
Neuroprotective and Regenerative Effects of Growth Hormone (GH) in the Embryonic Chicken Cerebral Pallium Exposed to Hypoxic–Ischemic (HI) Injury

 and
and 
Abstract
:1. Introduction
2. Results
2.1. Neuroprotective Effects of GH in Pallial Cell Cultures Exposed to HI Injury
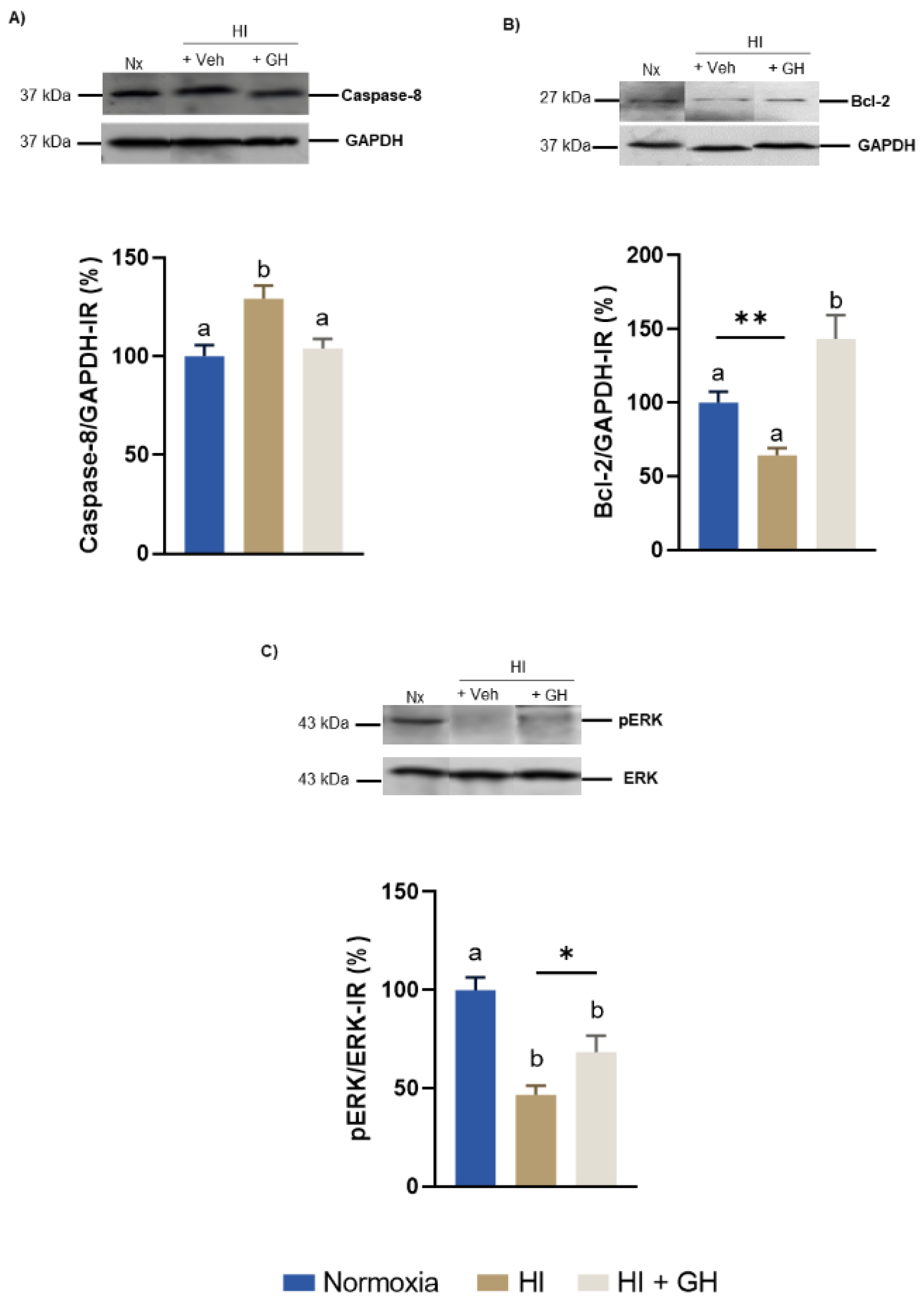
2.2. Effects of GH upon the Expression of Caspase-8, Bcl-2, ERK1/2 in Pallial Tissue after Exposing Chicken Embryos to HI Injury In Vivo
2.3. GH Increases Doublecortin IR and NeuN IR in Pallial Cultures Exposed to HI Injury
2.4. GH Promotes Cell Proliferation Both In Vitro and In Vivo after HI Injury
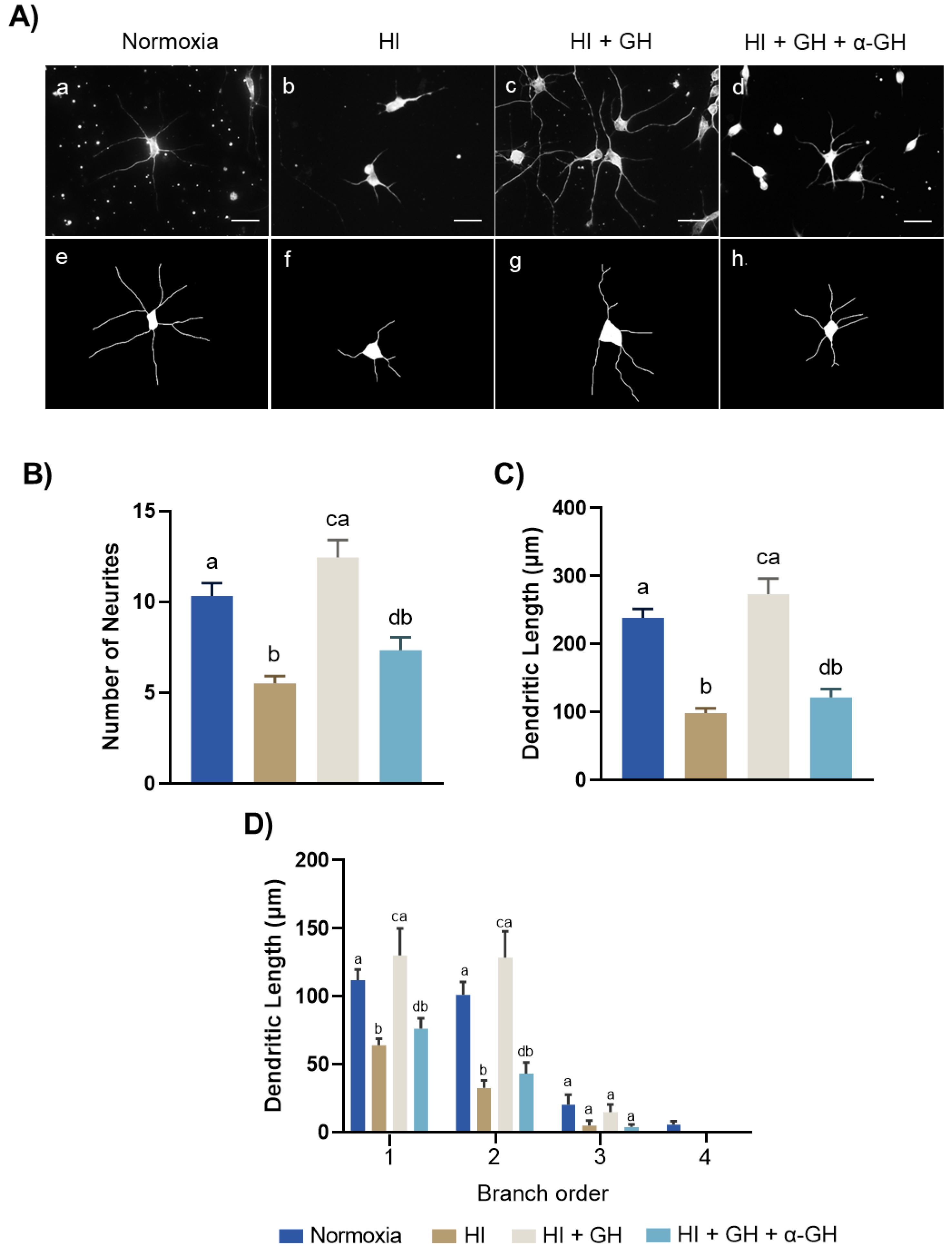
2.5. GH Promotes Neurite Outgrowths in Pallial Cell Cultures after HI Injury
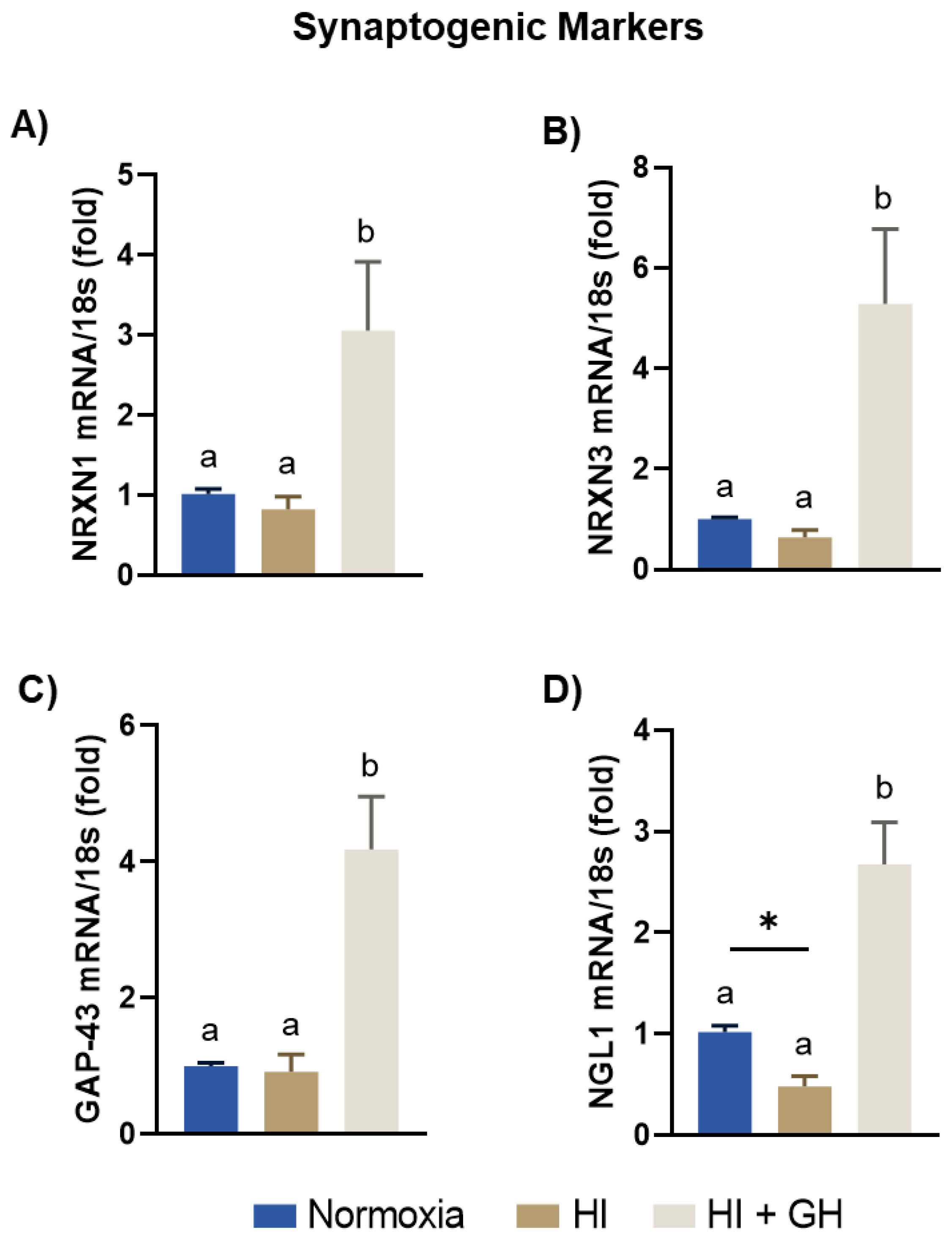
2.6. GH Promotes the Expression of Synaptogenic Markers in Pallial Cell Cultures after HI Injury
2.7. GH Regulates the Expression of Local GH, BDNF, IGF-1, NT-3, BMP4, and GHR mRNAs in Pallial Cultures after HI Injury
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Primary Pallial Cell Cultures
4.3. Treatments
4.3.1. In Vitro Experiments
4.3.2. In Vivo Experiments
4.4. Determination of Cell Survival
4.5. Immunofluorescent Staining
4.6. Cell Quantification
4.7. SDS-PAGE and Western Blot Analysis
4.8. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.9. Quantitative PCR (qPCR)
4.10. Determination of Apoptosis by Caspase-3 Activity
4.11. Neurite Growth Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahearne, C.E.; Boylan, G.B.; Murray, D.M. Short- and long-term prognosis in perinatal asphyxia: An update. World J. Clin. Pediatr. 2016, 5, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Ischemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef]
- Chen, H.; Lin, W.; Zhang, Y.; Lin, L.; Chen, J.; Zeng, Y.; Liu, N. IL-10 promotes neurite outgrowth and synapse formation in cultured cortical neurons after the oxygen-glucose deprivation via JAK1/STAT3 pathway. Sci. Rep. 2016, 6, 30459. [Google Scholar] [CrossRef]
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell death pathways in ischemic stroke and targeted pharmacotherapy. Transl. Stroke Res. 2020, 11, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Wassink, G.; van den Heuij, L.G.; Bennet, L.; Gunn, A.J. Therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy—Where to from here? Front. Neurol. 2015, 6, 198. [Google Scholar] [CrossRef]
- Arteaga, O.; Álvarez, A.; Revuelta, M.; Santaolalla, F.; Urtasun, A.; Hilario, E. Role of antioxidants in neonatal hypoxic-ischemic brain injury: New therapeutic approaches. Int. J. Mol. Sci. 2017, 18, 265. [Google Scholar] [CrossRef] [PubMed]
- Åberg, N.D.; Brywe, K.G.; Isgaard, J. Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. Sci. World J. 2006, 6, 53–80. [Google Scholar] [CrossRef]
- Arámburo, C.; Alba-Betancourt, C.; Luna, M.; Harvey, S. Expression and function of growth hormone in the nervous system: A brief review. Gen. Comp. Endocrinol. 2014, 203, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.; Baudet, M.L.; Sanders, E.J. Growth hormone-induced neuroprotection in the neural retina during chick embryogenesis. Ann. N. Y. Acad. Sci. 2009, 1163, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Regalado-Santiago, C.; López-Meraz, M.L.; Santiago-García, J.; Fernández–Pomares, C.; Juárez-Aguilar, E. Growth hormone (GH) is a survival rather than a proliferative factor for embryonic striatal neural precursor cells. Growth Horm. IGF Res. 2013, 23, 179–186. [Google Scholar] [CrossRef]
- Ajo, R.; Cacicedo, L.; Navarro, C.; Sánchez-Franco, F. Growth hormone action on proliferation and differentiation of cerebral cortical cells from fetal rat. Endocrinology 2003, 144, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, D.G.; Reynolds, B.A.; Golmohammadi, M.G.; Large, B.; Aguilar, R.M.; Haro, L.; Waters, M.J.; Rietze, R.L. Growth hormone responsive neural precursor cells reside within the adult mammalian brain. Sci. Rep. 2012, 2, 250. [Google Scholar] [CrossRef]
- Devesa, P.; Agasse, F.; Xapelli, S.; Almengló, C.; Devesa, J.; Malva, J.O.; Arce, V.M. Growth hormone pathways signaling for cell proliferation and survival in hippocampal neural precursors from postnatal mice. BMC Neurosci. 2014, 15, 100. [Google Scholar] [CrossRef]
- Nylander, E.; Zelleroth, S.; Stam, F.; Nyberg, F.; Grönbladh, A.; Hallberg, M. Growth hormone increases dendritic spine density in primary hippocampal cell cultures. Growth Horm. IGF Res. 2020, 50, 42–47. [Google Scholar] [CrossRef]
- Olivares-Hernández, J.D.; Balderas-Márquez, J.E.; Carranza, M.; Luna, M.; Martínez-Moreno, C.G.; Arámburo, C. Growth hormone (GH) enhances endogenous mechanisms of neuroprotection and neuroplasticity after oxygen and glucose deprivation injury (OGD) and reoxygenation (OGD/R) in chicken hippocampal cell cultures. Neural Plast. 2021, 2021, 9990166. [Google Scholar] [CrossRef]
- Fleming, T.; Martinez-Moreno, C.G.; Carranza, M.; Luna, M.; Harvey, S.; Arámburo, C. Growth hormone promotes synaptogenesis and protects neuroretinal dendrites against kainic acid (KA) induced damage. Gen. Comp. Endocrinol. 2018, 265, 111–120. [Google Scholar] [CrossRef]
- Jung, S.; Boie, G.; Doerr, H.G.; Trollmann, R. Oxygen-sensitive regulation and neuroprotective effects of growth hormone-dependent growth factors during early postnatal development. Am. J. Physiol-Reg. I. 2017, 312, R539–R548. [Google Scholar] [CrossRef]
- Jung, S.; Terörde, K.; Dörr, H.G.; Trollmann, R. Recombinant human growth hormone activates neuroprotective growth factors in hypoxic brain injury in neonatal mice. Endocrinology 2021, 162, bqab008. [Google Scholar] [CrossRef]
- Baltazar-Lara, R.; Ávila-Mendoza, J.; Martínez-Moreno, C.G.; Carranza, M.; Pech-Pool, S.; Vázquez-Martínez, O.; Díaz-Muñoz, M.; Luna, M.; Arámburo, C. Neuroprotective effects of growth hormone (GH) and insulin-like growth factor type 1 (IGF-1) after hypoxic-ischemic injury in chicken cerebellar cell cultures. Int. J. Mol. Sci. 2021, 22, 256. [Google Scholar] [CrossRef]
- Johnston, M.V.; Trescher, W.H.; Ishida, A.; Nakajima, W. Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr. Res. 2001, 49, 735–741. [Google Scholar] [CrossRef]
- McQuillen, P.S.; Sheldon, R.A.; Shatz, C.J.; Ferriero, D.M. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J. Neurosci. 2003, 23, 3308–3315. [Google Scholar] [CrossRef] [PubMed]
- Sanches, E.F.; Arteni, N.S.; Nicola, F.; Boisserand, L.; Willborn, S.; Netto, C.A. Early hypoxia–ischemia causes hemisphere and sex-dependent cognitive impairment and histological damage. Neuroscience 2013, 237, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R. Genomic approach to selective vulnerability of the hippocampus in brain ischemia–hypoxia. Neuroscience 2015, 309, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Muntsant, A.; Shrivastava, K.; Recasens, M.; Giménez-Llort, L. Severe perinatal hypoxic-ischemic brain injury induces long-term sensorimotor deficits, anxiety-like behaviors and cognitive impairment in a sex-, age-and task-selective manner in C57BL/6 mice but can be modulated by neonatal handling. Front. Behav. Neurosci. 2019, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Scheepens, A.; Sirimanne, E.; Beilharz, E.; Breier, B.H.; Waters, M.J.; Gluckman, P.D.; Williams, C.E. Alterations in the neural growth hormone axis following hypoxic–ischemic brain injury. Mol. Brain Res. 1999, 68, 88–100. [Google Scholar] [CrossRef]
- Scheepens, A.; Sirimanne, E.S.; Breier, B.H.; Clark, R.G.; Gluckman, P.D.; Williams, C.E. Growth hormone as a neuronal rescue factor during recovery from CNS injury. Neuroscience 2001, 104, 677–687. [Google Scholar] [CrossRef]
- Christophidis, L.J.; Gorba, T.; Gustavsson, M.; Williams, C.E.; Werthe, G.A.; Russo, V.C.; Scheepens, A. Growth hormone receptor immunoreactivity is increased in the subventricular zone of juvenile rat brain after focal ischemia: A potential role for growth hormone in injury-induced neurogenesis. Growth Horm. IGF Res. 2009, 19, 497–506. [Google Scholar] [CrossRef]
- Zhang, H.; Han, M.; Zhang, X.; Sun, X.; Ling, F. The effect and mechanism of growth hormone replacement on cognitive function in rats with traumatic brain injury. PLoS ONE 2014, 9, e108518. [Google Scholar] [CrossRef]
- Ong, L.K.; Chow, W.Z.; TeBay, C.; Kluge, M.; Pietrogrande, G.; Zalewska, K.; Isgaard, J. Growth hormone improves cognitive function after experimental stroke. Stroke. 2018, 49, 1257–1266. [Google Scholar] [CrossRef]
- Sanchez-Bezanilla, S.; Åberg, N.D.; Crock, P.; Walker, F.R.; Nilsson, M.; Isgaard, J.; Ong, L.K. Growth hormone treatment promotes remote hippocampal plasticity after experimental cortical stroke. Int. J. Mol. Sci. 2020, 21, 4563. [Google Scholar] [CrossRef]
- Li, R.C.; Guo, S.Z.; Raccurt, M.; Moudilou, E.; Morel, G.; Brittian, K.R.; Gozal, D. Exogenous growth hormone attenuates cognitive deficits induced by intermittent hypoxia in rats. Neuroscience 2011, 196, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Arce, V.M.; Devesa, P.; Devesa, J. Role of growth hormone (GH) in the treatment on neural diseases: From neuroprotection to neural repair. Neurosci. Res. 2013, 76, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Díaz-Getino, G.; Rey, P.; García-Cancela, J.; Loures, I.; Nogueiras, S.; Hurtado de Mendoza, A.; Salgado, L.; González, M.; Pablos, T.; et al. Brain recovery after a plane crash: Treatment with growth hormone (GH) and neurorehabilitation. A case report. Int. J. Mol. Sci. 2015, 16, 30470–30482. [Google Scholar] [CrossRef] [PubMed]
- Mossberg, K.A.; Durham, W.J.; Zgaljardic, D.J.; Gilkison, C.R.; Danesi, C.P.; Sheffield-Moore, M.; Masel, B.E.; Urban, R.J. Functional changes after recombinant human growth hormone replacement in patients with chronic traumatic brain injury and abnormal growth hormone secretion. J. Neurotrauma. 2017, 34, 845–852. [Google Scholar] [CrossRef]
- Heredia, M.; Palomero, J.; De La Fuente, A.; Criado, J.M.; Yajeya, J.; Devesa, J.; Devesa, P.; Vicente-Villardón, J.L.; Riolobos, A.S. Motor improvement of skilled forelimb use induced by treatment with growth hormone and rehabilitation is dependent on the onset of the treatment after cortical ablation. Neural Plast. 2018, 2018, 6125901. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Reimunde, P.; Devesa, A.; Souto, S.; Lopez-Amado, M.; Devesa, P.; Arce, V.; Arce, V. Recovery from neurological sequelae secondary to oncological brain surgery in an adult growth hormone-deficient patient after growth hormone treatment. J. Rehabil. Med. 2009, 41, 775–777. [Google Scholar] [CrossRef] [PubMed]
- High, W.M.; Briones-Galang, M.; Clark, J.A.; Gilkison, C.; Mossberg, K.A.; Zgaljardic, D.J.; Masel, B.E.; Urban, R.J. Effect of growth hormone replacement therapy on cognition after traumatic brain injury. J. Neurotrauma 2010, 27, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Reimunde, P.; Quintana, A.; Castañón, B.; Casteleiro, N.; Vilarnovo, Z.; Otero, A.; Devesa, A.; Otero-Cepeda, X.L.; Devesa, J. Effects of growth hormone (GH) replacement and cognitive rehabilitation in patients with cognitive disorders after traumatic brain injury. Brain Inj. 2011, 25, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Park, K.; Lee, H.; Kim, M. The effect of recombinant human growth hormone therapy in patients with completed stroke: A Pilot Trial. Ann. Rehabil. Med. 2012, 36, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Devesa, P.; Reimunde, P.; Arce, V. Growth hormone and kynesitherapy for brain injury recovery. In Brain Injury—Pathogenesis, Monitoring, Recovery and Management; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Moreau, O.K.; Cortet-Rudelli, C.; Yollin, E.; Merlen, E.; Daveluy, W.; Rousseaux, M. Growth hormone replacement therapy in patients with traumatic brain injury. J. Neurotrauma 2013, 30, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Perkel, D.J.; Bruce, L.L.; Butler, A.B.; Csillag, A.; Kuenzel, W.; Medina, L.; Paxinos, G.; Shimizu, T.; Striedter, G.; et al. Revised nomenclature for avian telencephalon and some related brainstem nuclei. J. Comp. Neurol. 2004, 473, 377–414. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Güntürkün, O.; Bruce, L.; Csillag, A.; Karten, H.; Kuenzel, W.; Medina, L.; Paxinos, G.; Perkel, D.J.; Shimizu, T.; et al. Avian brain nomenclature consortium. Avian brains and a new understanding of vertebrate brain evolution. Nat. Rev. Neurosci. 2005, 6, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kuenzel, W.J. Research advances made in the avian brain and their relevance to poultry scientists. Poult. Sci. 2014, 93, 2945–2952. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain injury in the premature infant: Neuropathology, clinical aspects, and pathogenesis. Dev. Disabil. Res. Rev. 1997, 3, 3–12. [Google Scholar] [CrossRef]
- Sie, L.T.L.; Van der Knaap, M.S.; Oosting, J.; De Vries, L.S.; Lafeber, H.N.; Valk, J. MR patterns of hypoxic-ischemic brain damage after prenatal, perinatal or postnatal asphyxia. Neuropediatrics 2000, 31, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, A.; Wilson, M.A.; Johnston, M.V. Hypoxic-ischemic encephalopathy in the term infant. Clin. Perinatol. 2009, 36, 835–858. [Google Scholar] [CrossRef] [PubMed]
- Koehler, R.C.; Yang, Z.J.; Lee, J.K.; Martin, L.J. Perinatal hypoxic-ischemic brain injury in large animal models: Relevance to human neonatal encephalopathy. J. Cereb. Blood Flow Metab. 2018, 38, 2092–2111. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; Freund, T.F. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 1991, 40, 599–636. [Google Scholar] [CrossRef]
- Bernaudin, M.; Nouvelot, A.; MacKenzie, E.T.; Petit, E. Selective neuronal vulnerability and specific glial reactions in hippocampal and neocortical organotypic cultures submitted to ischemia. Exp. Neurol. 1998, 150, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Gailus, B.; Naundorf, H.; Welzel, L.; Johne, M.; Römermann, K.; Kaila, K.; Löscher, W. Long-term outcome in a noninvasive rat model of birth asphyxia with neonatal seizures: Cognitive impairment, anxiety, epilepsy, and structural brain alterations. Epilepsia 2021, 62, 2826–2844. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, K.; Hagberg, H.; Bengtsson, B.A.; Brantsing, C.; Isgaard, J. Possible protective role of growth hormone in hypoxia-ischemia in neonatal rats. Pediatr. Res. 1999, 45, 318–323. [Google Scholar] [CrossRef]
- Alba-Betancourt, C.; Luna-Acosta, J.L.; Ramirez-Martínez, C.E.; Avila-Gonzalez, D.; Granados-Avalos, E.; Carranza, M.; Martínez-Coria, H.; Arámburo, C.; Luna, M. Neuro-protective effects of growth hormone (GH) after hypoxia–ischemia injury in embryonic chicken cerebellum. Gen. Comp. Endocrinol. 2013, 183, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Mendoza, J.; Mora, J.; Carranza, M.; Luna, M.; Arámburo, C. Growth hormone reverses excitotoxic damage induced by kainic acid in the green iguana neuroretina. Gen. Comp. Endocrinol. 2016, 234, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moreno, C.G.; Ávila-Mendoza, J.; Wu, Y.; Arellanes-Licea, E.C.; Louie, M.; Luna, M.; Arámburo, C.; Harvey, S. Neuroprotection by GH against excitotoxic-induced cell death in retinal ganglion cells. Gen. Comp. Endocrinol. 2016, 234, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moreno, C.G.; Fleming, T.; Carranza, M.; Ávila-Mendoza, J.; Luna, M.; Harvey, S.; Arámburo, C. Growth hormone protects against kainate excitotoxicity and induces BDNF and NT3 expression in chicken neuroretinal cells. Exp. Eye Res. 2018, 166, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sanders, E.J.; Lin, W.Y.; Parker, E.; Harvey, S. Growth hormone promotes the survival of retinal cells in vivo. Gen. Comp. Endocrinol. 2011, 172, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Lee, E.; Kim, J.W.; Kwon, B.S.; Jung, M.K.; Jee, Y.H.; Kim, J.; Bae, S.R.; Chang, Y.P. Protective effect of growth hormone on neuronal apoptosis after hypoxia-ischemia in the neonatal rat brain. Neurosci. Lett. 2004, 354, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Control of oxygen homeostasis in health and disease. Pediatr. Res. 2001, 49, 614–617. [Google Scholar] [CrossRef]
- Giusti, S.; Fiszer de Plazas, S. Neuroprotection by hypoxic preconditioning involves upregulation of hypoxia-inducible factor-1 in a prenatal model of acute hypoxia. J. Neurosci. Res. 2012, 90, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, M.; Gidday, J.M.; Yu, A.Y.; Semenza, G.L.; Ferriero, D.M.; Sharp, F.R. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann. Neurol. 2000, 48, 285–296. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Cho, M.; Kim, J.; Kaeberlein, M.; Lee, S.J.; Suh, Y. Syringaresinol protects against hypoxia/reoxygenation-induced cardiomyocytes injury and death by destabilization of HIF-1α in a FOXO3- dependent mechanism. Oncotarget 2014, 6, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, W.; Kang, Y.J. Copper affects the binding of HIF-1α to the critical motifs of its target genes. Metallomics 2019, 11, 429–438. [Google Scholar] [CrossRef]
- Amin, N.; Chen, S.; Ren, Q.; Tan, X.; Botchway, B.; Hu, Z.; Chen, F.; Ye, S.; Du, X.; Chen, Z.; et al. Hypoxia inducible factor-1α attenuates ischemic brain damage by modulating inflammatory response and glial activity. Cells 2021, 10, 1359. [Google Scholar] [CrossRef]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-inducible factors (HIFs) and phosphorylation: Impact on stability, localization, and transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Han, Y.; Leaman, D.W.; Watling, D.; Rogers, N.C.; Groner, B.; Kerr, I.M.; Wood, W.I.; Stark, G.R. Participation of JAK and STAT proteins in growth hormone-induced signaling. J. Biol. Chem. 1996, 271, 5947–5952. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Youle, R.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Chen, J.; Graham, S.H.; Chan, P.H.; Lan, J.; Zhou, R.L.; Simon, R.P. Bcl-2 is expressed in neurons that survive focal ischemia in the rat. Neuroreport 1995, 6, 394–398. [Google Scholar] [CrossRef]
- Clark, R.S.; Chen, J.; Watkins, S.C.; Kochanek, P.M.; Chen, M.; Stetler, R.A.; Graham, S.H. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J. Neurosci. 1997, 17, 9172–9182. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moreno, C.G.; Epardo, D.; Balderas-Márquez, J.E.; Fleming, T.; Carranza, M.; Luna, M.; Arámburo, C. Regenerative effect of growth hormone (GH) in the retina after kainic acid excitotoxic damage. Int. J. Mol. Sci. 2019, 20, 4433. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, X.O.; Zhu, Y.; Greenberg, D.A. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J. Neurochem. 2002, 80, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Yang, S.N. Perinatal hypoxic-ischemic encephalopathy. J. Biomed. Biotechnol. 2011, 2011, 609813. [Google Scholar] [CrossRef]
- Gleeson, J.G.; Lin, P.T.; Flanagan, L.A.; Walsh, C.A. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 1999, 23, 257–271. [Google Scholar] [CrossRef]
- Schaar, B.T.; Kinoshita, K.; McConnell, S.K. Doublecortin micro-tubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron 2004, 41, 203–213. [Google Scholar] [CrossRef]
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development 1992, 116, 201–211. [Google Scholar] [CrossRef]
- Jones, K.A.; Baughman, R.W. Both NMDA and non-NMDA subtypes of glutamate receptors are concentrated at synapses on cerebral cortical neurons in culture. Neuron 1991, 7, 593–603. [Google Scholar] [CrossRef]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Kumagai, A.; Sasaki, T.; Matsuoka, K.; Abe, M.; Tabata, T.; Itoh, Y.; Fuchino, H.; Wugangerile, S.; Suga, M.; Yamaguchi, T.; et al. Monitoring of glutamate-induced excitotoxicity by mitochondrial oxygen consumption. Synapse 2019, 73, e22067. [Google Scholar] [CrossRef]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Van Marle, G.; Antony, J.M.; Silva, C.; Sullivan, A.; Power, C. Aberrant cortical neurogenesis in a pediatric neuroAIDS model: Neurotrophic effects of growth hormone. AIDS 2005, 19, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Möderscheim, T.A.; Christophidis, L.J.; Williams, C.E.; Scheepens, A. Distinct neuronal growth hormone receptor ligand specificity in the rat brain. Brain Res. 2007, 1137, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Wang, X.; Xie, L.; Mao, X.O.; Zhu, W.; Wang, Y.; Shen, J.; Mao, Y.; Banwait, S.; Greenberg, D.A. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13198–13202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Y.; Li, J.; Kang, Q.; Tian, Y.; Chen, X.; Shi, Q.; Song, T. Cell proliferation in ependymal/subventricular zone and nNOS expression following focal cerebral ischemia in adult rats. Neurol. Res. 2006, 28, 91–96. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, Y.; Li, J.; Kang, Q.; Tian, Y.; Chen, X.; Shi, Q.; Song, T. Decreased neuronal nitric oxide synthase expression and cell migration in the peri-infarction after focal cerebral ischemia in rats. Neuropathology 2007, 27, 347–354. [Google Scholar] [CrossRef]
- Santilli, G.; Lamorte, G.; Carlessi, L.; Ferrari, D.; Rota, N.L.; Binda, E.; Delia, D.; Vescovi, A.L.; De Filippis, L. Mild hypoxia enhances proliferation and multipotency of human neural stem cells. PLoS ONE 2010, 5, e8575. [Google Scholar] [CrossRef]
- Ohira, K. Injury-induced neurogenesis in the mammalian forebrain. Cell Mol. Life Sci. 2011, 68, 1645–1656. [Google Scholar] [CrossRef]
- Ohab, J.J.; Fleming, S.; Blesch, A.; Carmichael, S.T. A neurovascular niche for neurogenesis after stroke. J. Neurosci. 2006, 26, 13007–13016. [Google Scholar] [CrossRef]
- Olivares-Hernández, J.D.; García-García, F.; Camacho-Abrego, I.; Flores, G.; Juárez-Aguilar, E. Intracerebroventricular administration of growth hormone induces morphological changes in pyramidal neurons of the hippocampus and prefrontal cortex in adult rats. Synapse 2018, 72, e22030. [Google Scholar] [CrossRef]
- Benowitz, L.I.; Routtenberg, A. GAP-43: An intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997, 20, 84–91. [Google Scholar] [CrossRef]
- Kawasaki, T.; Nishio, T.; Kawaguchi, S.; Kurosawa, H. Spatiotemporal distribution of GAP-43 in the developing rat spinal cord: A histological and quantitative immunofluorescence study. Neurosci Res. 2001, 39, 347–358. [Google Scholar] [CrossRef]
- Aigner, L.; Arber, S.; Kapfhammer, J.P.; Laux, T.; Schneider, C.; Botteri, F.; Brenner, H.R.; Caroni, P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell 1995, 83, 269–278. [Google Scholar] [CrossRef]
- Reissner, C.; Runkel, F.; Missler, M. Neurexins. Genome Biol. 2013, 14, 213. [Google Scholar] [CrossRef]
- Futai, K.; Kim, M.J.; Hashikawa, T.; Scheiffele, P.; Sheng, M.; Hayashi, Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95–neuroligin. Nat. Neurosci. 2007, 10, 186–195. [Google Scholar] [CrossRef]
- Gómez-Palacio-Schjetnan, A.; Escobar, M.L. Neurotrophins and synaptic plasticity. Curr. Top. Behav. Neurosci. 2013, 15, 117–136. [Google Scholar] [CrossRef]
- Conner, J.M.; Lauterborn, J.C.; Yan, Q.; Gall, C.M.; Varon, S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: Evidence for anterograde axonal transport. J. Neurosci. 1997, 17, 2295–2313. [Google Scholar] [CrossRef]
- Lindén, A.M.; Väisänen, J.; Lakso, M.; Nawa, H.; Wong, G.; Castrén, E. Expression of neurotrophins BDNF and NT-3, and their receptors in rat brain after administration of antipsychotic and psychotrophic agents. J. Mol. Neurosci. 2000, 14, 27–37. [Google Scholar] [CrossRef]
- Sharma, H.S. A select combination of neurotrophins enhances neuroprotection and functional recovery following spinal cord injury. Ann. N. Y. Acad. Sci. 2007, 1122, 95–111. [Google Scholar] [CrossRef]
- Musarò, A.; Dobrowolny, G.; Rosenthal, N. The neuroprotective effects of a locally acting IGF-1 isoform. Exp. Gerontol. 2007, 42, 76–80. [Google Scholar] [CrossRef]
- Ma, K.; Xu, H.; Zhang, J.; Zhao, F.; Liang, H.; Sun, H.; Li, P.; Zhang, S.; Wang, R.; Chen, X. Insulin-like growth factor-1 enhances neuroprotective effects of neural stem cell exosomes after spinal cord injury via an miR-219a-2-3p/YY1 mechanism. Aging 2019, 11, 12278–12294. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.S.; Yoon, J.Y.; Kim, M.Y.; Lee, S.H.; Choi, T.; Choi, K.Y. Bone morphogenetic protein 4 stimulates neuronal differentiation of neuronal stem cells through the ERK pathway. Exp. Mol. Med. 2009, 41, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef] [PubMed]
- Arámburo, C.; Navarrete, S.; Montiel, J.; Sánchez, R.; Berghman, L.R. Purification and electrophoretic analysis of glycosylated chicken growth hormone (G-cGH): Evidence of G-cGH isoforms. Gen. Comp. Endocrinol. 1999, 84, 135–146. [Google Scholar] [CrossRef]
- Fernandez, A.S.; Pieau, C.; Repérant, J.; Boncinelli, E.; Wassef, M. Expression of the Emx-1 and Dlx-1 homeobox genes define three molecularly distinct domains in the telencephalon of mouse, chick, turtle and frog embryos: Implications for the evolution of telencephalic subdivisions in amniotes. Development 1998, 125, 2099–2111. [Google Scholar] [CrossRef]
- Puelles, L.; Kuwana, E.; Puelles, E.; Bulfone, A.; Shimamura, K.; Keleher, J.; Smiga, S.; Rubenstein, J.L. Pallial and subpallial derivatives in the embryonic chick and mouse telencephalon, traced by the expression of the genes Dlx-2, Emx-1, Nkx-2.1, Pax-6, and Tbr-1. J. Comp. Neurol. 2000, 424, 409–438. [Google Scholar] [CrossRef]
- Fujita, T.; Aoki, N.; Fujita, E.; Matsushima, T.; Homma, K.J.; Yamaguchi, S. The chick pallium displays divergent expression patterns of chick orthologues of mammalian neocortical deep layer-specific genes. Sci. Rep. 2019, 9, 20400. [Google Scholar] [CrossRef]
- Kuenzel, W.J.; Masson, M. A Stereotaxic Atlas of the Brain of the Chick (Gallus domesticus), 1st ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 1988; 152p, ISBN 0-8018-3700-6. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) method. Methods 2001, 408, 402–408. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef]
- Bas, A.; Forsberg, G.; Hammarström, S.; Hammarström, M.L. Utility of the housekeeping genes 18S rRNA, β-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand. J. Immunol. 2004, 59, 566–573. [Google Scholar] [CrossRef]
- Luna-Acosta, J.L.; Alba-Betancourt, C.; Martínez-Moreno, C.G.; Ramírez, C.; Carranza, M.; Luna, M.; Arámburo, C. Direct antiapoptotic effects of growth hormone are mediated by PI3K/Akt pathway in the chicken bursa of Fabricius. Gen. Comp. Endocrinol. 2015, 224, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Host/Type | Dilution | Source | Cat. No. |
|---|---|---|---|---|
| DCX | Guinea pig/polyclonal | 1:250 (IF) 1:1000 (WB) | Millipore | AB2253 |
| NeuN | Mouse/monoclonal | 1:250 (IF) 1:1000 (WB) | Millipore | MAB377 |
| BrdU | Rat/monoclonal | 1:50 | Abcam | AB-6326 |
| β-III-tubulin | Mouse/monoclonal | 1:250 | Abcam | Ab78078 |
| HIF-1 α | Rabbit monoclonal | 1:1000 | Abcam | Ab2185 |
| Caspase-8 | Rabbit/polyclonal | 1:500 | Santa Cruz | SC-6134 |
| Bcl-2 | Rabbit/polyclonal | 1:500 | Santa Cruz | SC-492 |
| p44/42 MAPK (Erk1/2) | Mouse/monoclonal | 1:1000 | Cell signaling | 91065 |
| P-p44/42 MAPK (Erk1/2) | Mouse/monoclonal | 1:1000 | Cell signaling | 4370S |
| GAPDH | Rabbit/monoclonal | 1:3000 | Cell signaling | 14C10 |
| Guinea pig IgG | Goat/Alexa fluor 488 | 1:1000 | Invitrogen | A-11073 |
| Mouse IgG | Goat/Alexa fluor 595 | 1:1000 | Invitrogen | A-11032 |
| Rat IgG | Goat/Alexa fluor 488 | 1:1000 | Invitrogen | A-11029 |
| Guinea pig IgG | Goat/HRPconjugated | 1:3000 | Millipore | AP108P |
| Mouse IgG | Goat/HRPconjugated | 1:3000 | Abcam | AB-20043 |
| Rabbit IgG | Goat/HRPconjugated | 1:3000 | Invitrogen | A-65611 |
| Target | Primer | Sequence (5′-3′) | Size | Accesion # |
|---|---|---|---|---|
| cGH | Fwd Rev | CGCACCTATATTCCGGAGGAC GGCAGCTCCATGTCTGACT | 128 bp | NM_204359 |
| cBDNF | Fwd Rev | AGCAGTCAAGTGCCTTTGGA TCCGCTGCTGTTACCCACTCG | 167 bp | NM_001031616 |
| cIGF1 | Fwd Rev | TACCTTGGCCTGTGTTTGCT CCCTTGTGGTGTAAGCGTCT | 170 bp | NM_001004384 |
| cNT3 | Fwd Rev | AGGCAGCAGAGACGCTACAAC AGCACAGTTACCTGGTGTCCT | 248 bp | NM_001109762 |
| cBMP4 | Fwd Rev | CGCTGGGAGACCTTTGATGT CCCCTGAGGTAAAGATCGGC | 153 bp | NM_205237.3 |
| cGHR | Fwd Rev | ACTTCACCATGGACAATGCCTA GGGGTTTCTGCCATTGAAGCTC | 180 bp | NM_001001293.1 |
| cNRXN1 | Fwd Rev | CCACTCTGATCATTGACCGGG CGCCAGACCTTCCACATAGT | 392 bp | NM_001198975.1 |
| cNRXN3 | Fwd Rev | GCTGGGTCTCTCTTTGGGTC CACCCACAA AAAGGTCGCTG | 394 bp | NM_001271923.1 |
| cNLG1 | Fwd Rev | CTCCAGTGTGTCCCCAGA AC CATCACAGGCTTAGGTCCCC | 170 bp | NM_001081502.1 |
| cGAP43 | Fwd Rev | AGGAGCCTAAACAAGCCGAC TGCTGGGCACTTTCAGTAGG | 178 bp | NM_001305054.1 |
| C18s | Fwd Rev | CTCTTTCTCGATTCCGTGGGT TTAGCATGCCAGAGTCTCGT | 100 bp | M59389 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivares-Hernández, J.D.; Carranza, M.; Balderas-Márquez, J.E.; Epardo, D.; Baltazar-Lara, R.; Ávila-Mendoza, J.; Martínez-Moreno, C.G.; Luna, M.; Arámburo, C. Neuroprotective and Regenerative Effects of Growth Hormone (GH) in the Embryonic Chicken Cerebral Pallium Exposed to Hypoxic–Ischemic (HI) Injury. Int. J. Mol. Sci. 2022, 23, 9054. https://doi.org/10.3390/ijms23169054
Olivares-Hernández JD, Carranza M, Balderas-Márquez JE, Epardo D, Baltazar-Lara R, Ávila-Mendoza J, Martínez-Moreno CG, Luna M, Arámburo C. Neuroprotective and Regenerative Effects of Growth Hormone (GH) in the Embryonic Chicken Cerebral Pallium Exposed to Hypoxic–Ischemic (HI) Injury. International Journal of Molecular Sciences. 2022; 23(16):9054. https://doi.org/10.3390/ijms23169054
Chicago/Turabian StyleOlivares-Hernández, Juan David, Martha Carranza, Jerusa Elienai Balderas-Márquez, David Epardo, Rosario Baltazar-Lara, José Ávila-Mendoza, Carlos G. Martínez-Moreno, Maricela Luna, and Carlos Arámburo. 2022. "Neuroprotective and Regenerative Effects of Growth Hormone (GH) in the Embryonic Chicken Cerebral Pallium Exposed to Hypoxic–Ischemic (HI) Injury" International Journal of Molecular Sciences 23, no. 16: 9054. https://doi.org/10.3390/ijms23169054