

CRISPR/nCas9-Based Genome Editing on GM2 Gangliosidoses Fibroblasts via Non-Viral Vectors

 ,
,  , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Molecular Validation of the CRISPR/nCas9 System on HEK293 Cells
CRISPR/nCas9 Leads the Integration of Expression Cassettes Containing HEXA or HEXB cDNA into the AAVS1 Locus
2.2. CRISPR/nCas9-Based Genome Editing on GM2 Fibroblasts
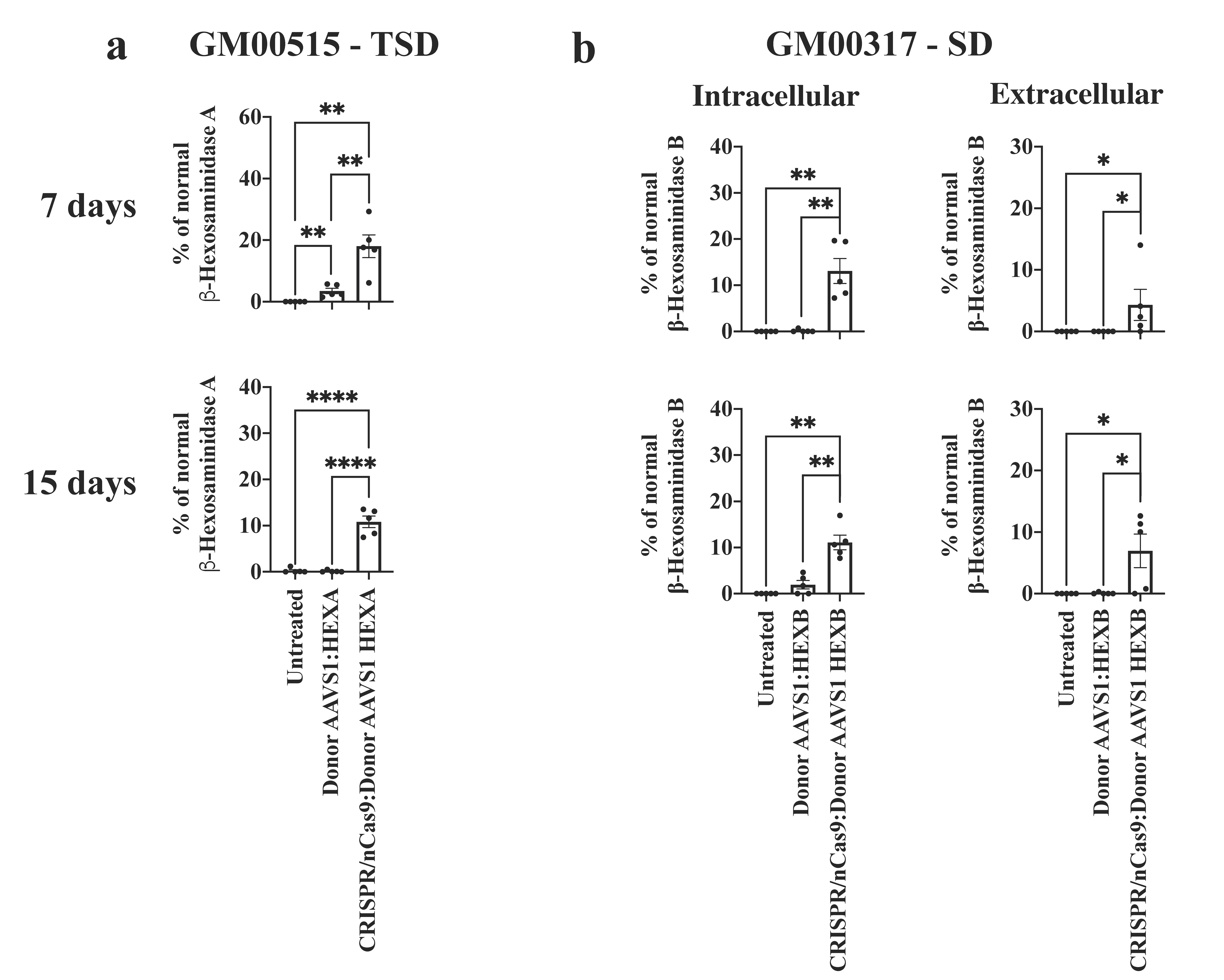
2.2.1. Stable Genome Edition Is Achieved on TSD and SD Fibroblasts upon CRISPR/nCas9-Based Treatment
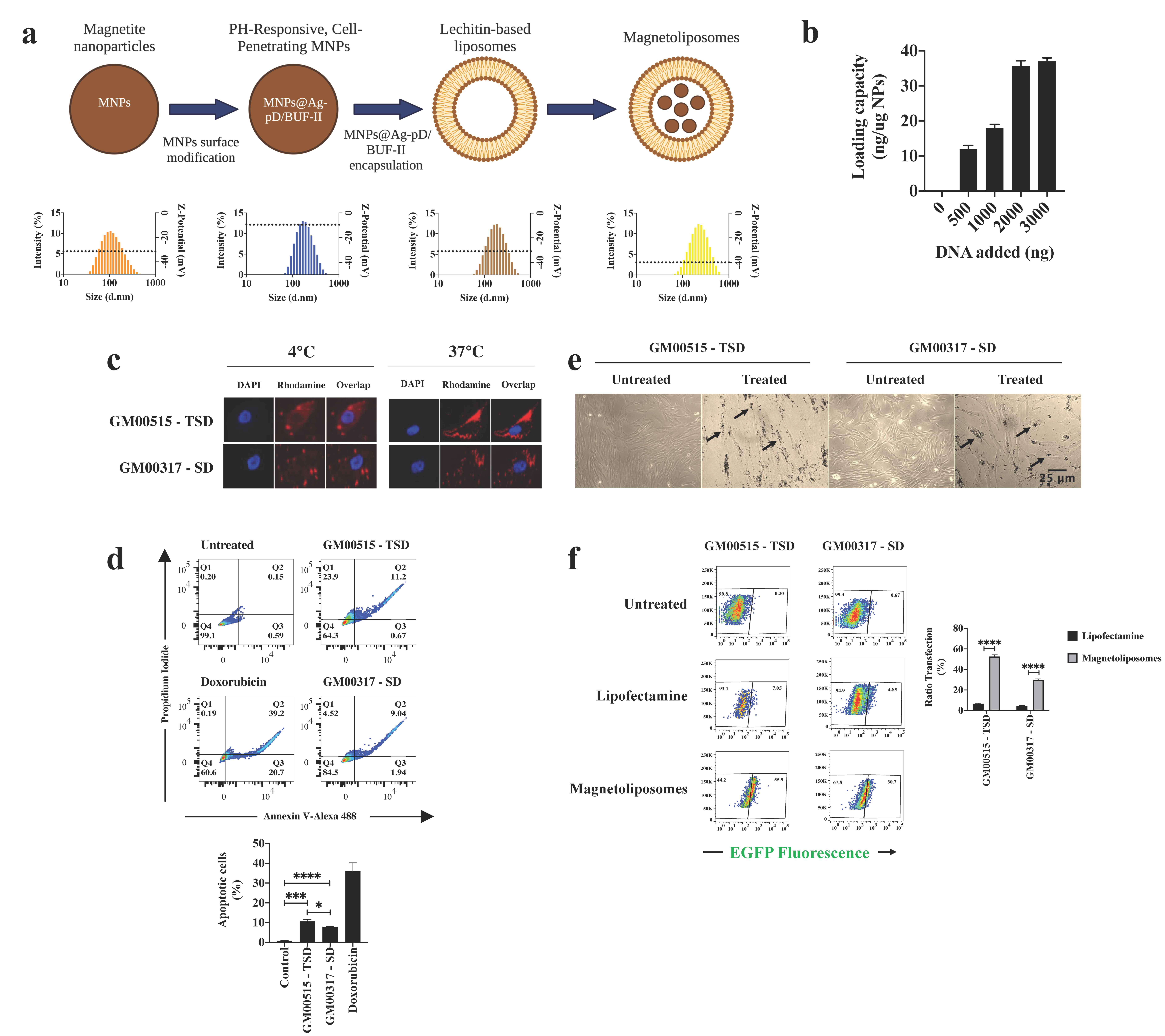
2.2.2. MLPs Increase the Transfection of CRISPR/nCas9-Related Plasmids with Non-Cytotoxic Effects in SD and TSD Fibroblasts
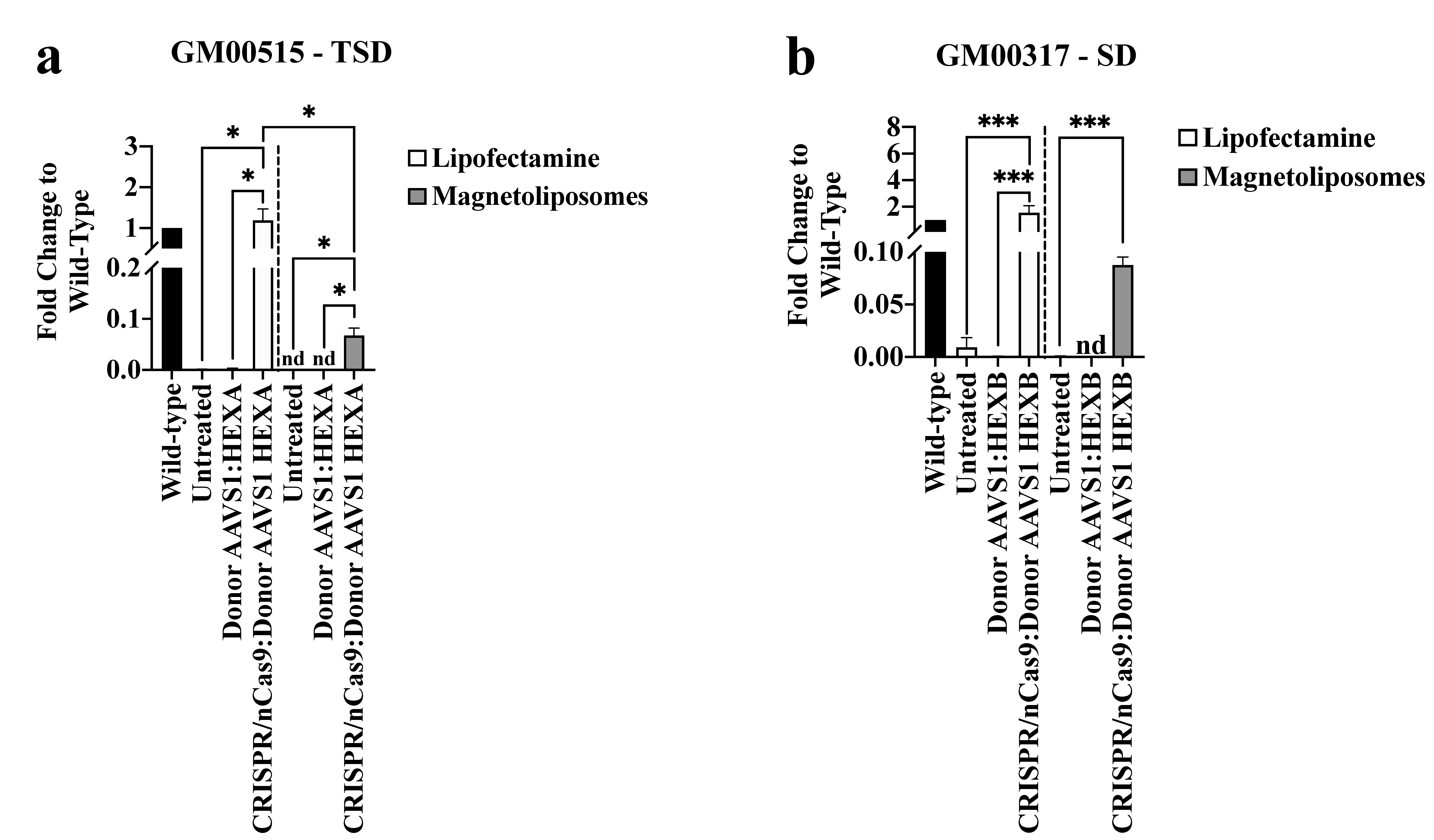
2.2.3. LPs-Assisted CRISPR/nCas9 Delivery Elicits a Better Response for Long-Term Edition Than MLPs on TSD and SD Fibroblasts
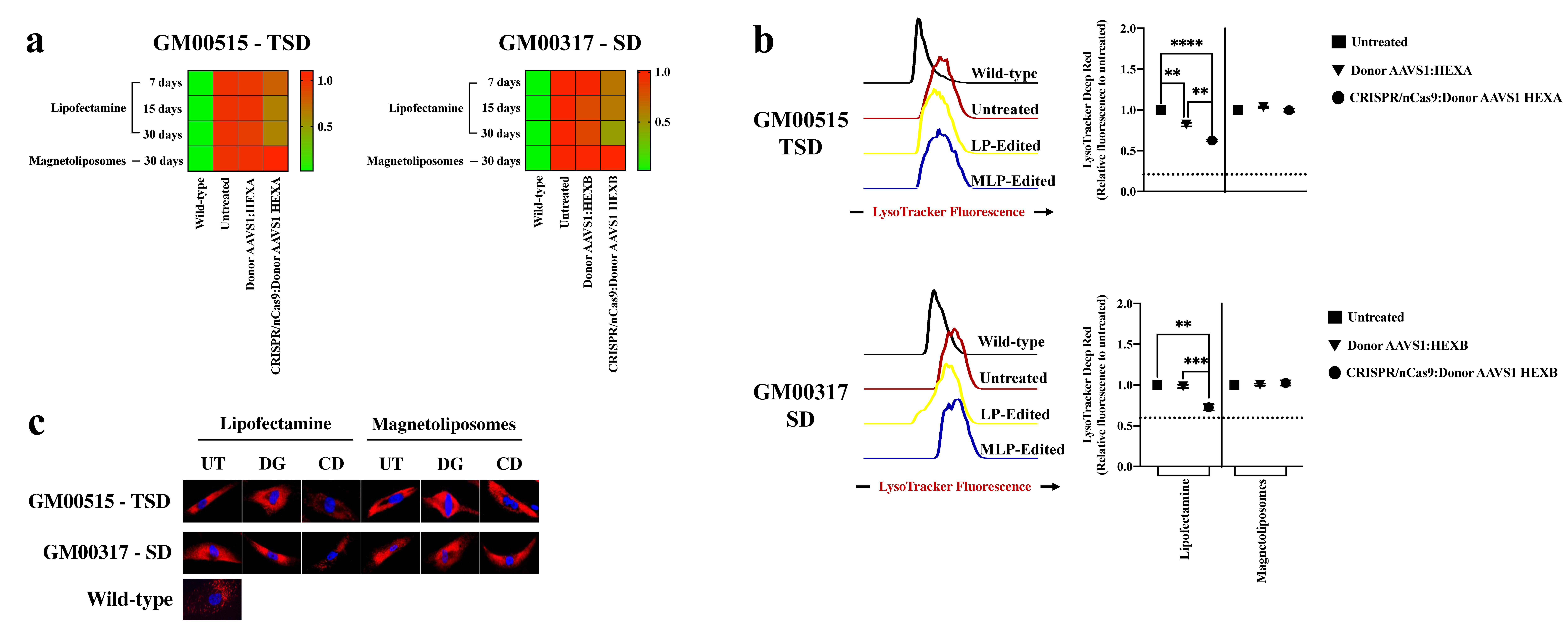
2.2.4. Lysosomal Storage Significantly Decreases in TSD and SD Fibroblasts after LPs-Assisted CRISPR/nCas9 System Delivery
2.2.5. Global Oxidative Stress Is Positively Impacted by CRISPR/nCas9-Based Genome Editing
3. Discussion
4. Materials and Methods
4.1. Molecular Validation on HEK293 Cells
4.2. Delivery of the CRISPR/nCas9 System and Donor AAVS1 to GM2 Fibroblasts
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leal, A.F.; Suarez, D.A.; Echeverri-PeñA, O.Y.; Albarracín, S.L.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. Sphingolipids and their role in health and disease in the central nervous system. Adv. Biol. Regul. 2022, 85, 100900. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Garzón Jaramillo, R.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Brunel-Guitton, C.; Lortie, A.; Gauvin, F.; Morales, C.R.; Mitchell, G.A.; Pshezhetsky, A.V. Atypical Juvenile Presentation of G M2 Gangliosidosis AB in a Patient Compound-Heterozygote for c.259G ≫ T and c.164C ≫ T Mutations in the GM2A Gene. Mol. Genet. Metab. Rep. 2017, 11, 24–29. [Google Scholar] [CrossRef]
- Sheth, J.; Datar, C.; Mistri, M.; Bhavsar, R.; Sheth, F.; Shah, K. GM2 gangliosidosis AB variant: Novel mutation from India—A case report with a review. BMC Pediatr. 2016, 16, 88. [Google Scholar] [CrossRef]
- Sandhoff, R.; Sandhoff, K. Emerging concepts of ganglioside metabolism. FEBS Lett. 2018, 592, 3835–3864. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, A.E.; Brockhausen, I.; Walia, J.S. Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders. Int. J. Mol. Sci. 2020, 21, 6881. [Google Scholar] [CrossRef]
- Cachon-Gonzalez, M.B.; Zaccariotto, E.; Cox, T.M. Genetics and Therapies for GM2 Gangliosidosis. Curr. Gene Ther. 2018, 18, 68–89. [Google Scholar] [CrossRef]
- Virgolini, M.J.; Feliziani, C.; Cambiasso, M.J.; Lopez, P.H.; Bollo, M. Neurite atrophy and apoptosis mediated by PERK signaling after accumulation of GM2-ganglioside. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 225–239. [Google Scholar] [CrossRef]
- Allende, M.L.; Cook, E.K.; Larman, B.C.; Nugent, A.; Brady, J.M.; Golebiowski, D.; Sena-Esteves, M.; Tifft, C.J.; Proia, R.L. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 2018, 59, 550–563. [Google Scholar] [CrossRef]
- Espejo-Mojica, A.J.; Rodríguez-López, A.; Li, R.; Zheng, W.; Alméciga-Díaz, C.J.; Dulcey-Sepúlveda, C.; Combariza, G.; Barrera, L.A. Human recombinant lysosomal β-Hexosaminidases produced in Pichia pastoris efficiently reduced lipid accumulation in Tay-Sachs fibroblasts. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 885–895. [Google Scholar] [CrossRef]
- Espejo-Mojica, A.J.; Mosquera, A.; Rodrfguez-Lopez, A.; Diaz, D.; Beltran, L.; Hernandez, F.L.; Alméciga-Diaz, C.J.; Barrera, L.A. Characterization of recombinant human lysosomal beta-hexosaminidases produced in the methylotrophic yeast Pichia pastoris. Univ. Sci. 2016, 21, 195–217. [Google Scholar] [CrossRef]
- Osher, E.; Fattal-Valevski, A.; Sagie, L.; Urshanski, N.; Sagiv, N.; Peleg, L.; Lerman-Sagie, T.; Zimran, A.; Elstein, D.; Navon, R.; et al. Effect of cyclic, low dose pyrimethamine treatment in patients with Late Onset Tay Sachs: An open label, extended pilot study. Orphanet J. Rare Dis. 2015, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, E.; Niemir, N.; Aureli, M.; Magini, A.; Loberto, N.; Prinetti, A.; Bassi, R.; Polchi, A.; Emiliani, C.; Caillaud, C.; et al. Chaperone therapy for GM2 gangliosidosis: Effects of pyrimethamine on β-hexosaminidase activity in Sandhoff fibroblasts. Mol. Neurobiol. 2014, 50, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Masciullo, M.; Santoro, M.; Modoni, A.; Ricci, E.; Guitton, J.; Tonali, P.; Silvestri, G. Substrate reduction therapy with miglustat in chronic GM2 gangliosidosis type Sandhoff: Results of a 3-year follow-up. J. Inherit. Metab. Dis. 2010, 33, 355–361. [Google Scholar] [CrossRef]
- Ornaghi, F.; Sala, D.; Tedeschi, F.; Maffia, M.C.; Bazzucchi, M.; Morena, F.; Valsecchi, M.; Aureli, M.; Martino, S.; Gritti, A. Novel bicistronic lentiviral vectors correct β-Hexosaminidase deficiency in neural and hematopoietic stem cells and progeny: Implications for in vivo and ex vivo gene therapy of GM2 gangliosidosis. Neurobiol. Dis. 2020, 134, 104667. [Google Scholar] [CrossRef]
- Flotte, T.R.; Cataltepe, O.; Puri, A.; Batista, A.R.; Moser, R.; McKenna-Yasek, D.; Douthwright, C.; Gernoux, G.; Blackwood, M.; Mueller, C.; et al. AAV gene therapy for Tay-Sachs disease. Nat. Med. 2022, 28, 251–259. [Google Scholar] [CrossRef]
- Toro, C.; Zainab, M.; Tifft, C.J. The GM2 gangliosidoses: Unlocking the mysteries of pathogenesis and treatment. Neurosci. Lett. 2021, 764, 136195. [Google Scholar] [CrossRef]
- Caruso, S.M.; Quinn, P.M.; da Costa, B.L.; Tsang, S.H. CRISPR/Cas therapeutic strategies for autosomal dominant disorders. J. Clin. Investig. 2022, 132, e158287. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes 2020, 11, 704. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Tăbăran, A.F.; Overn, P.; O’Sullivan, M.G.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; Whitley, C.B. A novel gene editing system to treat both Tay-Sachs and Sandhoff diseases. Gene Ther. 2020, 27, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Tropak, M.B.; Yonekawa, S.; Karumuthil-Melethil, S.; Thompson, P.; Wakarchuk, W.; Gray, S.J.; Walia, J.S.; Mark, B.L.; Mahuran, D. Construction of a hybrid β-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol. Ther. Methods Clin. Dev. 2016, 3, 15057. [Google Scholar] [CrossRef]
- Kot, S.; Karumuthil-Melethil, S.; Woodley, E.; Zaric, V.; Thompson, P.; Chen, Z.; Lykken, E.; Keimel, J.G.; Kaemmerer, W.F.; Gray, S.J.; et al. Investigating Immune Responses to the scAAV9-HEXM Gene Therapy Treatment in Tay-Sachs Disease and Sandhoff Disease Mouse Models. Int. J. Mol. Sci. 2021, 22, 6751. [Google Scholar] [CrossRef]
- Leal, A.F.; Alméciga-Díaz, C.J. Efficient CRISPR/Cas9 nickase-mediated genome editing in an in vitro model of mucopolysaccharidosis IVA. Gene Ther. 2022, 1–8. [Google Scholar] [CrossRef]
- Leal, A.F.; Cifuentes, J.; Torres, C.E.; Suarez, D.; Quezada, V.; Gomez, S.C.; Cruz, J.C.; Reyes, L.H.; Espejo-Mojica, A.J.; Almeciga-Diaz, C.J. Delivery and assessment of a CRISPR/nCas9-based genome editing system on in vitro models of mucopolysaccharidoses IVA assisted by magnetite-based nanoparticles. Sci. Rep. 2022, 12, 15045. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Przybilla, M.J.; Whitley, C.B. Metabolomics profiling reveals profound metabolic impairments in mice and patients with Sandhoff disease. Mol. Genet. Metab. 2019, 126, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, R.; Schulze, H.; Sandhoff, K. Ganglioside Metabolism in Health and Disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 1–62. [Google Scholar] [CrossRef] [PubMed]
- Vu, M.; Li, R.; Baskfield, A.; Lu, B.; Farkhondeh, A.; Gorshkov, K.; Motabar, O.; Beers, J.; Chen, G.; Zou, J.; et al. Neural stem cells for disease modeling and evaluation of therapeutics for Tay-Sachs disease. Orphanet J. Rare Dis. 2018, 13, 152. [Google Scholar] [CrossRef]
- Mahuran, D.J. The biochemistry of HEXA and HEXB gene mutations causing GM2 gangliosidosis. Biochim. Biophys. Acta 1991, 1096, 87–94. [Google Scholar] [CrossRef]
- Dersh, D.; Iwamoto, Y.; Argon, Y. Tay-Sachs disease mutations in HEXA target the α chain of hexosaminidase A to endoplasmic reticulum-associated degradation. Mol. Biol. Cell 2016, 27, 3813–3827. [Google Scholar] [CrossRef]
- Wendeler, M.; Sandhoff, K. Hexosaminidase assays. Glycoconj. J. 2009, 26, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Sala, D.; Ornaghi, F.; Morena, F.; Argentati, C.; Valsecchi, M.; Alberizzi, V.; Di Guardo, R.; Bolino, A.; Aureli, M.; Martino, S.; et al. Therapeutic advantages of combined gene/cell therapy strategies in a murine model of GM2 gangliosidosis. Mol. Ther. Methods Clin. Dev. 2022, 25, 170–189. [Google Scholar] [CrossRef]
- Lemieux, M.J.; Mark, B.L.; Cherney, M.M.; Withers, S.G.; Mahuran, D.J.; James, M.N. Crystallographic structure of human beta-hexosaminidase A: Interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis. J. Mol. Biol. 2006, 359, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Proia, R.L.; Soravia, E. Organization of the gene encoding the human beta-hexosaminidase alpha-chain. J. Biol. Chem. 1987, 262, 5677–5681. [Google Scholar] [CrossRef]
- Proia, R.L.; d’Azzo, A.; Neufeld, E.F. Association of alpha- and beta-subunits during the biosynthesis of beta-hexosaminidase in cultured human fibroblasts. J. Biol. Chem. 1984, 259, 3350–3354. [Google Scholar] [CrossRef]
- Rivera-Colón, Y.; Schutsky, E.K.; Kita, A.Z.; Garman, S.C. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A. J. Mol. Biol. 2012, 423, 736–751. [Google Scholar] [CrossRef]
- Zito, E.; Fraldi, A.; Pepe, S.; Annunziata, I.; Kobinger, G.; Di Natale, P.; Ballabio, A.; Cosma, M.P. Sulphatase activities are regulated by the interaction of sulphatase-modifying factor 1 with SUMF2. EMBO Rep. 2005, 6, 655–660. [Google Scholar] [CrossRef]
- de Carvalho, T.G.; Schuh, R.; Pasqualim, G.; Pellenz, F.M.; Filippi-Chiela, E.C.; Giugliani, R.; Baldo, G.; Matte, U. CRISPR-Cas9-mediated gene editing in human MPS I fibroblasts. Gene 2018, 678, 33–37. [Google Scholar] [CrossRef]
- Schuh, R.S.; de Carvalho, T.G.; Giugliani, R.; Matte, U.; Baldo, G.; Teixeira, H.F. Gene editing of MPS I human fibroblasts by co-delivery of a CRISPR/Cas9 plasmid and a donor oligonucleotide using nanoemulsions as nonviral carriers. Eur. J. Pharm. Biopharm. 2018, 122, 158–166. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Poletto, E.; Baldo, G.; Gomez-Ospina, N. Genome Editing for Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 500. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Antequera, F. Structure, function and evolution of CpG island promoters. Cell. Mol. Life Sci. 2003, 60, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.R.; Harkins, R.N.; Wang, P.; Qian, H.S.; Liu, P.; Rubanyi, G.M. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med. 2004, 6, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. CRISPR therapies march into clinic, but genotoxicity concerns linger. Nat. Biotechnol. 2021, 39, 897–899. [Google Scholar] [CrossRef]
- Chiang, T.W.; le Sage, C.; Larrieu, D.; Demir, M.; Jackson, S.P. CRISPR-Cas9(D10A) nickase-based genotypic and phenotypic screening to enhance genome editing. Sci. Rep. 2016, 6, 24356. [Google Scholar] [CrossRef]
- Ramírez-Acosta, C.M.; Cifuentes, J.; Castellanos, M.C.; Moreno, R.J.; Muñoz-Camargo, C.; Cruz, J.C.; Reyes, L.H. PH-Responsive, Cell-Penetrating, Core/Shell Magnetite/Silver Nanoparticles for the Delivery of Plasmids: Preparation, Characterization, and Preliminary. Pharmaceutics 2020, 12, 561. [Google Scholar] [CrossRef]
- Patil, Y.P.; Jadhav, S. Novel methods for liposome preparation. Chem. Phys. Lipids 2014, 177, 8–18. [Google Scholar] [CrossRef]
- Aranda, P.S.; LaJoie, D.M.; Jorcyk, C.L. Bleach gel: A simple agarose gel for analyzing RNA quality. Electrophoresis 2012, 33, 366–369. [Google Scholar] [CrossRef]
- Tropak, M.B.; Reid, S.P.; Guiral, M.; Withers, S.G.; Mahuran, D. Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff Patients. J. Biol. Chem. 2004, 279, 13478–13487. [Google Scholar] [CrossRef] [Green Version]
- Mauri, V.; Lotfi, P.; Segatori, L.; Sardiello, M. A rapid and sensitive method for measuring N-acetylglucosaminidase activity in cultured cells. PLoS ONE 2013, 8, e68060. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Toma, L.; Pinto, W.; Rodrigues, V.C.; Dietrich, C.P.; Nader, H.B. Impaired sulphated glycosaminoglycan metabolism in a patient with GM-2 gangliosidosis (Tay-Sachs disease). J. Inherit. Metab. Dis. 1990, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.S.; Grisham, M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, A.F.; Cifuentes, J.; Quezada, V.; Benincore-Flórez, E.; Cruz, J.C.; Reyes, L.H.; Espejo-Mojica, A.J.; Alméciga-Díaz, C.J. CRISPR/nCas9-Based Genome Editing on GM2 Gangliosidoses Fibroblasts via Non-Viral Vectors. Int. J. Mol. Sci. 2022, 23, 10672. https://doi.org/10.3390/ijms231810672
Leal AF, Cifuentes J, Quezada V, Benincore-Flórez E, Cruz JC, Reyes LH, Espejo-Mojica AJ, Alméciga-Díaz CJ. CRISPR/nCas9-Based Genome Editing on GM2 Gangliosidoses Fibroblasts via Non-Viral Vectors. International Journal of Molecular Sciences. 2022; 23(18):10672. https://doi.org/10.3390/ijms231810672
Chicago/Turabian StyleLeal, Andrés Felipe, Javier Cifuentes, Valentina Quezada, Eliana Benincore-Flórez, Juan Carlos Cruz, Luis Humberto Reyes, Angela Johana Espejo-Mojica, and Carlos Javier Alméciga-Díaz. 2022. "CRISPR/nCas9-Based Genome Editing on GM2 Gangliosidoses Fibroblasts via Non-Viral Vectors" International Journal of Molecular Sciences 23, no. 18: 10672. https://doi.org/10.3390/ijms231810672