
A Functional Network Driven by MicroRNA-125a Regulates Monocyte Trafficking in Acute Inflammation
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. miR-125a Modulates the Expression of Genes Involved in Monocyte Adhesion
2.2. JAM-A and JAM-L Are Direct Targets of miR-125a
2.3. miR-125a Impairs Monocyte Adhesion to Inflammatory Endothelial Cells by Targeting JAM-A and JAM-L
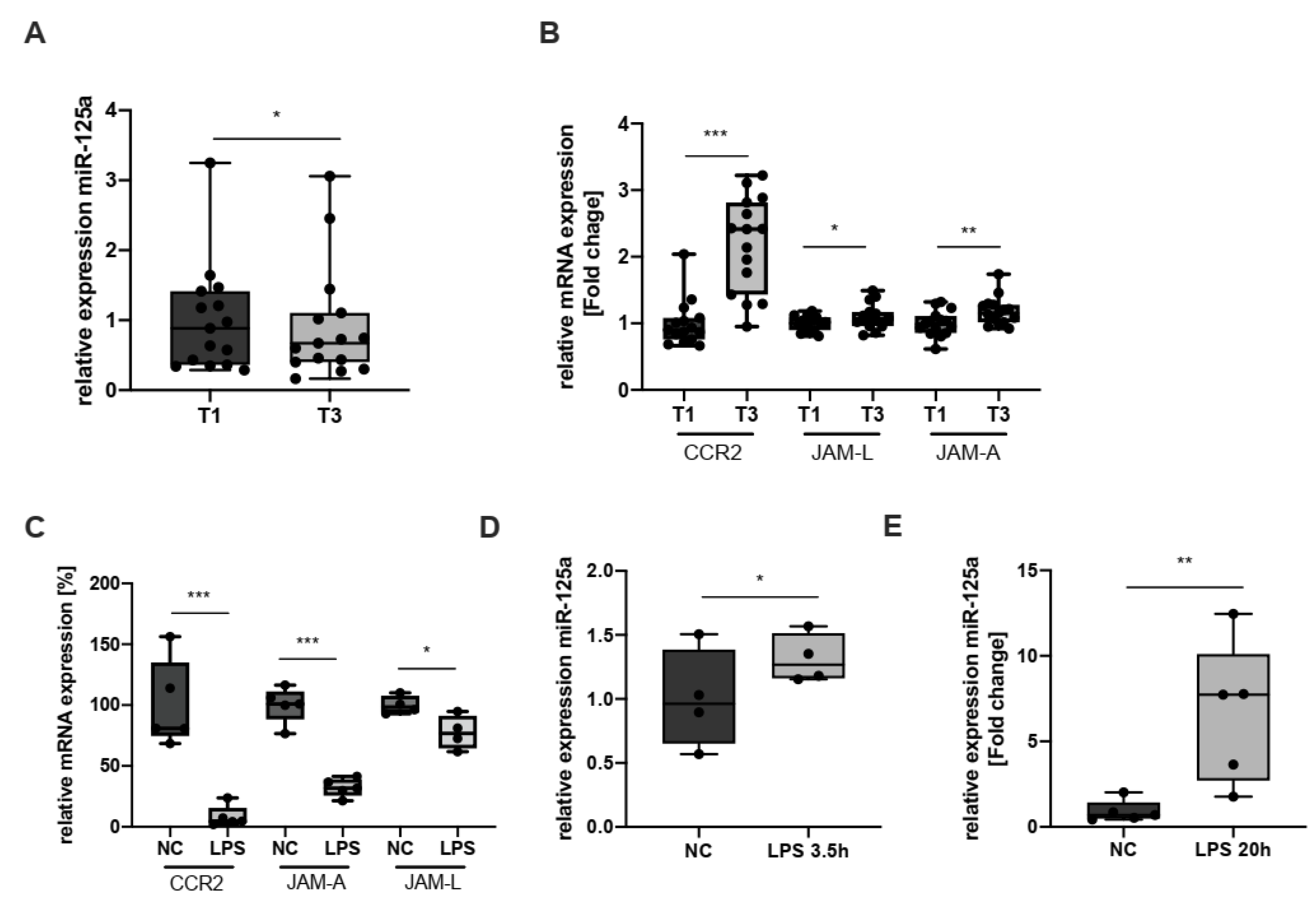
2.4. Expression of CCR2, JAM-L, JAM-A and miR-125a during Inflammation
3. Discussion
4. Materials and Methods
4.1. PBMC Isolation and Microbead-Based Monocyte Extraction
4.2. Transfection of Primary Human Monocytes
4.3. HUVEC Isolation and Cell Culture
4.4. DNA and RNA Extraction
4.5. cDNA Synthesis and Quantitative Real-Time PCR
4.6. Microarray
4.7. Cloning of Reporter Constructs
4.8. Reporter Gene Assays
4.9. Flow Cytometry
4.10. Adhesion Assays
4.11. Chemotaxis Assay
4.12. Ethics
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortega-Gomez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, R.A.; Gupta, M.; George, A.; Naqvi, A.R. MicroRNAs in shaping the resolution phase of inflammation. Semin. Cell Dev. Biol. 2021, 124, 48–62. [Google Scholar] [CrossRef]
- Feehan, K.T.; Gilroy, D.W. Is Resolution the End of Inflammation? Trends Mol. Med. 2019, 25, 198–214. [Google Scholar] [CrossRef] [PubMed]
- Doran, A.C. Inflammation Resolution: Implications for Atherosclerosis. Circ. Res. 2022, 130, 130–148. [Google Scholar] [CrossRef]
- Herrero-Cervera, A.; Soehnlein, O.; Kenne, E. Neutrophils in chronic inflammatory diseases. Cell. Mol. Immunol. 2022, 19, 177–191. [Google Scholar] [CrossRef]
- Müller, M.B.; Hübner, M.; Li, L.; Tomasi, S.; Ließke, V.; Effinger, D.; Hirschberger, S.; Pogoda, K.; Sperandio, M.; Kreth, S. Cell-Crossing Functional Network Driven by microRNA-125a Regulates Endothelial Permeability and Monocyte Trafficking in Acute Inflammation. Front. Immunol. 2022, 13, 826047. [Google Scholar] [CrossRef]
- Yi, L.; Chandrasekaran, P.; Venkatesan, S. TLR Signaling Paralyzes Monocyte Chemotaxis through Synergized Effects of p38 MAPK and Global Rap-1 Activation. PLoS ONE 2012, 7, e30404. [Google Scholar] [CrossRef]
- Li, L.; Huang, L.; Sung, S.-S.J.; Vergis, A.L.; Rosin, D.L.; Rose, C.E., Jr.; Lobo, P.I.; Okusa, M.D. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia–reperfusion injury. Kidney Int. 2008, 74, 1526–1537. [Google Scholar] [CrossRef]
- Lee, D.K.; Grantham, R.N.; Trachte, A.L.; Mannion, J.D.; Wilson, C.L. Activation of the canonical Wnt/β-catenin pathway enhances monocyte adhesion to endothelial cells. Biochem. Biophys. Res. Commun. 2006, 347, 109–116. [Google Scholar] [CrossRef]
- Al-Chaqmaqchi, H.A.M.; Moshfegh, A.; Dadfar, E.; Paulsson, J.; Hassan, M.; Jacobson, S.H.; Lundahl, J. Activation of Wnt/β-Catenin Pathway in Monocytes Derived from Chronic Kidney Disease Patients. PLoS ONE 2013, 8, e68937. [Google Scholar] [CrossRef] [PubMed]
- Tickenbrock, L.; Schwäble, J.; Strey, A.; Sargin, B.; Hehn, S.; Baas, M.; Gerke, V.; Serve, H.; Choudhary, C.; Berdel, W.E.; et al. Wnt signaling regulates transendothelial migration of monocytes. J. Leukoc. Biol. 2006, 79, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Lorenowicz, M.J.; Van Gils, J.; De Boer, M.; Hordijk, P.L.; Fernandez-Borja, M. Epac1-Rap1 signaling regulates monocyte adhesion and chemotaxis. J. Leukoc. Biol. 2006, 80, 1542–1552. [Google Scholar] [CrossRef]
- Ostermann, G.; Weber, K.S.; Zernecke, A.; Schröder, A.; Weber, C. JAM-1 Is a Ligand of the β2 Integrin LFA-1 Involved in Transendothelial Migration of Leukocytes. Nat. Immunol. 2002, 3, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Anastos, K.; Morgello, S.; Berman, J.W. JAM-A and ALCAM are therapeutic targets to inhibit diapedesis across the BBB of CD14+CD16+monocytes in HIV-infected individuals. J. Leukoc. Biol. 2014, 97, 401–412. [Google Scholar] [CrossRef]
- Ebnet, K. Junctional Adhesion Molecules (JAMs): Cell Adhesion Receptors With Pleiotropic Functions in Cell Physiology and Development. Physiol. Rev. 2017, 97, 1529–1554. [Google Scholar] [CrossRef]
- Luissint, A.-C.; Lutz, P.G.; Calderwood, D.A.; Couraud, P.-O.; Bourdoulous, S. JAM-L-Mediated Leukocyte Adhesion to En-dothelial Cells Is Regulated in Cis by α4β1 Integrin Activation. J. Gen. Physiol. 2009, 133, i1. [Google Scholar] [CrossRef]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Khan, H.N.; Perlee, D.; Schoenmaker, L.; Van Der Meer, A.-J.; Franitza, M.; Toliat, M.R.; Nürnberg, P.; Zwinderman, A.H.; Van Der Poll, T.; Scicluna, B.P.; et al. Leukocyte transcriptional signatures dependent on LPS dosage in human endotoxemia. J. Leukoc. Biol. 2019, 106, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Song, L.; Maurer, K.; Dou, Y.; Patel, V.R.; Su, C.; Leonard, M.E.; Lu, S.; Hodge, K.M.; Torres, A.; et al. IL-1 Transcrip-tional Responses to Lipopolysaccharides Are Regulated by a Complex of RNA Binding Proteins. J. Immunol. 2020, 204, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Koplev, S.; Fisher, E.A.; Tabas, I.; Björkegren, J.L.M.; Doran, A.C.; Kovacic, J.C. Macrophage Trafficking, In-flammatory Resolution, and Genomics in Atherosclerosis: JACC Macrophage in CVD Series (Part 2). J. Am. Coll. Cardiol. 2018, 72, 2181–2197. [Google Scholar] [CrossRef]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, H. The Roles of Junctional Adhesion Molecules (JAMs) in Cell Migration. Front. Cell Dev. Biol. 2022, 10, 843671. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-L.; Bai, R.; Chen, C.X.-J.; Liu, D.-Q.; Liu, Y.; Zhang, C.-Y.; Zen, K. Role of Junctional Adhesion Molecule-Like Protein in Mediating Monocyte Transendothelial Migration. Arter. Thromb. Vasc. Biol. 2009, 29, 75–83. [Google Scholar] [CrossRef]
- Cera, M.R.; Fabbri, M.; Molendini, C.; Corada, M.; Orsenigo, F.; Rehberg, M.; Reichel, C.A.; Krombach, F.; Pardi, R.; Dejana, E. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J. Cell Sci. 2009, 122, 268–277. [Google Scholar] [CrossRef]
- Martìn-Padura, I.; Lostaglio, S.; Schneemann, M.; Williams, L.; Romano, M.; Fruscella, P.; Panzeri, M.C.; Stoppacciaro, A.; Ruco, L.; Villa, A.; et al. Junctional Adhesion Molecule, a Novel Member of the Immunoglobulin Superfamily That Distributes at Intercellular Junctions and Modulates Monocyte Transmigration. J. Cell Biol. 1998, 142, 117–127. [Google Scholar] [CrossRef]
- Vestweber, D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol. Rev. 2007, 218, 178–196. [Google Scholar] [CrossRef]
- Ingersoll, M.A.; Platt, A.M.; Potteaux, S.; Randolph, G.J. Monocyte trafficking in acute and chronic inflammation. Trends Immunol. 2011, 32, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Imhof, B.A.; Aurrand-Lions, M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004, 4, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Rajakariar, R.; Lawrence, T.; Bystrom, J.; Hilliard, M.; Colville-Nash, P.; Bellingan, G.; Fitzgerald, D.; Yaqoob, M.M.; Gilroy, D.W. Novel biphasic role for lymphocytes revealed during resolving inflammation. Blood 2008, 111, 4184–4192. [Google Scholar] [CrossRef] [PubMed]
- Hirschberger, S.; Hinske, L.C.; Kreth, S. MiRNAs: Dynamic regulators of immune cell functions in inflammation and cancer. Cancer Lett. 2018, 431, 11–21. [Google Scholar] [CrossRef]
- Kreth, S.; Hübner, M.; Hinske, L.C. MicroRNAs as Clinical Biomarkers and Therapeutic Tools in Perioperative Medicine. Anesthesia Analg. 2018, 126, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Verdino, P.; Witherden, D.A.; Havran, W.L.; Wilson, I.A. The Molecular Interaction of CAR and JAML Recruits the Central Cell Signal Transducer PI3K. Science 2010, 329, 1210–1214. [Google Scholar] [CrossRef]
- Witherden, D.A.; Verdino, P.; Rieder, S.E.; Garijo, O.; Mills, R.E.; Teyton, L.; Fischer, W.H.; Wilson, I.A.; Havran, W.L. The Junctional Adhesion Molecule JAML Is a Costimulatory Receptor for Epithelial γδ T Cell Activation. Science 2010, 329, 1205–1210. [Google Scholar] [CrossRef]
- Steinbacher, T.; Kummer, D.; Ebnet, K. Junctional adhesion molecule-A: Functional diversity through molecular promiscuity. Cell. Mol. Life Sci. 2017, 75, 1393–1409. [Google Scholar] [CrossRef]
- Hatami, S.; Hefler, J.; Freed, D.H. Inflammation and Oxidative Stress in the Context of Extracorporeal Cardiac and Pulmonary Support. Front. Immunol. 2022, 13, 831930. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [Green Version]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion During Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Cui, H.; Xie, N.; Tan, Z.; Yang, S.; Icyuz, M.; Thannickal, V.J.; Abraham, E.; Liu, G. miR-125a-5p Regulates Dif-ferential Activation of Macrophages and Inflammation. J. Biol. Chem. 2013, 288, 35428–35436. [Google Scholar] [CrossRef] [PubMed]
- Hübner, M.; Hinske, C.L.; Effinger, D.; Wu, T.; Thon, N.; Kreth, F.-W.; Kreth, S. Intronic miR-744 Inhibits Glioblastoma Migration by Functionally Antagonizing Its Host Gene MAP2K4. Cancers 2018, 10, 400. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Hinske, L.C.; Dos Santos, F.R.C.; Ohara, D.T.; Ohno-Machado, L.; Kreth, S.; Galante, P.A.F. MiRIAD update: Using alternative polyadenylation, protein interaction network analysis and additional species to enhance exploration of the role of intragenic miRNAs and their host genes. Database 2017, 2017, bax053. [Google Scholar] [CrossRef]
- Hübner, M.; Effinger, D.; Wu, T.; Strauß, G.; Pogoda, K.; Kreth, F.-W.; Kreth, S. The IL-1 Antagonist Anakinra Attenuates Glioblastoma Aggressiveness by Dampening Tumor-Associated Inflammation. Cancers 2020, 12, 433. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomasi, S.; Li, L.; Hinske, L.C.; Tomasi, R.; Amini, M.; Strauß, G.; Müller, M.B.; Hirschberger, S.; Peterss, S.; Effinger, D.; et al. A Functional Network Driven by MicroRNA-125a Regulates Monocyte Trafficking in Acute Inflammation. Int. J. Mol. Sci. 2022, 23, 10684. https://doi.org/10.3390/ijms231810684
Tomasi S, Li L, Hinske LC, Tomasi R, Amini M, Strauß G, Müller MB, Hirschberger S, Peterss S, Effinger D, et al. A Functional Network Driven by MicroRNA-125a Regulates Monocyte Trafficking in Acute Inflammation. International Journal of Molecular Sciences. 2022; 23(18):10684. https://doi.org/10.3390/ijms231810684
Chicago/Turabian StyleTomasi, Stephanie, Lei Li, Ludwig Christian Hinske, Roland Tomasi, Martina Amini, Gabriele Strauß, Martin Bernhard Müller, Simon Hirschberger, Sven Peterss, David Effinger, and et al. 2022. "A Functional Network Driven by MicroRNA-125a Regulates Monocyte Trafficking in Acute Inflammation" International Journal of Molecular Sciences 23, no. 18: 10684. https://doi.org/10.3390/ijms231810684