
Compound C Inhibits Renca Renal Epithelial Carcinoma Growth in Syngeneic Mouse Models by Blocking Cell Cycle Progression, Adhesion and Invasion

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. CompC Reduces the Viability of Renca Cells via G2/M Cell Cycle Arrest
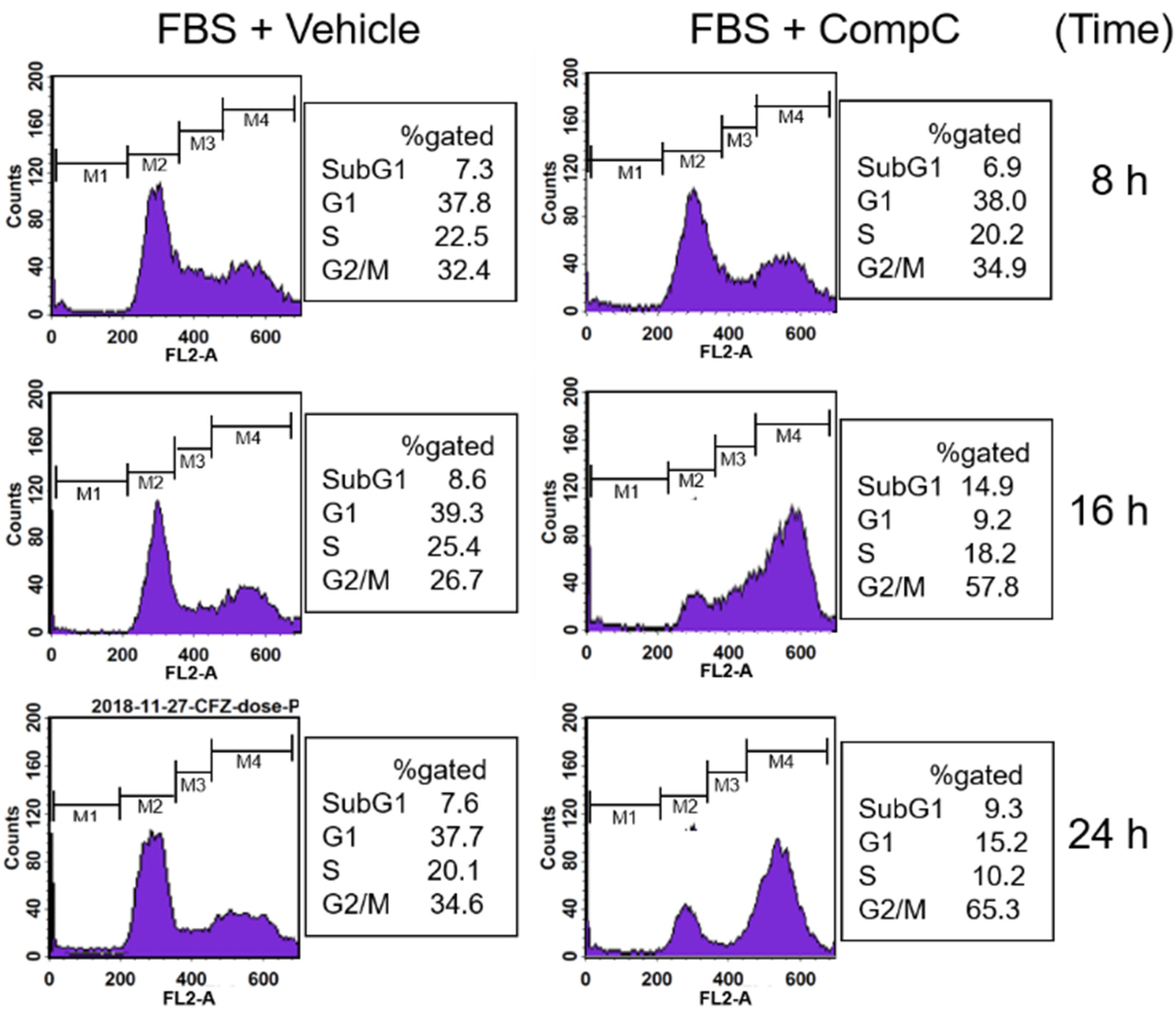
2.2. CompC Induces G2/M Cell Cycle Arrest via ROS Production in Renca Cells
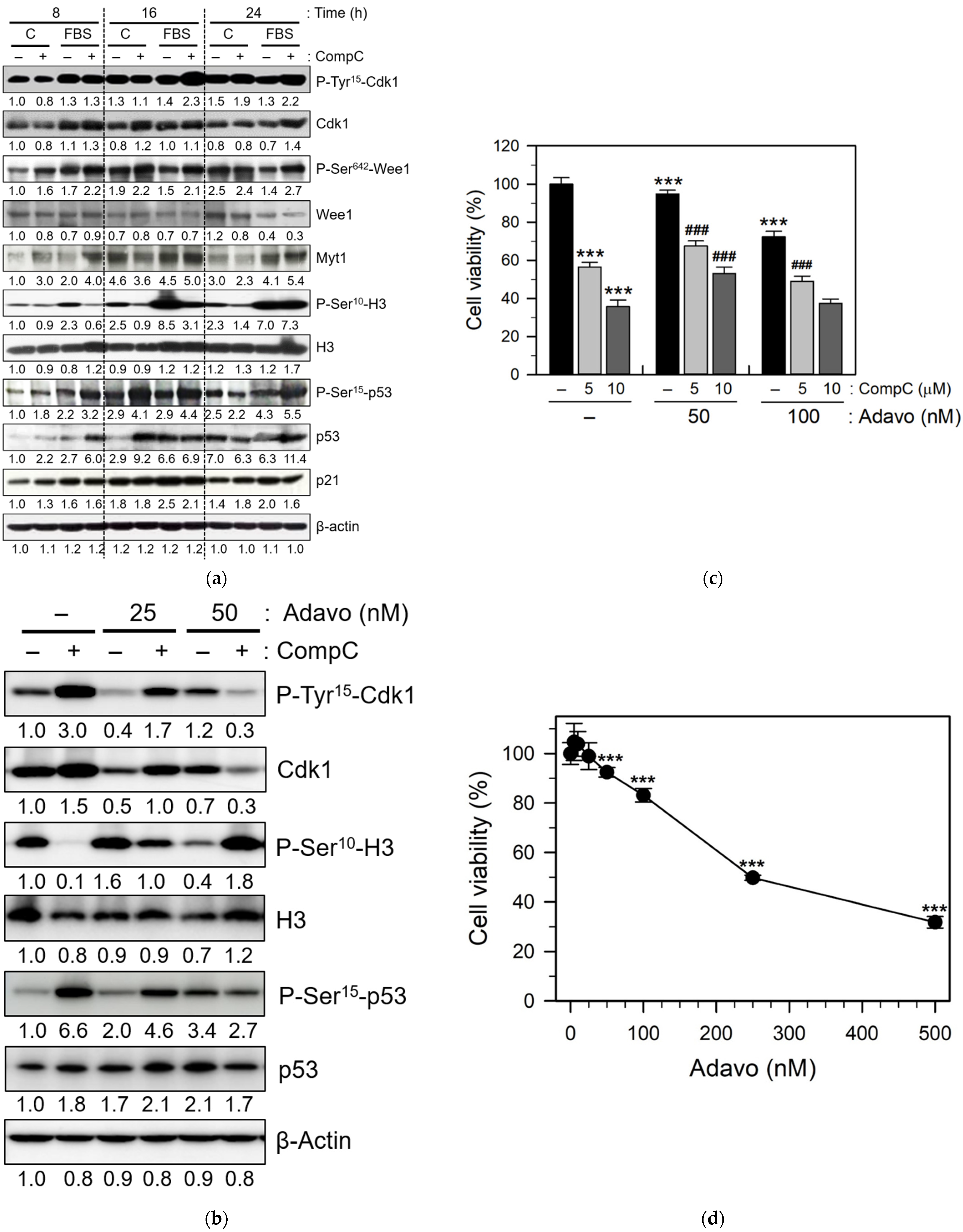
2.3. CompC Inhibits the Phosphorylation of Histone H3 via the Inhibitory Tyrosine Phosphorylation of Cdk1 by Wee1
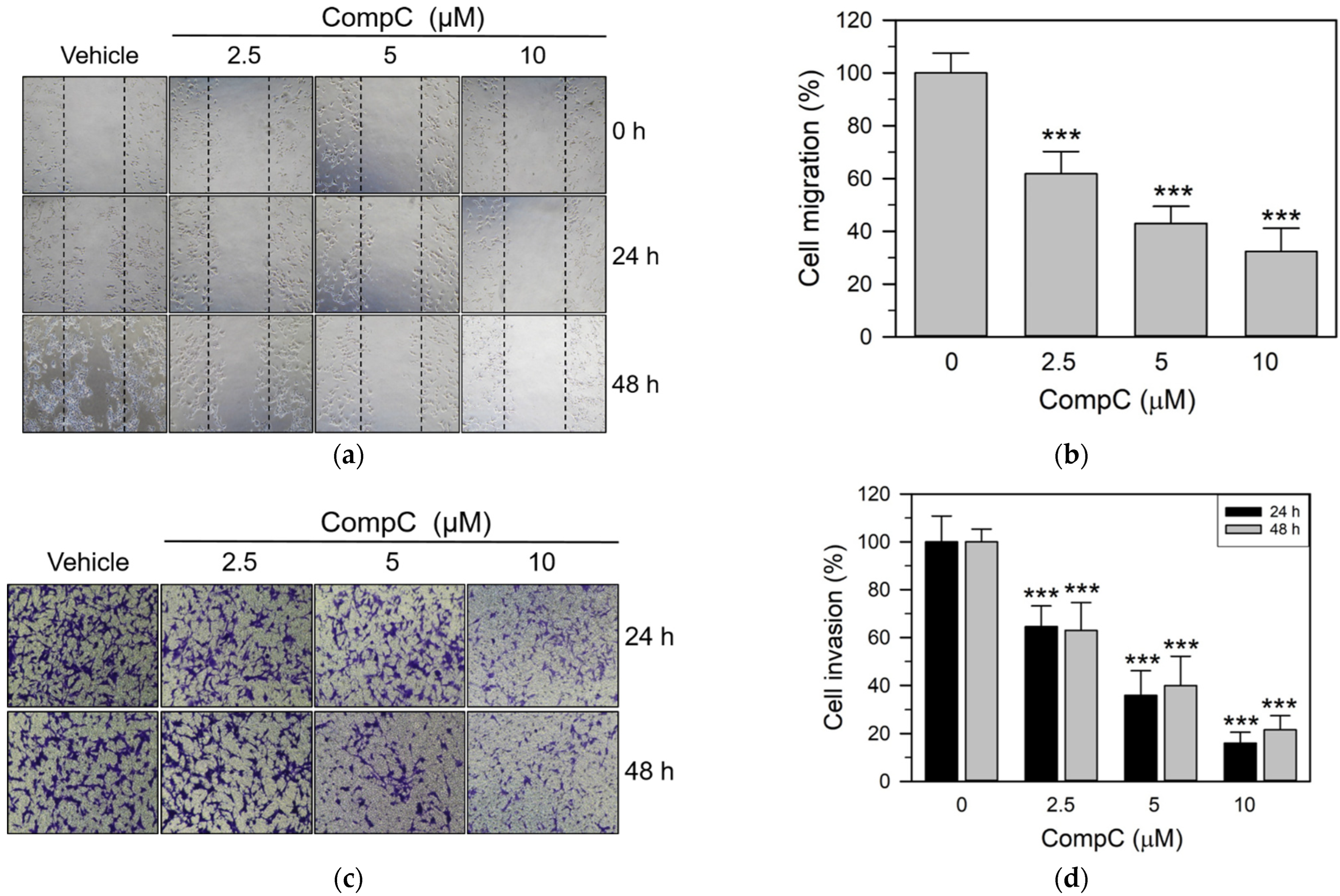
2.4. CompC Reduces the Cell Adhesion, Migration and Invasion of Renca Cells
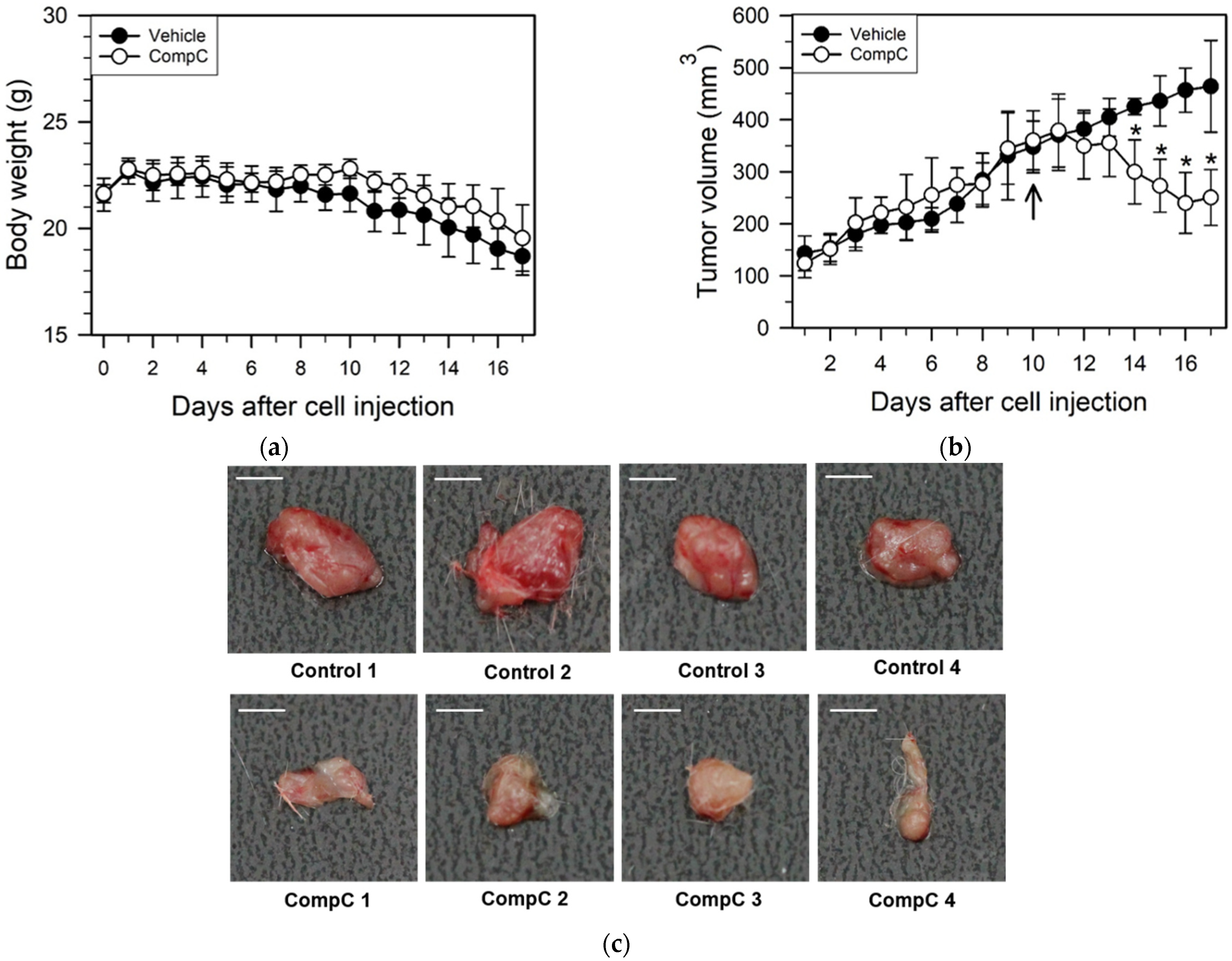
2.5. CompC Inhibits Renca Tumor Growth in BALB/c Syngeneic Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. MTT Assay
4.4. Flow Cytometry
4.5. Western Blot
4.6. Measurement of Intracellular ROS
4.7. Matrigel Adhesion Assay
4.8. Cell Migration Assay
4.9. Cell Invasion Assay
4.10. In Vivo Xenograft Experiment
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Osca-Verdegal, R.; Beltran-Garcia, J.; Gorriz, J.L.; Martinez Jabaloyas, J.M.; Pallardo, F.V.; Garcia-Gimenez, J.L. Use of Circular RNAs in Diagnosis, Prognosis and Therapeutics of Renal Cell Carcinoma. Front. Cell Dev. Biol. 2022, 10, 879814. [Google Scholar] [CrossRef] [PubMed]
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of Renal Cell Carcinoma. World J. Oncol. 2020, 11, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Li, R.G.; Deng, H.; Liu, X.H.; Chen, Z.Y.; Wan, S.S.; Wang, L. Histone Methyltransferase G9a Promotes the Development of Renal Cancer through Epigenetic Silencing of Tumor Suppressor Gene SPINK5. Oxid. Med. Cell Longev. 2021, 2021, 6650781. [Google Scholar] [CrossRef]
- Valladares Ayerbes, M.; Aparicio Gallego, G.; Diaz Prado, S.; Jimenez Fonseca, P.; Garcia Campelo, R.; Anton Aparicio, L.M. Origin of renal cell carcinomas. Clin. Transl. Oncol. 2008, 10, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Leppert, J.; Patel, C. Perspective: Beyond the genome. Nature 2016, 537, S105. [Google Scholar] [CrossRef] [PubMed]
- Bahadoram, S.; Davoodi, M.; Hassanzadeh, S.; Bahadoram, M.; Barahman, M.; Mafakher, L. Renal cell carcinoma: An overview of the epidemiology, diagnosis, and treatment. G Ital. Nefrol. 2022, 39, 2022-vol3. [Google Scholar]
- Cairns, P. Renal cell carcinoma. Cancer Biomark 2010, 9, 461–473. [Google Scholar] [CrossRef]
- Lieder, A.; Guenzel, T.; Lebentrau, S.; Schneider, C.; Franzen, A. Diagnostic relevance of metastatic renal cell carcinoma in the head and neck: An evaluation of 22 cases in 671 patients. Int. Braz. J. Urol. 2017, 43, 202–208. [Google Scholar] [CrossRef]
- Lara, P.N., Jr.; Evans, C.P. Cytoreductive Nephrectomy in Metastatic Renal Cell Cancer: Not All That It’s Cut Out to Be. JAMA Oncol. 2019, 5, 171–172. [Google Scholar] [CrossRef]
- Pal, S.K.; Agarwal, N. Kidney cancer: Finding a niche for girentuximab in metastatic renal cell carcinoma. Nat. Rev. Urol. 2016, 13, 442–443. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK inhibitor Compound C is a potent AMPK-independent anti-glioma agent. Mol. Cancer 2014, 13, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Kim, G.E.; Lee, Y.T.; Jeong, M.S.; Kang, I.; Yang, D.; Yeo, E.J. Inhibition of platelet-derived growth factor receptor tyrosine kinase and downstream signaling pathways by Compound, C. Cell Signal. 2013, 25, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Lim, S.H.; Lee, B.; Kang, I.; Yeo, E.J. Compound C Inhibits B16-F1 Tumor Growth in a Syngeneic Mouse Model Via the Blockage of Cell Cycle Progression and Angiogenesis. Cancers 2019, 11, 823. [Google Scholar] [CrossRef]
- Park, J.B.; Lee, M.S.; Cha, E.Y.; Lee, J.S.; Sul, J.Y.; Song, I.S.; Kim, J.Y. Magnolol-induced apoptosis in HCT-116 colon cancer cells is associated with the AMP-activated protein kinase signaling pathway. Biol. Pharm. Bull. 2012, 35, 1614–1620. [Google Scholar] [CrossRef]
- Yi, B.; Liu, D.; He, M.; Li, Q.; Liu, T.; Shao, J. Role of the ROS/AMPK signaling pathway in tetramethylpyrazine-induced apoptosis in gastric cancer cells. Oncol. Lett. 2013, 6, 583–589. [Google Scholar] [CrossRef]
- Vucicevic, L.; Misirkic, M.; Janjetovic, K.; Harhaji-Trajkovic, L.; Prica, M.; Stevanovic, D.; Isenovic, E.; Sudar, E.; Sumarac-Dumanovic, M.; Micic, D.; et al. AMP-activated protein kinase-dependent and -independent mechanisms underlying in vitro antiglioma action of compound C. Biochem. Pharm. 2009, 77, 1684–1693. [Google Scholar] [CrossRef]
- Chua, Y.L.; Hagen, T. Compound C prevents Hypoxia-Inducible Factor-1alpha protein stabilization by regulating the cellular oxygen availability via interaction with Mitochondrial Complex, I. BMC Res. Notes 2011, 4, 117. [Google Scholar] [CrossRef]
- Emerling, B.M.; Viollet, B.; Tormos, K.V.; Chandel, N.S. Compound C inhibits hypoxic activation of HIF-1 independent of AMPK. FEBS Lett. 2007, 581, 5727–5731. [Google Scholar] [CrossRef]
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Thyagarajan-Sahu, A.; Krchnak, V.; Jedinak, A.; Sandusky, G.E.; Sliva, D. NAHA, a novel hydroxamic acid-derivative, inhibits growth and angiogenesis of breast cancer in vitro and in vivo. PLoS ONE 2012, 7, e34283. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, W.; Zhang, B.; Liu, Y.Q.; Wang, Z.Y.; Wu, Y.P.; Yu, X.J.; Zhang, X.D.; Ming, P.H.; Zhou, G.B.; et al. The natural compound magnolol inhibits invasion and exhibits potential in human breast cancer therapy. Sci. Rep. 2013, 3, 3098. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.C.; Tsai, M.H.; Chiu, C.F.; Lu, C.C.; Kuo, S.C.; Chang, N.W.; Yang, J.S. AMPK-dependent signaling modulates the suppression of invasion and migration by fenofibrate in CAL 27 oral cancer cells through NF-kappaB pathway. Environ. Toxicol. 2014, 31, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, C.R.; Ribatti, D. Animal models of spontaneous renal carcinoma: A possible rebirth? Urol. Oncol. 2020, 38, 174–183. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Chen, Q.; Song, S.; Wang, Z.; Shen, Y.; Xie, L.; Li, J.; Jiang, L.; Zhao, H.; Feng, X.; Zhou, Y.; et al. Isorhamnetin induces the paraptotic cell death through ROS and the ERK/MAPK pathway in OSCC cells. Oral. Dis. 2021, 27, 240–250. [Google Scholar] [CrossRef]
- Zhang, G.; He, J.; Ye, X.; Zhu, J.; Hu, X.; Shen, M.; Ma, Y.; Mao, Z.; Song, H.; Chen, F. beta-Thujaplicin induces autophagic cell death, apoptosis, and cell cycle arrest through ROS-mediated Akt and p38/ERK MAPK signaling in human hepatocellular carcinoma. Cell Death Dis. 2019, 10, 255. [Google Scholar] [CrossRef]
- Poon, R.Y.; Chau, M.S.; Yamashita, K.; Hunter, T. The role of Cdc2 feedback loop control in the DNA damage checkpoint in mammalian cells. Cancer Res. 1997, 57, 5168–5178. [Google Scholar]
- Wang, J.; Zhang, Y.; Xu, S.; Li, W.; Chen, Z.; Wang, Z.; Han, X.; Zhao, Y.; Li, S. Prognostic significance of G2/M arrest signaling pathway proteins in advanced non-small cell lung cancer patients. Oncol. Lett. 2015, 9, 1266–1272. [Google Scholar] [CrossRef]
- Jang, J.H.; Lee, T.J.; Yang, E.S.; Min do, S.; Kim, Y.H.; Kim, S.H.; Choi, Y.H.; Park, J.W.; Choi, K.S.; Kwon, T.K. Compound C sensitizes Caki renal cancer cells to TRAIL-induced apoptosis through reactive oxygen species-mediated down-regulation of c-FLIPL and Mcl-1. Exp. Cell Res. 2010, 316, 2194–2203. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [CrossRef]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharm. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.Y.; Choi, J.H.; Yoon, H.; Hwang, K.Y.; Noh, M.H.; Choe, W.; Yoon, K.S.; Ha, J.; Yeo, E.J.; Kang, I. Luteolin induces apoptosis through endoplasmic reticulum stress and mitochondrial dysfunction in Neuro-2a mouse neuroblastoma cells. Eur. J. Pharm. 2011, 668, 115–126. [Google Scholar] [CrossRef]
- Chen, H.; Liu, R.H. Potential Mechanisms of Action of Dietary Phytochemicals for Cancer Prevention by Targeting Cellular Signaling Transduction Pathways. J. Agric. Food Chem 2018, 66, 3260–3276. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- Persons, D.L.; Yazlovitskaya, E.M.; Pelling, J.C. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J. Biol. Chem 2000, 275, 35778–35785. [Google Scholar] [CrossRef]
- Torres, M.; Forman, H.J. Redox signaling and the MAP kinase pathways. Biofactors 2003, 17, 287–296. [Google Scholar] [CrossRef]
- Kim, H.S.; Hwang, J.T.; Yun, H.; Chi, S.G.; Lee, S.J.; Kang, I.; Yoon, K.S.; Choe, W.J.; Kim, S.S.; Ha, J. Inhibition of AMP-activated protein kinase sensitizes cancer cells to cisplatin-induced apoptosis via hyper-induction of p53. J. Biol. Chem. 2008, 283, 3731–3742. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.X.; Domingo-Sananes, M.R.; Huzarska, M.; Novak, B.; Gould, K.L. Multisite phosphoregulation of Cdc25 activity refines the mitotic entrance and exit switches. Proc. Natl. Acad. Sci. USA 2012, 109, 9899–9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Sagata, N. Mechanism for inactivation of the mitotic inhibitory kinase Wee1 at M phase. Proc. Natl. Acad. Sci. USA 2007, 104, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.S.; Ruderman, J.V. Changes in regulatory phosphorylation of Cdc25C Ser287 and Wee1 Ser549 during normal cell cycle progression and checkpoint arrests. Mol. Biol. Cell 2005, 16, 5749–5760. [Google Scholar] [CrossRef]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. 2010, 9, 514–522. [Google Scholar] [CrossRef]
- Chera, B.S.; Sheth, S.H.; Patel, S.A.; Goldin, D.; Douglas, K.E.; Green, R.L.; Shen, C.J.; Gupta, G.P.; Moore, D.T.; Grilley Olson, J.E.; et al. Phase 1 trial of adavosertib (AZD1775) in combination with concurrent radiation and cisplatin for intermediate-risk and high-risk head and neck squamous cell carcinoma. Cancer 2021, 127, 4447–4454. [Google Scholar] [CrossRef]
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring. J. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Luciani, M.G.; Hutchins, J.R.; Zheleva, D.; Hupp, T.R. The C-terminal regulatory domain of p53 contains a functional docking site for cyclin A. J. Mol. Biol. 2000, 300, 503–518. [Google Scholar] [CrossRef]
- Ababneh, M.; Gotz, C.; Montenarh, M. Downregulation of the cdc2/cyclin B protein kinase activity by binding of p53 to p34(cdc2). Biochem Biophys Res. Commun. 2001, 283, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Delassus, G.S.; Cho, H.; Park, J.; Eliceiri, G.L. New pathway links from cancer-progression determinants to gene expression of matrix metalloproteinases in breast cancer cells. J. Cell. Physiol. 2008, 217, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lee, M.; Choi, K.C.; Shin, D.M.; Ko, J.; Jang, S.W. N-(4-hydroxyphenyl)retinamide inhibits breast cancer cell invasion through suppressing NF-KB activation and inhibiting matrix metalloproteinase-9 expression. J. Cell. Biochem. 2012, 113, 2845–2855. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.; Ham, N.Y.; Hwang, C.Y.; Jang, J.; Lee, B.; Jeong, J.-W.; Kang, I.; Yeo, E.-J. Compound C Inhibits Renca Renal Epithelial Carcinoma Growth in Syngeneic Mouse Models by Blocking Cell Cycle Progression, Adhesion and Invasion. Int. J. Mol. Sci. 2022, 23, 9675. https://doi.org/10.3390/ijms23179675
Lee M, Ham NY, Hwang CY, Jang J, Lee B, Jeong J-W, Kang I, Yeo E-J. Compound C Inhibits Renca Renal Epithelial Carcinoma Growth in Syngeneic Mouse Models by Blocking Cell Cycle Progression, Adhesion and Invasion. International Journal of Molecular Sciences. 2022; 23(17):9675. https://doi.org/10.3390/ijms23179675
Chicago/Turabian StyleLee, Myungyeon, Na Yeon Ham, Chi Yeon Hwang, Jiwon Jang, Boram Lee, Joo-Won Jeong, Insug Kang, and Eui-Ju Yeo. 2022. "Compound C Inhibits Renca Renal Epithelial Carcinoma Growth in Syngeneic Mouse Models by Blocking Cell Cycle Progression, Adhesion and Invasion" International Journal of Molecular Sciences 23, no. 17: 9675. https://doi.org/10.3390/ijms23179675