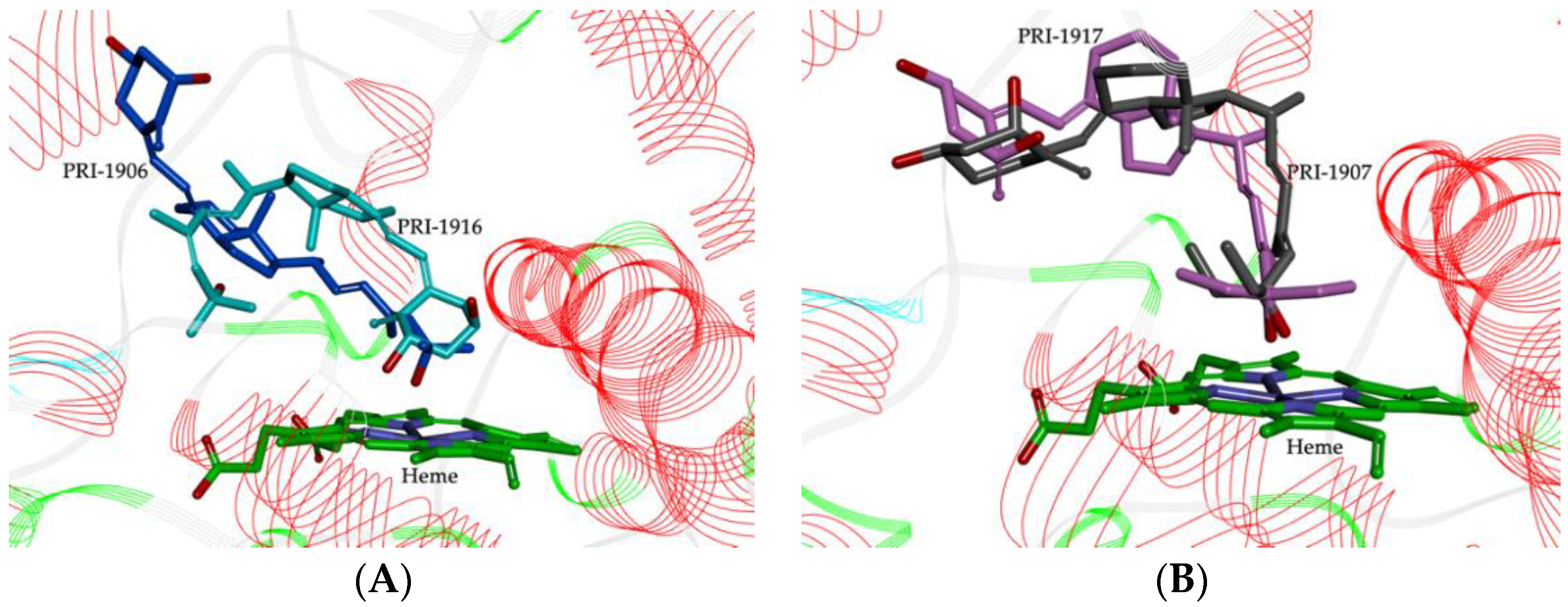
2.1. Potential Binding Site of 1,25D Analogs in CYP3A4
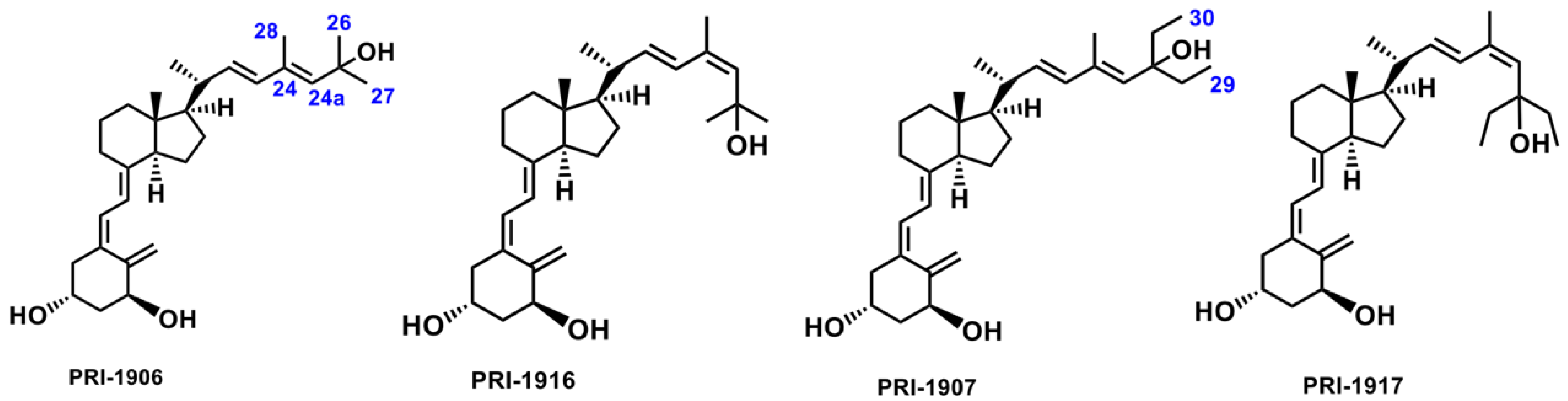
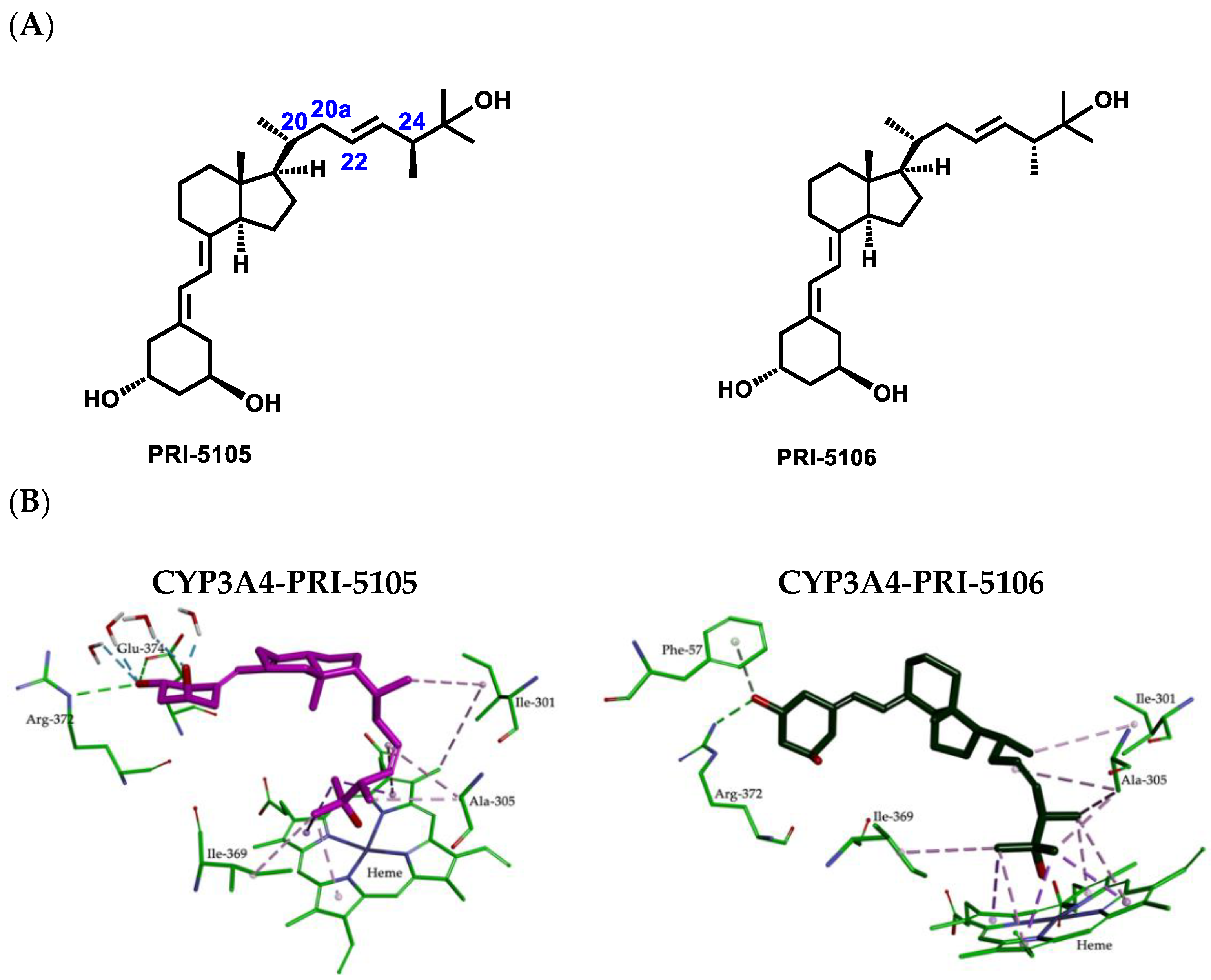
2.2. Importance of Side-Chain Geometry of 1,25D2 Analogs in the CYP3A4 Binding Site
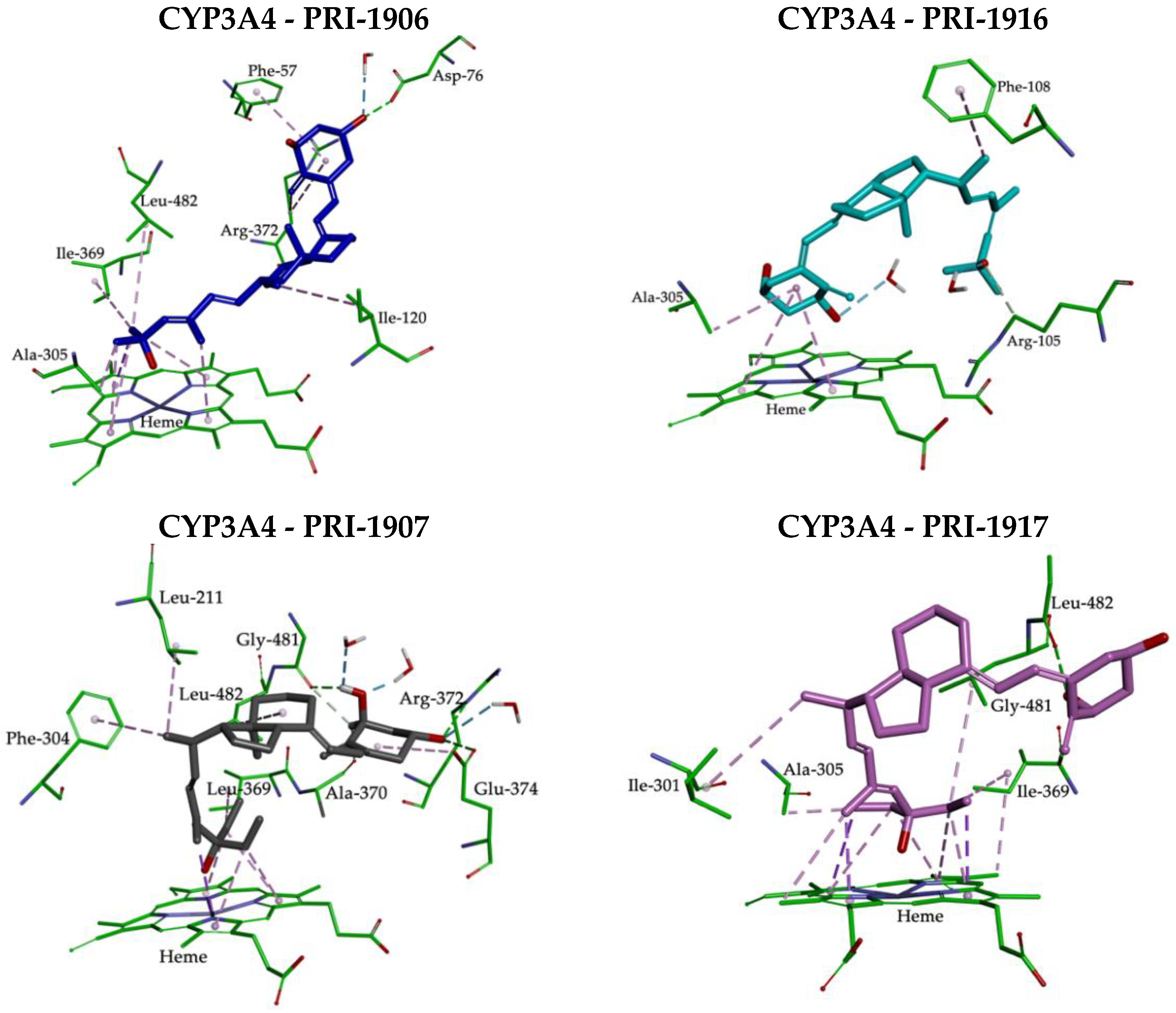
2.3. Molecular Interactions between the CYP3A4 Binding Site and 1,25D2 Analogs
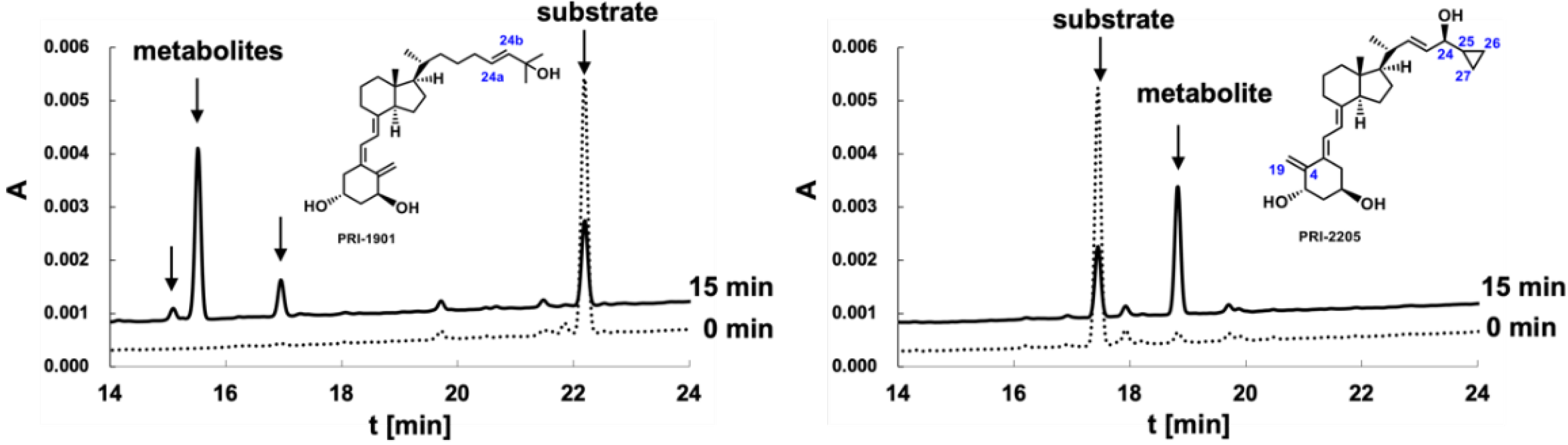
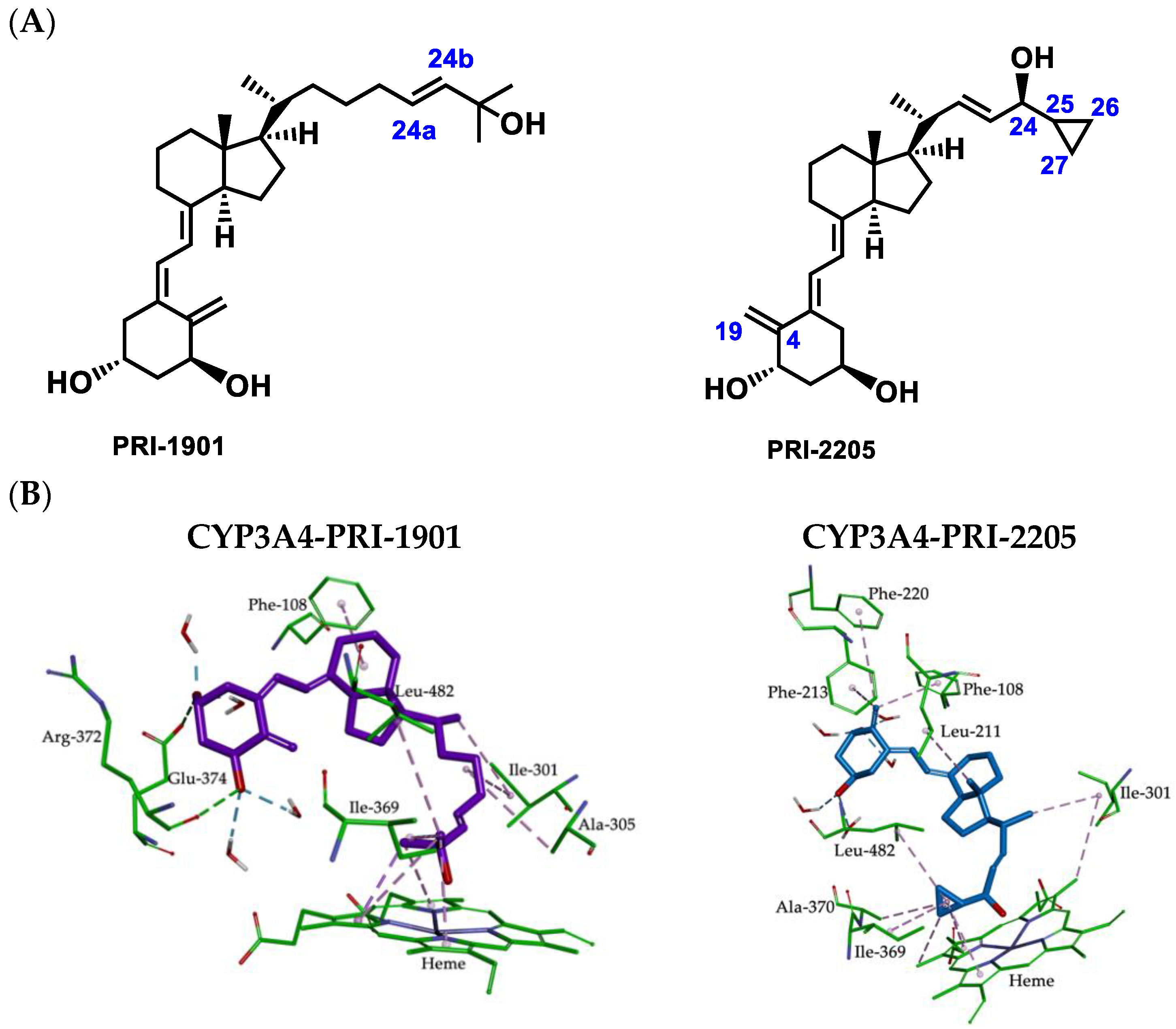
2.4. Metabolic Conversion of 1,25D3 Analogs PRI-1901 and PRI-2205 to hCYP24A1-Mediated Degradation
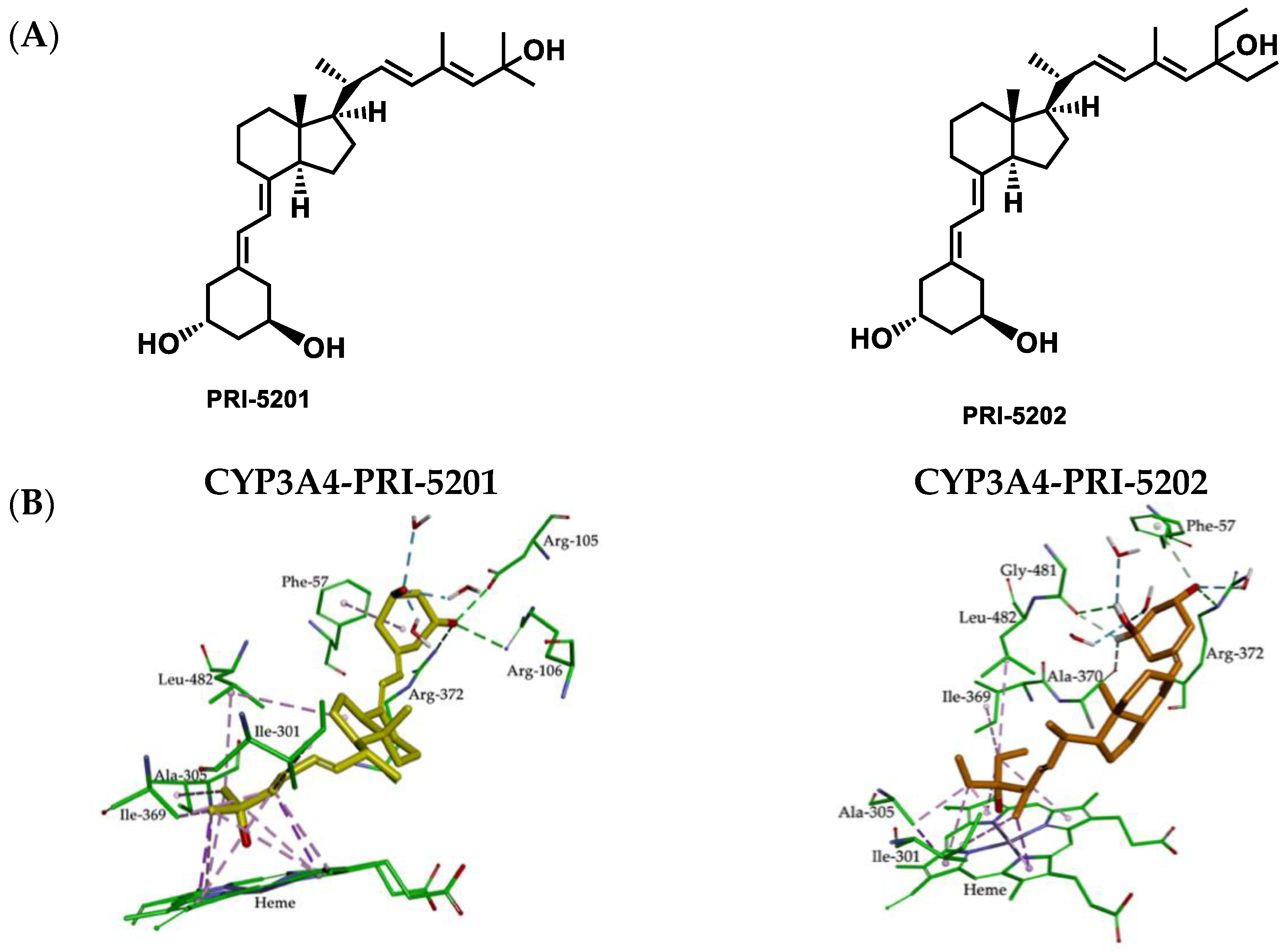
2.5. Molecular Interactions between CYP3A4 Binding Site and 1,25D3 Analogs
3. Materials and Methods
3.1. Theoretical Calculations
3.1.1. Dataset Preparation
3.1.2. Molecular Docking and Dynamic Simulation
3.1.3. Binding Free Enthalpy Calculation
3.2. Metabolic Conversion of 1,25D3 Analogs PRI-1901 and PRI-2205 to CYP24A1-Mediated Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mozolowski, W. Jędrzej Sniadecki (1768–1838) on the Cure of Rickets. Nature 1939, 3612, 121. [Google Scholar] [CrossRef]
- Funk, C. The etiology of the deficiency diseases. J. State Med. 1912, 20, 341–368. [Google Scholar]
- Bikle, D.D. Vitamin D: Newer Concepts of Its Metabolism and Function at the Basic and Clinical Level. J. Endocr. Soc. 2020, 4, bvz038. [Google Scholar] [CrossRef] [PubMed]
- Cancela, L.; Theofan, G.; Norman, A.W. The Pleiotropic Vitamin D Hormone; Cooke, B.A., van der Molen, H.J., Eds.; New Comprehensive Biochemistry; Elsevier: Amsterdam, The Netherlands, 1988; Volume 18, Part A. [Google Scholar]
- Carlberg, C.; Muñoz, A. An update on vitamin D signaling and cancer. Semin. Cancer Biol. 2020, 79, 217–230. [Google Scholar] [CrossRef]
- Pike, J.W.; Meyer, M.B. New Approaches to Assess Mechanisms of Action of Selective Vitamin D Analogues. Int. J. Mol. Sci. 2021, 22, 12352. [Google Scholar] [CrossRef] [PubMed]
- Kutner, A.; Brown, G. Vitamins D: Relationship between Structure and Biological Activity. Int. J. Mol. Sci. 2018, 19, 2119. [Google Scholar] [CrossRef] [Green Version]
- Bouillon, R.; Marcocci, C.; Carmeliet, G.; Bikle, D.; White, J.H.; Dawson-Hughes, B.; Lips, P.; Munns, C.F.; Lazaretti-Castro, M.; Giustina, A.; et al. Skeletal and Extraskeletal Actions of Vitamin D: Current Evidence and Outstanding Questions. Endocr. Rev. 2018, 40, 1109–1151. [Google Scholar] [CrossRef] [Green Version]
- Milczarek, M.; Rossowska, J.; Klopotowska, D.; Stachowicz, M.; Kutner, A.; Wietrzyk, J. Tacalcitol increases the sensitivity of colorectal cancer cells to 5-fluorouracil by downregulating the thymidylate synthase. J. Steroid Biochem. Mol. Biol. 2019, 190, 139–151. [Google Scholar] [CrossRef]
- Nachliely, M.; Trachtenberg, A.; Khalfin, B.; Nalbandyan, K.; Cohen-Lahav, M.; Yasuda, K.; Sakaki, T.; Kutner, A.; Danilenko, M. Dimethyl fumarate and vitamin D derivatives cooperatively enhance VDR and Nrf2 signaling in differentiating AML cells in vitro and inhibit leukemia progression in a xenograft mouse model. J. Steroid Biochem. Mol. Biol. 2019, 188, 8–16. [Google Scholar] [CrossRef]
- Trynda, J.; Turlej, E.; Milczarek, M.; Pietraszek, A.; Chodyński, M.; Kutner, A.; Wietrzyk, J. Antiproliferative Activity and in Vivo Toxicity of Double-Point Modified Analogs of 1,25-Dihydroxyergocalciferol. Int. J. Mol. Sci. 2015, 16, 24873–24894. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, A.; Nadkarni, S.; Yasuda, K.; Sakaki, T.; Brown, G.; Kutner, A.; Marcinkowska, E. Biological Evaluation of Double Point Modified Analogues of 1,25-Dihydroxyvitamin D2 as Potential Anti-Leukemic Agents. Int. J. Mol. Sci. 2016, 17, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filip-Psurska, B.; Psurski, M.; Anisiewicz, A.; Libako, P.; Zbrojewicz, E.; Maciejewska, M.; Chodyński, M.; Kutner, A.; Wietrzyk, J. Vitamin D Compounds PRI-2191 and PRI-2205 Enhance Anastrozole Activity in Human Breast Cancer Models. Int. J. Mol. Sci. 2021, 22, 2781. [Google Scholar] [CrossRef]
- Maj, E.; Maj, B.; Bobak, K.; Gos, M.; Chodyński, M.; Kutner, A.; Wietrzyk, J. Differential Response of Lung Cancer Cells, with Various Driver Mutations, to Plant Polyphenol Resveratrol and Vitamin D Active Metabolite PRI-2191. Int. J. Mol. Sci. 2021, 22, 2354. [Google Scholar] [CrossRef] [PubMed]
- Baurska, H.; Klopot, A.; Kielbinski, M.; Chrobak, A.; Wijas, E.; Kutner, A.; Marcinkowska, E. Structure–function analysis of vitamin D2 analogs as potential inducers of leukemia differentiation and inhibitors of prostate cancer proliferation. J. Steroid Biochem. Mol. Biol. 2011, 126, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Milczarek, M.; Chodyński, M.; Pietraszek, A.; Stachowicz-Suhs, M.; Yasuda, K.; Sakaki, T.; Wietrzyk, J.; Kutner, A. Synthesis, CYP24A1-Dependent Metabolism and Antiproliferative Potential against Colorectal Cancer Cells of 1,25-Dihydroxyvitamin D2 Derivatives Modified at the Side Chain and the A-Ring. Int. J. Mol. Sci. 2020, 21, 642. [Google Scholar] [CrossRef] [Green Version]
- Kotlarz, A.; Przybyszewska, M.; Swoboda, P.; Miłoszewska, J.; Grygorowicz, M.A.; Kutner, A.; Markowicz, S. Differential interference of vitamin D analogs PRI-1906, PRI-2191, and PRI-2205 with the renewal of human colon cancer cells refractory to treatment with 5-fluorouracil. Tumor Biol. 2015, 37, 4699–4709. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, A.; Wierzbicka, J.; Kwiatkowska, K.; Chodyński, M.; Kutner, A.; Żmijewski, M.A. Antiproliferative activity of side-chain truncated vitamin D analogs (PRI-1203 and PRI-1204) against human malignant melanoma cell lines. Eur. J. Pharmacol. 2020, 881, 173170. [Google Scholar] [CrossRef] [PubMed]
- Piątek, K.; Kutner, D.A.; Castillo-Tong, D.C.; Manhardt, T.; Kupper, N.; Nowak, U.; Chodyński, M.; Marcinkowska, E.; Kallay, E.; Schepelmann., M. Vitamin D Analogs Regulate the Vitamin D System and Proliferation in Ovarian Cancer Cells. Int. J. Mol. Sci. 2022, 23, 172. [Google Scholar] [CrossRef] [PubMed]
- Rochel, N.; Wurtz, J.; Mitschler, A.; Klaholz, B.; Moras, D. The Crystal Structure of the Nuclear Receptor for Vitamin D Bound to Its Natural Ligand. Mol. Cell 2000, 5, 173–179. [Google Scholar] [CrossRef]
- Tocchini-Valentini, G.; Rochel, N.; Wurtz, J.M.; Mitschler, A.; Moras, D. Crystal structures of the vitamin D receptor complexed to superagonist 20-epi ligands. Proc. Natl. Acad. Sci. USA 2001, 98, 5491–5496. [Google Scholar] [CrossRef] [Green Version]
- Brélivet, Y.; Rochel, N.; Moras, D. Structural analysis of nuclear receptors: From isolated domains to integral proteins. Mol. Cell. Endocrinol. 2012, 348, 466–473. [Google Scholar] [CrossRef]
- Kotlarz, A.; Przybyszewska, M.; Swoboda, P.; Neska, J.; Miłoszewska, J.; Grygorowicz, M.A.; Kutner, A.; Markowicz, S. Imatinib inhibits the regrowth of human colon cancer cells after treatment with 5-FU and cooperates with vitamin D analogue PRI-2191 in the downregulation of expression of stemness-related genes in 5-FU refractory cells. J. Steroid Biochem. Mol. Biol. 2019, 189, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Ben-Eltriki, M.; Deb, S.; Guns, E.S.T. 1α,25-Dihydroxyvitamin D3 synergistically enhances anticancer effects of ginsenoside Rh2 in human prostate cancer cells. J. Steroid Biochem. Mol. Biol. 2021, 209, 105828. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, F.; Mototani, S.; Yasuda, K.; Mano, H.; Sakaki, T.; Kittaka, A. Stereoselective Synthesis of 24-Fluoro-25-Hydroxyvitamin D3 Analogues and Their Stability to hCYP24A1-Dependent Catabolism. Int. J. Mol. Sci. 2021, 22, 11863. [Google Scholar] [CrossRef]
- Urushino, N.; Yasuda, K.; Ikushiro, S.; Kamakura, M.; Ohta, M.; Sakaki, T. Metabolism of 1α,25-dihydroxyvitamin D2 by human CYP24A1. Biochem. Biophys. Res. Commun. 2009, 384, 144–148. [Google Scholar] [CrossRef]
- Annalora, A.J.; Goodin, D.B.; Hong, W.-X.; Zhang, Q.; Johnson, E.F.; Stout, C.D. Crystal Structure of CYP24A1, a Mitochondrial Cytochrome P450 Involved in Vitamin D Metabolism. J. Mol. Biol. 2010, 396, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltis, S.M.; Cohen, A.E.; Deacon, A.; Eriksson, T.; González, A.; McPhillips, S.; Chui, H.; Dunten, P.; Hollenbeck, M.; Mathews, I.; et al. New paradigm for macromolecular crystallography experiments at SSRL: Automated crystal screening and remote data collection. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 1210–1221. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.A.; Cosme, J.; Vinković, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal Structures of Human Cytochrome P450 3A4 Bound to Metyrapone and Progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-ray Crystallography to 2.05-Å Resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef] [Green Version]
- Ekroos, M.; Sjögren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.P.; He, Y.A.; Patrick, K.S.; Halpert, J.R.; Bell, N.H. CYP3A4 Is a Vitamin D-24- and 25-Hydroxylase: Analysis of Structure Function by Site-Directed Mutagenesis. J. Clin. Endocrinol. Metab. 2005, 90, 1210–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Hashizume, T.; Shuhart, M.C.; Davis, C.L.; Nelson, W.L.; Sakaki, T.; Kalhorn, T.F.; Watkins, P.B.; Schuetz, E.G.; Thummel, K.E. Intestinal and Hepatic CYP3A4 Catalyze Hydroxylation of 1α,25-Dihydroxyvitamin D3: Implications for Drug-Induced Osteomalacia. Mol. Pharmacol. 2005, 69, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P.; Dusek, J.; Smutny, T.; Lochman, L.; Kucera, R.; Skoda, J.; Smutna, L.; Kamaraj, R.; Soucek, P.; Vrzal, R.; et al. Gene Expression Profiling of 1. Mol. Nutr. Food Res. 2022, 66, 2200070. [Google Scholar] [CrossRef]
- Wang, Z.; Schuetz, E.G.; Xu, Y.; Thummel, K.E. Interplay between vitamin D and the drug metabolizing enzyme CYP3A4. J. Steroid. Biochem. Mol. Biol. 2012, 136, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomaa, M.S.; Simons, C.; Brancale, A. Homology model of 1α,25-dihydroxyvitamin D3 24-hydroxylase cytochrome P450 24A1 (CYP24A1): Active site architecture and ligand binding. J. Steroid Biochem. Mol. Biol. 2007, 104, 53–60. [Google Scholar] [CrossRef]
- Stjernschantz, E.; Vermeulen, N.P.; Oostenbrink, C. Computational prediction of drug binding and rationalisation of selectivity towards cytochromes P450. Expert Opin. Drug Metab. Toxicol. 2008, 4, 513–527. [Google Scholar] [CrossRef]
- Zołek, T.; Dömötör, O.; Ostrowska, K.; Enyedy, É.A.; Maciejewska, D. Evaluation of blood-brain barrier penetration and examination of binding to human serum albumin of 7-O-arylpiperazinylcoumarins as potential antipsychotic agents. Bioorg. Chem. 2019, 84, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Żołek, T.; Dömötör, O.; Rezler, M.; Enyedy, A.; Maciejewska, D. Deposition of pentamidine analogues in the human body—Spectroscopic and computational approaches. Eur. J. Pharm. Sci. 2021, 161, 105779. [Google Scholar] [CrossRef]
- Bolla, N.R.; Corcoran, A.; Yasuda, K.; Chodyński, M.; Krajewski, K.; Cmoch, P.; Marcinkowska, E.; Brown, G.; Sakaki, T.; Kutner, A. Synthesis and evaluation of geometric analogs of 1α,25-dihydroxyvitamin D2 as potential therapeutics. J. Steroid. Biochem. Mol. Biol. 2015, 164, 50–55. [Google Scholar] [CrossRef]
- Pietraszek, A.; Malińska, M.; Chodyński, M.; Krupa, M.; Krajewski, K.; Cmoch, P.; Woźniak, K.; Kutner, A. Synthesis and crystallographic study of 1,25-dihydroxyergocalciferol analogs. Steroids 2013, 78, 1003–1014. [Google Scholar] [CrossRef]
- Suwinska, K.; Kutner, A. Crystal and molecular structure of 1,25-dihydroxycholecalciferol. Acta Crystallogr. Sect. B Struct. Sci. 1996, 52, 550–554. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, Inc., Wallingford CT. 2016. Available online: https://gaussian.com/citation/ (accessed on 13 July 2022).
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systèmes, Discovery Studio Modeling Environment, San Diego: Dassault Systèmes. 2021. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 13 July 2022).
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Sagui, C.; Darden, T.A. MOLECULAR DYNAMICS SIMULATIONS OF BIOMOLECULES: Long-Range Electrostatic Effects. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 155–179. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vitamin D Metabolite and Its Analogs | Metabolic Conversion by CYP24A1 (%) ± SD | Free Enthalpy of Binding to CYP3A4 * ΔGbind (kcal/mol) |
|---|---|---|
| 1,25D2 | 34 ± 5 | −24 |
| PRI-5106 | 26 ± 3 | −37 |
| PRI-5105 | 25 ± 2 | −27 |
| PRI-1916 | 13 ± 4 | −52 |
| PRI-1917 | 10 ± 1 | −65 |
| PRI-1906 | 1.9 ± 0.7 | −76 |
| PRI-1907 | 0.9 ± 0.1 | −120 |
| PRI-5201 | 1.9 ± 0.1 | −91 |
| PRI-5202 | 2.0 ± 0.2 | −100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żołek, T.; Yasuda, K.; Brown, G.; Sakaki, T.; Kutner, A. In Silico Prediction of the Metabolic Resistance of Vitamin D Analogs against CYP3A4 Metabolizing Enzyme. Int. J. Mol. Sci. 2022, 23, 7845. https://doi.org/10.3390/ijms23147845
Żołek T, Yasuda K, Brown G, Sakaki T, Kutner A. In Silico Prediction of the Metabolic Resistance of Vitamin D Analogs against CYP3A4 Metabolizing Enzyme. International Journal of Molecular Sciences. 2022; 23(14):7845. https://doi.org/10.3390/ijms23147845
Chicago/Turabian StyleŻołek, Teresa, Kaori Yasuda, Geoffrey Brown, Toshiyuki Sakaki, and Andrzej Kutner. 2022. "In Silico Prediction of the Metabolic Resistance of Vitamin D Analogs against CYP3A4 Metabolizing Enzyme" International Journal of Molecular Sciences 23, no. 14: 7845. https://doi.org/10.3390/ijms23147845