
Home Dust Mites Promote MUC5AC Hyper-Expression by Modulating the sNASP/TRAF6 Axis in the Airway Epithelium

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TLR Signaling Is Required for HDM-Induced Elevation of MUC5AC Levels in 16HBE Cells
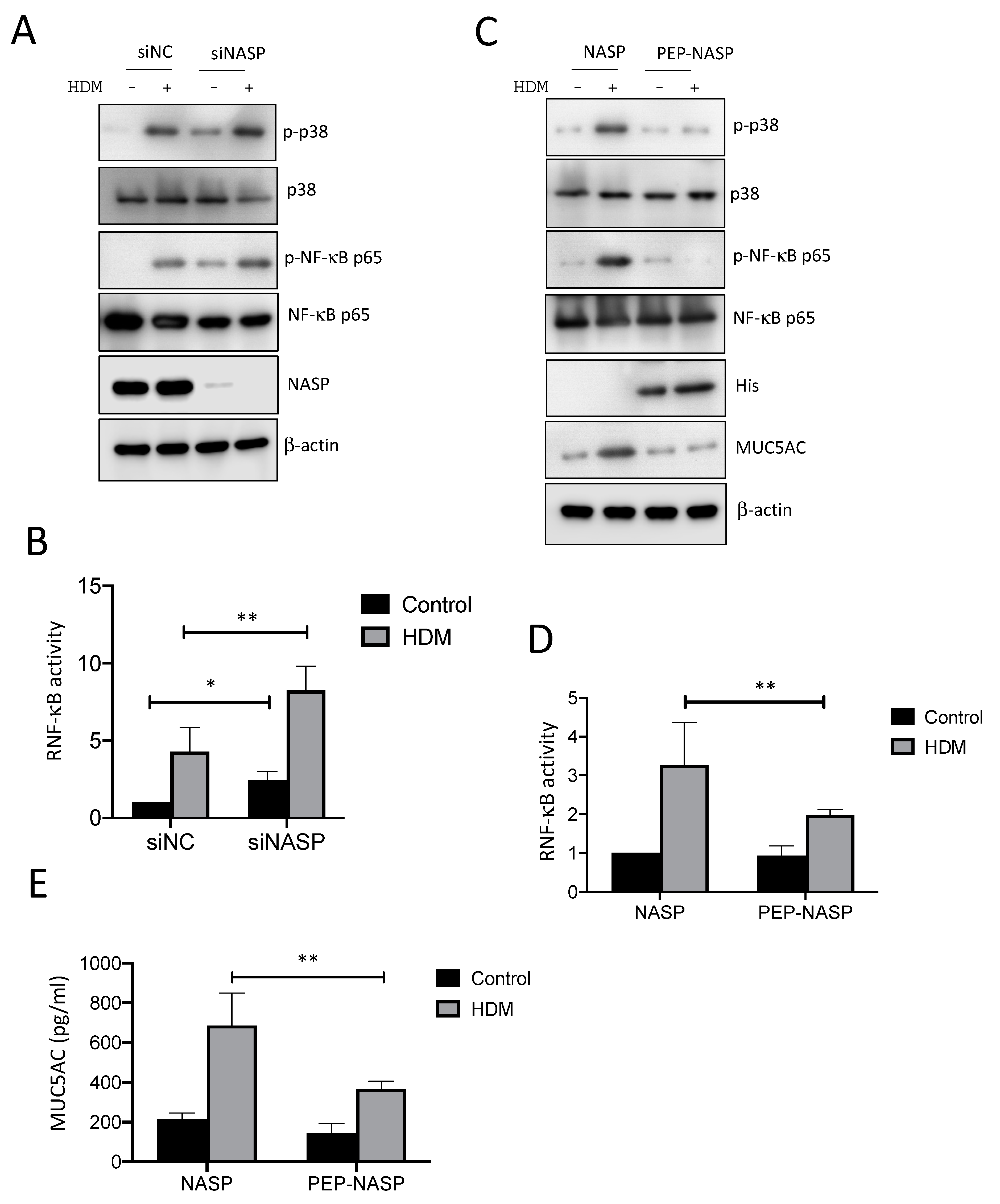
2.2. NASP-Knockdown Enhanced MUC5AC Production by HDMs
2.3. The PEP-NASP Peptide Negatively Regulates sNASP/TRAF6-Meidiated NF-κB Signaling Stimulated by the HDM Extract
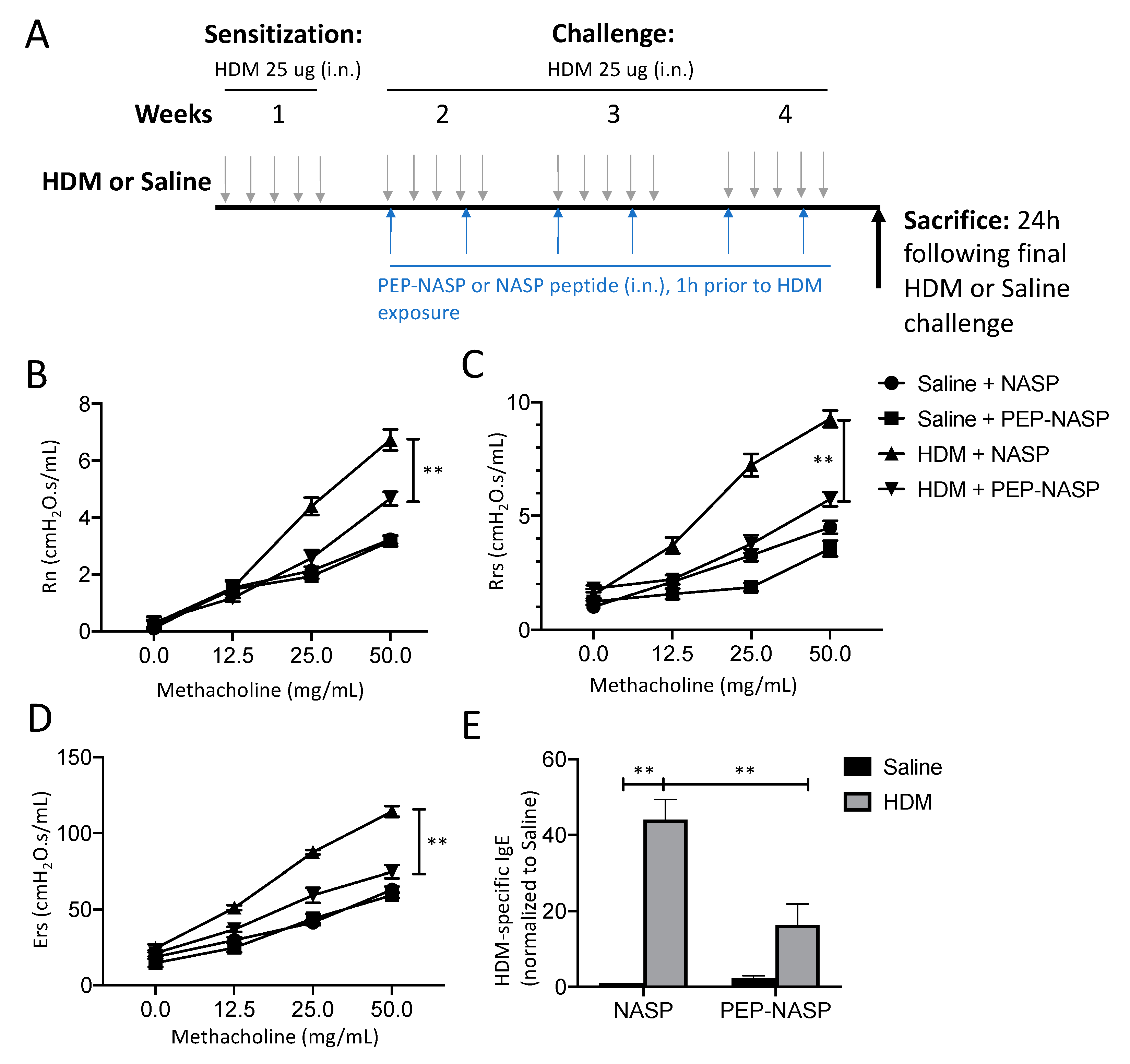
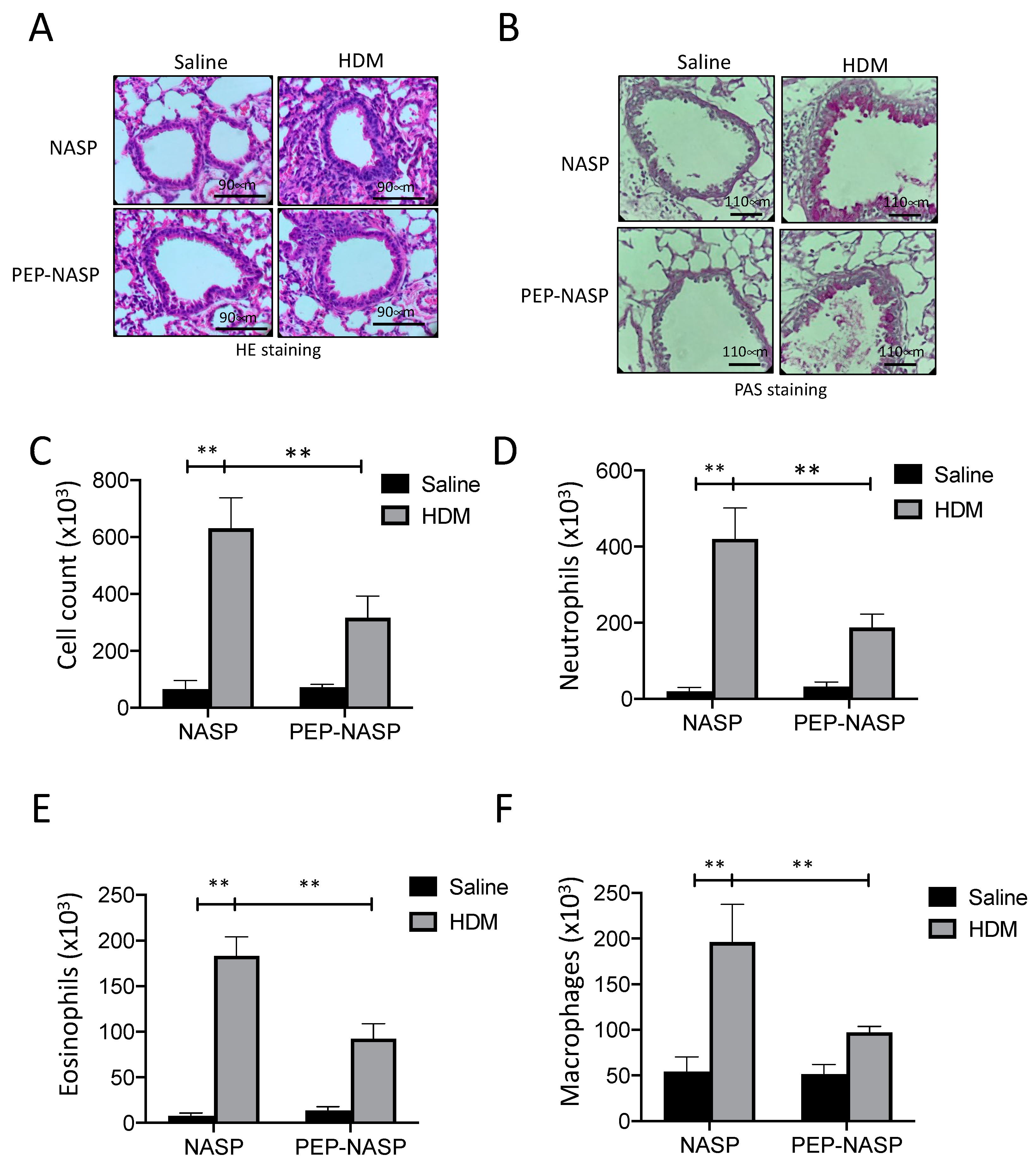
2.4. The PEP-NASP Peptide Attenuates HDM-Induced Airway Inflammation
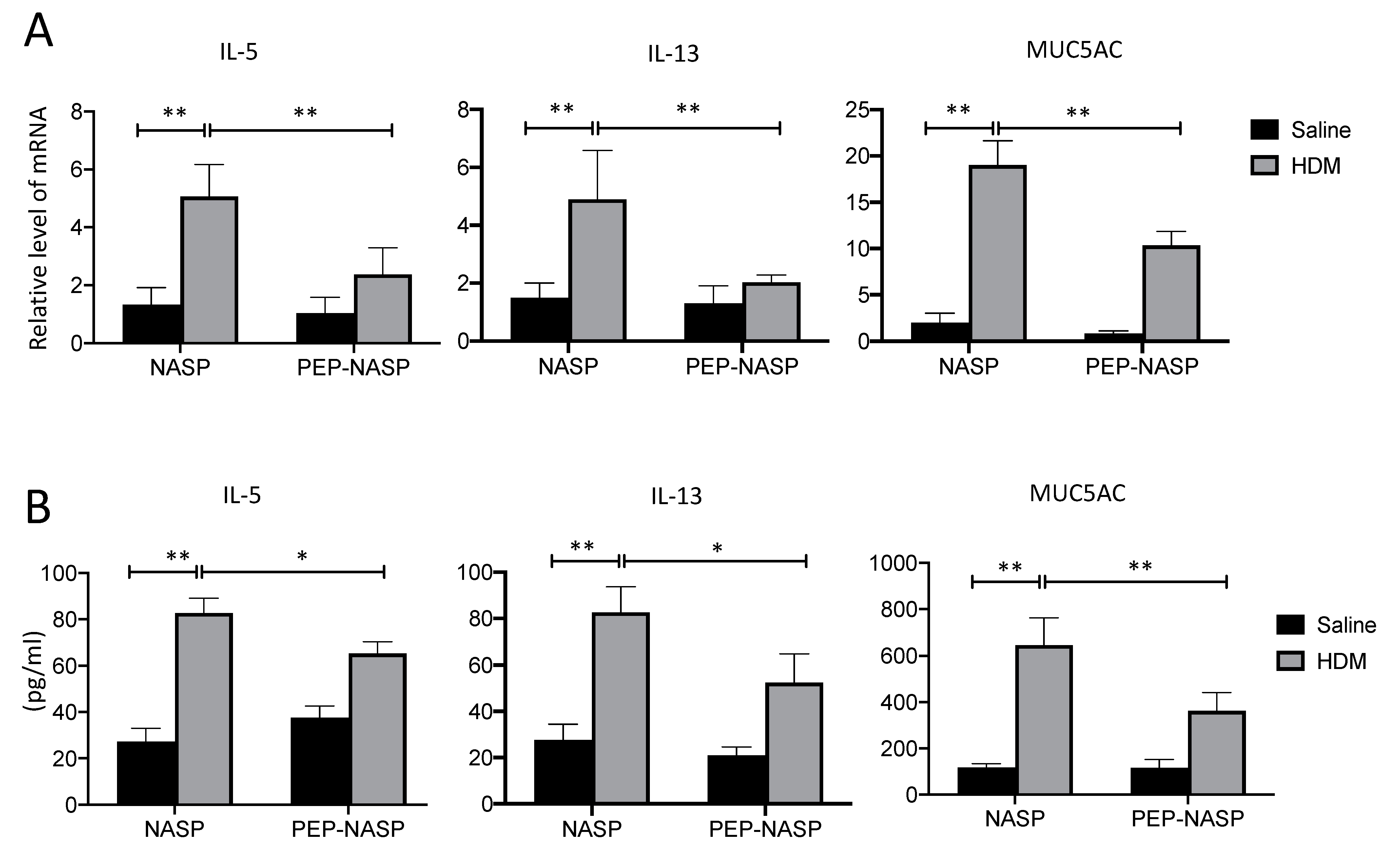
2.5. The PEP-NASP Peptide Inhibits Th2 Inflammation and MUC5AC Expression in Mice with HDM-Induced Asthma
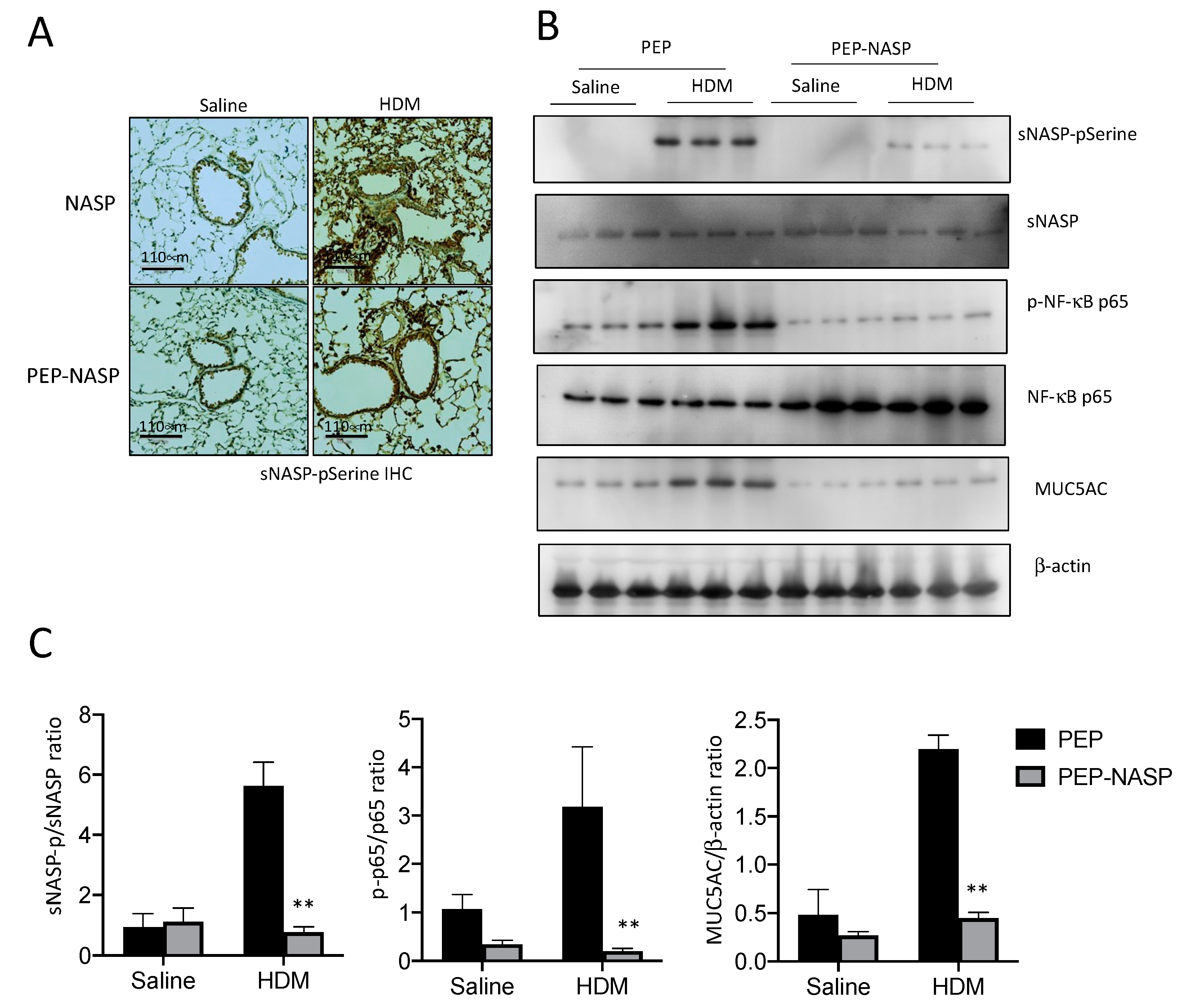
2.6. The PEP-NASP Peptide Inhibits the sNASP/TRAF6 Signaling in HDM-Induced Mice Asthma
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. HDM-Induced Asthma Model
4.3. Bronchoalveolar Lavage Fluid (BALF)
4.4. Measurement of AHR
4.5. Plasmids, siRNA, and Transfection
4.6. Real-Time Polymerase Chain Reaction (RT-PCR)
4.7. Co-Immunoprecipitation (IP) and Western Blot Analysis
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. NF-κB Reporter Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ray, A.; Raundhal, M.; Oriss, T.B.; Ray, P.; Wenzel, S.E. Current concepts of severe asthma. J. Clin. Investig. 2016, 126, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.R.; Hales, B.J.; Smith, W.A. House dust mite allergens in asthma and allergy. Trends Mol. Med. 2010, 16, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.R. The advent of recombinant allergens and allergen cloning. J. Allergy Clin. Immunol. 2011, 127, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Wiley, R.E.; Fattouh, R.; Swirski, F.K.; Gajewska, B.U.; Coyle, A.J.; Gutierrez-Ramos, J.C.; Ellis, R.; Inman, M.D.; Jordana, M. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am. J. Respir. Crit. Care Med. 2004, 169, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.; Elias, J.A.; Chupp, G.L. Asthma: Mechanisms of disease persistence and progression. Annu. Rev. Immunol. 2004, 22, 789–815. [Google Scholar] [CrossRef] [PubMed]
- Minnicozzi, M.; Sawyer, R.T.; Fenton, M.J. Innate immunity in allergic disease. Immunol. Rev. 2011, 242, 106–127. [Google Scholar] [CrossRef]
- Gregory, L.G.; Lloyd, C.M. Orchestrating house dust mite-associated allergy in the lung. Trends Immunol. 2011, 32, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, K.; Mitra, S.; Gavrilin, M.A.; Wewers, M.D. House Dust Mite Allergens and the Induction of Monocyte Interleukin 1beta Production That Triggers an IkappaBzeta-Dependent Granulocyte Macrophage Colony-Stimulating Factor Release from Human Lung Epithelial Cells. Am. J. Respir Cell Mol. Biol. 2015, 53, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, D. Dust mites’ dirty dealings with dendritic cells. Nat. Med. 2008, 14, 487–488. [Google Scholar] [CrossRef]
- Liu, C.F.; Drocourt, D.; Puzo, G.; Wang, J.Y.; Riviere, M. Innate immune response of alveolar macrophage to house dust mite allergen is mediated through TLR2/-4 co-activation. PLoS ONE 2013, 8, e75983. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, M.; Rochman, Y.; Spolski, R.; Samsel, L.; Leonard, W.J. Thymic stromal lymphopoietin is produced by dendritic cells. J. Immunol. 2011, 187, 1207–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.; Lunding, L.P.; Ehlers, J.C.; Weckmann, M.; Zissler, U.M.; Wegmann, M. More Than Just a Barrier: The Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front. Immunol. 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Lachowicz-Scroggins, M.E.; Yuan, S.; Kerr, S.C.; Dunican, E.M.; Yu, M.; Carrington, S.D.; Fahy, J.V. Abnormalities in MUC5AC and MUC5B Protein in Airway Mucus in Asthma. Am. J. Respir. Crit. Care Med. 2016, 194, 1296–1299. [Google Scholar] [CrossRef] [Green Version]
- Welsh, K.G.; Rousseau, K.; Fisher, G.; Bonser, L.R.; Bradding, P.; Brightling, C.E.; Thornton, D.J.; Gaillard, E.A. MUC5AC and a Glycosylated Variant of MUC5B Alter Mucin Composition in Children with Acute Asthma. Chest 2017, 152, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordonez, C.L.; Khashayar, R.; Wong, H.H.; Ferrando, R.; Wu, R.; Hyde, D.M.; Hotchkiss, J.A.; Zhang, Y.; Novikov, A.; Dolganov, G.; et al. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am. J. Respir. Crit. Care Med. 2001, 163, 517–523. [Google Scholar] [CrossRef]
- Evans, C.M.; Raclawska, D.S.; Ttofali, F.; Liptzin, D.R.; Fletcher, A.A.; Harper, D.N.; McGing, M.A.; McElwee, M.M.; Williams, O.W.; Sanchez, E.; et al. The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Nat. Commun. 2015, 6, 6281. [Google Scholar] [CrossRef] [Green Version]
- Lachowicz-Scroggins, M.E.; Finkbeiner, W.E.; Gordon, E.D.; Yuan, S.; Zlock, L.; Bhakta, N.R.; Woodruff, P.G.; Fahy, J.V.; Boushey, H.A. Corticosteroid and long-acting ss-agonist therapy reduces epithelial goblet cell metaplasia. Clin. Exp. Allergy 2017, 47, 1534–1545. [Google Scholar] [CrossRef]
- Hu, H.; Li, H. Prunetin inhibits lipopolysaccharide-induced inflammatory cytokine production and MUC5AC expression by inactivating the TLR4/MyD88 pathway in human nasal epithelial cells. Biomed. Pharmacother. 2018, 106, 1469–1477. [Google Scholar] [CrossRef]
- Na, H.G.; Kim, Y.D.; Choi, Y.S.; Bae, C.H.; Song, S.Y. Diesel exhaust particles elevate MUC5AC and MUC5B expression via the TLR4-mediated activation of ERK1/2, p38 MAPK, and NF-kappaB signaling pathways in human airway epithelial cells. Biochem. Biophys. Res. Commun. 2019, 512, 53–59. [Google Scholar] [CrossRef]
- Shen, W.; Yin, Y.; Li, T.; Cao, G. Euxanthone inhibits lipopolysaccharide-induced injury, inflammatory response, and MUC5AC hypersecretion in human airway epithelial cells by the TLR4/MyD88 pathway. J. Appl. Toxicol. 2022, 42, 671–682. [Google Scholar] [CrossRef]
- Dickinson, J.D.; Sweeter, J.M.; Staab, E.B.; Nelson, A.J.; Bailey, K.L.; Warren, K.J.; Jaramillo, A.M.; Dickey, B.F.; Poole, J.A. MyD88 controls airway epithelial Muc5ac expression during TLR activation conditions from agricultural organic dust exposure. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L334–L347. [Google Scholar] [CrossRef] [PubMed]
- Ha, U.H.; Lim, J.H.; Kim, H.J.; Wu, W.; Jin, S.; Xu, H.; Li, J.D. MKP1 regulates the induction of MUC5AC mucin by Streptococcus pneumoniae pneumolysin by inhibiting the PAK4-JNK signaling pathway. J. Biol. Chem. 2008, 283, 30624–30631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jono, H.; Lim, J.H.; Xu, H.; Li, J.D. PKCtheta synergizes with TLR-dependent TRAF6 signaling pathway to upregulate MUC5AC mucin via CARMA1. PLoS ONE 2012, 7, e31049. [Google Scholar] [CrossRef]
- Richardson, R.T.; Alekseev, O.M.; Grossman, G.; Widgren, E.E.; Thresher, R.; Wagner, E.J.; Sullivan, K.D.; Marzluff, W.F.; O’Rand, M.G. Nuclear autoantigenic sperm protein (NASP), a linker histone chaperone that is required for cell proliferation. J. Biol. Chem. 2006, 281, 21526–21534. [Google Scholar] [CrossRef] [Green Version]
- Richardson, R.T.; Batova, I.N.; Widgren, E.E.; Zheng, L.X.; Whitfield, M.; Marzluff, W.F.; O’Rand, M.G. Characterization of the histone H1-binding protein, NASP, as a cell cycle-regulated somatic protein. J. Biol. Chem. 2000, 275, 30378–30386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, A.J.; Gurard-Levin, Z.A.; Vassias, I.; Almouzni, G. A specific function for the histone chaperone NASP to fine-tune a reservoir of soluble H3-H4 in the histone supply chain. Mol. Cell 2011, 44, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.C.; Hsu, S.P.; Hu, M.C.; Lan, Y.T.; Yeh, E.T.H.; Yang, F.M. PEP-sNASP Peptide Alleviates LPS-Induced Acute Lung Injury Through the TLR4/TRAF6 Axis. Front. Med. 2022, 9, 832713. [Google Scholar] [CrossRef]
- Yang, F.M.; Zuo, Y.; Zhou, W.; Xia, C.; Hahm, B.; Sullivan, M.; Cheng, J.; Chang, H.M.; Yeh, E.T. sNASP inhibits TLR signaling to regulate immune response in sepsis. J. Clin. Invest. 2018, 128, 2459–2472. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.M.; Chang, H.M.; Yeh, E.T.H. Regulation of TLR4 signaling through the TRAF6/sNASP axis by reversible phosphorylation mediated by CK2 and PP4. Proc. Natl. Acad. Sci. USA 2021, 118, e2107044118. [Google Scholar] [CrossRef]
- Morgan, L.E.; Jaramillo, A.M.; Shenoy, S.K.; Raclawska, D.; Emezienna, N.A.; Richardson, V.L.; Hara, N.; Harder, A.Q.; NeeDell, J.C.; Hennessy, C.E.; et al. Disulfide disruption reverses mucus dysfunction in allergic airway disease. Nat. Commun. 2021, 12, 249. [Google Scholar] [CrossRef]
- Jia, Z.; Bao, K.; Wei, P.; Yu, X.; Zhang, Y.; Wang, X.; Wang, X.; Yao, L.; Li, L.; Wu, P.; et al. EGFR activation-induced decreases in claudin1 promote MUC5AC expression and exacerbate asthma in mice. Mucosal. Immunol. 2021, 14, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Ferkol, T.; Schraufnagel, D. The global burden of respiratory disease. Ann. Am. Thorac. Soc. 2014, 11, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Fuhlbrigge, A.L.; Adams, R.J.; Guilbert, T.W.; Grant, E.; Lozano, P.; Janson, S.L.; Martinez, F.; Weiss, K.B.; Weiss, S.T. The burden of asthma in the United States: Level and distribution are dependent on interpretation of the national asthma education and prevention program guidelines. Am. J. Respir. Crit. Care Med. 2002, 166, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Athari, S.S. Targeting cell signaling in allergic asthma. Signal Transduct. Target. Ther. 2019, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Santus, P.; Radovanovic, D.; Chiumello, D.A. Mucins and Asthma: Are We Headed to the Revolutionary Road? J. Clin. Med. 2019, 8, 1955. [Google Scholar] [CrossRef] [Green Version]
- Bonser, L.R.; Erle, D.J. Airway Mucus and Asthma: The Role of MUC5AC and MUC5B. J. Clin. Med. 2017, 6, 112. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Koth, L.L.; Arron, J.R.; Fahy, J.V. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [Green Version]
- Bonser, L.R.; Zlock, L.; Finkbeiner, W.; Erle, D.J. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J. Clin. Investig. 2016, 126, 2367–2371. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Luo, D.; Wang, X.; Zhang, Y.; Liu, Z.; Zhong, N.; Wu, M.; Li, G. Lyn regulates mucus secretion and MUC5AC via the STAT6 signaling pathway during allergic airway inflammation. Sci. Rep. 2017, 7, 42675. [Google Scholar] [CrossRef] [Green Version]
- Dillard, P.; Wetsel, R.A.; Drouin, S.M. Complement C3a regulates Muc5ac expression by airway Clara cells independently of Th2 responses. Am. J. Respir. Crit. Care Med. 2007, 175, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova, J.L.; Abel, L.; Quintana-Murci, L. Human TLRs and IL-1Rs in host defense: Natural insights from evolutionary, epidemiological, and clinical genetics. Annu. Rev. Immunol. 2011, 29, 447–491. [Google Scholar] [CrossRef] [PubMed]
- Eder, W.; Klimecki, W.; Yu, L.; von Mutius, E.; Riedler, J.; Braun-Fahrlander, C.; Nowak, D.; Martinez, F.D.; The ALEX Study Team. Toll-like receptor 2 as a major gene for asthma in children of European farmers. J. Allergy Clin. Immunol. 2004, 113, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Sackesen, C.; Karaaslan, C.; Keskin, O.; Tokol, N.; Tahan, F.; Civelek, E.; Soyer, O.U.; Adalioglu, G.; Tuncer, A.; Birben, E.; et al. The effect of polymorphisms at the CD14 promoter and the TLR4 gene on asthma phenotypes in Turkish children with asthma. Allergy 2005, 60, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Lauener, R.P.; Birchler, T.; Adamski, J.; Braun-Fahrlander, C.; Bufe, A.; Herz, U.; von Mutius, E.; Nowak, D.; Riedler, J.; Waser, M.; et al. Expression of CD14 and Toll-like receptor 2 in farmers’ and non-farmers’ children. Lancet 2002, 360, 465–466. [Google Scholar] [CrossRef]
- Trompette, A.; Divanovic, S.; Visintin, A.; Blanchard, C.; Hegde, R.S.; Madan, R.; Thorne, P.S.; Wills-Karp, M.; Gioannini, T.L.; Weiss, J.P.; et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature 2009, 457, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Chiou, Y.L.; Lin, C.Y. Der p2 activates airway smooth muscle cells in a TLR2/MyD88-dependent manner to induce an inflammatory response. J. Cell. Physiol. 2009, 220, 311–318. [Google Scholar] [CrossRef]
- Cook, D.N.; Pisetsky, D.S.; Schwartz, D.A. Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 2004, 5, 975–979. [Google Scholar] [CrossRef]
- Ha, U.; Lim, J.H.; Jono, H.; Koga, T.; Srivastava, A.; Malley, R.; Pages, G.; Pouyssegur, J.; Li, J.D. A novel role for IkappaB kinase (IKK) alpha and IKKbeta in ERK-dependent up-regulation of MUC5AC mucin transcription by Streptococcus pneumoniae. J. Immunol. 2007, 178, 1736–1747. [Google Scholar] [CrossRef] [Green Version]
- Zhong, T.; Perelman, J.M.; Kolosov, V.P.; Zhou, X.D. MiR-146a negatively regulates neutrophil elastase-induced MUC5AC secretion from 16HBE human bronchial epithelial cells. Mol. Cell. Biochem. 2011, 358, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Sadowska, A.M. N-Acetylcysteine mucolysis in the management of chronic obstructive pulmonary disease. Ther. Adv. Respir. Dis. 2012, 6, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, D.F.; Barnes, P.J. Treatment of airway mucus hypersecretion. Ann. Med. 2006, 38, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Lafkas, D.; Shelton, A.; Chiu, C.; de Leon Boenig, G.; Chen, Y.; Stawicki, S.S.; Siltanen, C.; Reichelt, M.; Zhou, M.; Wu, X.; et al. Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature 2015, 528, 127–131. [Google Scholar] [CrossRef]
- Zakeri, A.; Yazdi, F.G. Toll-like receptor-mediated involvement of innate immune cells in asthma disease. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3270–3277. [Google Scholar] [CrossRef]
- Matesic, D.; Lenert, A.; Lenert, P. Modulating toll-like receptor 7 and 9 responses as therapy for allergy and autoimmunity. Curr. Allergy Asthma Rep. 2012, 12, 8–17. [Google Scholar] [CrossRef]
- Woo, L.N.; Guo, W.Y.; Wang, X.; Young, A.; Salehi, S.; Hin, A.; Zhang, Y.; Scott, J.A.; Chow, C.W. A 4-Week Model of House Dust Mite (HDM) Induced Allergic Airways Inflammation with Airway Remodeling. Sci. Rep. 2018, 8, 6925. [Google Scholar] [CrossRef] [Green Version]
- Khadangi, F.; Forgues, A.S.; Tremblay-Pitre, S.; Dufour-Mailhot, A.; Henry, C.; Boucher, M.; Beaulieu, M.J.; Morissette, M.; Fereydoonzad, L.; Brunet, D.; et al. Intranasal versus intratracheal exposure to lipopolysaccharides in a murine model of acute respiratory distress syndrome. Sci. Rep. 2021, 11, 7777. [Google Scholar] [CrossRef]
- Chen, J.Y.; Cheng, W.H.; Lee, K.Y.; Kuo, H.P.; Chung, K.F.; Chen, C.L.; Chen, B.C.; Lin, C.H. Abnormal ADAM17 expression causes airway fibrosis in chronic obstructive asthma. Biomed. Pharmacother. 2021, 140, 111701. [Google Scholar] [CrossRef]
- Yang, F.M.; Pan, C.T.; Tsai, H.M.; Chiu, T.W.; Wu, M.L.; Hu, M.C. Liver receptor homolog-1 localization in the nuclear body is regulated by sumoylation and cAMP signaling in rat granulosa cells. FEBS J. 2009, 276, 425–436. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.-Z.; Wang, S.-A.; Hsu, S.-C.; Silva, K.A.S.; Yang, F.-M. Home Dust Mites Promote MUC5AC Hyper-Expression by Modulating the sNASP/TRAF6 Axis in the Airway Epithelium. Int. J. Mol. Sci. 2022, 23, 9405. https://doi.org/10.3390/ijms23169405
Chen M-Z, Wang S-A, Hsu S-C, Silva KAS, Yang F-M. Home Dust Mites Promote MUC5AC Hyper-Expression by Modulating the sNASP/TRAF6 Axis in the Airway Epithelium. International Journal of Molecular Sciences. 2022; 23(16):9405. https://doi.org/10.3390/ijms23169405
Chicago/Turabian StyleChen, Ming-Zhen, Shao-An Wang, Shih-Chang Hsu, Kleiton Augusto Santos Silva, and Feng-Ming Yang. 2022. "Home Dust Mites Promote MUC5AC Hyper-Expression by Modulating the sNASP/TRAF6 Axis in the Airway Epithelium" International Journal of Molecular Sciences 23, no. 16: 9405. https://doi.org/10.3390/ijms23169405