
Epilepsy Characteristics in Neurodevelopmental Disorders: Research from Patient Cohorts and Animal Models Focusing on Autism Spectrum Disorder

Abstract
:1. Introduction
2. Causes of Epilepsy
2.1. Genetic Causes of Epilepsy
2.1.1. Ion Channel and Receptor-Mediated Causes
Voltage-Gated Sodium Ion (NaV) Channels
Voltage-Gated Potassium Ion (Kv) Channels
GABA Receptors
Glutamate Receptors
2.1.2. Metabolic Causes
2.2. Nongenetic Causes of Epilepsy
3. Patient Studies of Epilepsy and ASD
3.1. Mechanisms of Epilepsy–ASD Comorbidity
3.2. Hallmark EEG Signatures in Epilepsy–ASD Comorbidity
3.2.1. MeCP2
3.2.2. SYNGAP1
3.2.3. FMR1
3.2.4. SHANK1-3
3.2.5. TSC1
4. Animal Models of Epilepsy and NDDs
4.1. Nongenetic Models of Epilepsy
4.1.1. Chemical Convulsant
4.1.2. Electrical Stimulation
4.1.3. Traumatic Brain Injury
4.2. Genetic Models of Epilepsy
4.2.1. SCN1A
4.2.2. Syngap1
4.2.3. Fmr1
4.2.4. Shank3
5. Current Therapeutic Management and Antiepileptic Drugs in Clinical Trials
5.1. Species-Specific Differences between Rodent and Human-Derived Models of Epilepsy
5.2. Informed Modeling Approaches to Tackle the Species-Specific Challenges in the Development and Testing of Epileptic Models
5.2.1. Human Pluripotent Stem Cells (hPSCs)
5.2.2. Brain Organoids
5.2.3. Humanized Rodent Model
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Falco-Walter, J.J.; Scheffer, I.E.; Fisher, R.S. The new definition and classification of seizures and epilepsy. Epilepsy Res. 2018, 139, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Annu. Rev. Genom. Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef] [PubMed]
- Pack, A.M. Epilepsy Overview and Revised Classification of Seizures and Epilepsies. Contin. Minneap. Minn. 2019, 25, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Proposal for revised clinical and electroencephalographic classification of epileptic seizures. From the Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1981, 22, 489–501. [CrossRef]
- Meisler, M.H.; Kearney, J.; Ottman, R.; Escayg, A. Identification of epilepsy genes in human and mouse. Annu. Rev. Genet. 2001, 35, 567–588. [Google Scholar] [CrossRef]
- Engel, T.; Alves, M.; Sheedy, C.; Henshall, D.C. ATPergic signalling during seizures and epilepsy. Neuropharmacology 2016, 104, 140–153. [Google Scholar] [CrossRef]
- Beamer, E.; Lacey, A.; Alves, M.; Conte, G.; Tian, F.; de Diego-Garcia, L.; Khalil, M.; Rosenow, F.; Delanty, N.; Dale, N.; et al. Elevated blood purine levels as a biomarker of seizures and epilepsy. Epilepsia 2021, 62, 817–828. [Google Scholar] [CrossRef]
- Diamond, M.L.; Ritter, A.C.; Failla, M.D.; Boles, J.A.; Conley, Y.P.; Kochanek, P.M.; Wagner, A.K. IL-1beta associations with posttraumatic epilepsy development: A genetics and biomarker cohort study. Epilepsia 2014, 55, 1109–1119. [Google Scholar] [CrossRef]
- Nass, R.D.; Akgun, K.; Dague, K.O.; Elger, C.E.; Reichmann, H.; Ziemssen, T.; Surges, R. CSF and Serum Biomarkers of Cerebral Damage in Autoimmune Epilepsy. Front. Neurol. 2021, 12, 647428. [Google Scholar] [CrossRef]
- Tang, H.; Wang, X. PD-1 Is an Immune-Inflammatory Potential Biomarker in Cerebrospinal Fluid and Serum of Intractable Epilepsy. BioMed Res. Int. 2021, 2021, 7973123. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, D.; Wang, Z.; Zhou, C.; Luo, J.; Xu, Y.; Shen, L.; Yin, H.; Tao, S.; Xiao, Z.; et al. Time-dependent decrease of clusterin as a potential cerebrospinal fluid biomarker for drug-resistant epilepsy. J. Mol. Neurosci. 2014, 54, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Oberman, L.M. mGluR antagonists and GABA agonists as novel pharmacological agents for the treatment of autism spectrum disorders. Expert Opin. Investig. Drugs 2012, 21, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Benke, T.A. Autism and Epilepsy: Exploring the Relationship Using Experimental Models. Epilepsy Curr. 2015, 15, 206–210. [Google Scholar] [CrossRef]
- Buckley, A.W.; Holmes, G.L. Epilepsy and Autism. Cold Spring Harb. Perspect. Med. 2016, 6, a022749. [Google Scholar] [CrossRef]
- Jeste, S.S.; Tuchman, R. Autism Spectrum Disorder and Epilepsy: Two Sides of the Same Coin? J. Child Neurol. 2015, 30, 1963–1971. [Google Scholar] [CrossRef]
- Meng, H.; Xu, H.Q.; Yu, L.; Lin, G.W.; He, N.; Su, T.; Shi, Y.W.; Li, B.; Wang, J.; Liu, X.R.; et al. The SCN1A mutation database: Updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum. Mutat. 2015, 36, 573–580. [Google Scholar] [CrossRef]
- Nellist, M.; van den Heuvel, D.; Schluep, D.; Exalto, C.; Goedbloed, M.; Maat-Kievit, A.; van Essen, T.; van Spaendonck-Zwarts, K.; Jansen, F.; Helderman, P.; et al. Missense mutations to the TSC1 gene cause tuberous sclerosis complex. Eur. J. Hum. Genet. 2009, 17, 319–328. [Google Scholar] [CrossRef]
- Gronskov, K.; Brondum-Nielsen, K.; Dedic, A.; Hjalgrim, H. A nonsense mutation in FMR1 causing fragile X syndrome. Eur. J. Hum. Genet. 2011, 19, 489–491. [Google Scholar] [CrossRef]
- Poirier, K.; Saillour, Y.; Bahi-Buisson, N.; Jaglin, X.H.; Fallet-Bianco, C.; Nabbout, R.; Castelnau-Ptakhine, L.; Roubertie, A.; Attie-Bitach, T.; Desguerre, I.; et al. Mutations in the neuronal ss-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 2010, 19, 4462–4473. [Google Scholar] [CrossRef]
- Yu, T.W.; Mochida, G.H.; Tischfield, D.J.; Sgaier, S.K.; Flores-Sarnat, L.; Sergi, C.M.; Topcu, M.; McDonald, M.T.; Barry, B.J.; Felie, J.M.; et al. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 2010, 42, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Z.J.; Liu, L.; Xu, H.Q.; Shi, Y.W.; Yi, Y.H.; He, N.; Liao, W.P. Epilepsy-associated genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kodera, H.; Ohba, C.; Kato, M.; Maeda, T.; Araki, K.; Tajima, D.; Matsuo, M.; Hino-Fukuyo, N.; Kohashi, K.; Ishiyama, A.; et al. De novo GABRA1 mutations in Ohtahara and West syndromes. Epilepsia 2016, 57, 566–573. [Google Scholar] [CrossRef]
- Oyrer, J.; Maljevic, S.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S.; Reid, C.A. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol. Rev. 2018, 70, 142–173. [Google Scholar] [CrossRef]
- Parihar, R.; Ganesh, S. The SCN1A gene variants and epileptic encephalopathies. J. Hum. Genet. 2013, 58, 573–580. [Google Scholar] [CrossRef]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, B.P.; Claes, L.R.; Lagae, L.G. Clinical correlations of mutations in the SCN1A gene: From febrile seizures to severe myoclonic epilepsy in infancy. Pediatr. Neurol. 2004, 30, 236–243. [Google Scholar] [CrossRef]
- Claes, L.; Ceulemans, B.; Audenaert, D.; Smets, K.; Lofgren, A.; Del-Favero, J.; Ala-Mello, S.; Basel-Vanagaite, L.; Plecko, B.; Raskin, S.; et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum. Mutat. 2003, 21, 615–621. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Harkin, L.A.; Grinton, B.E.; Dibbens, L.M.; Turner, S.J.; Zielinski, M.A.; Xu, R.; Jackson, G.; Adams, J.; Connellan, M.; et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 2007, 130, 100–109. [Google Scholar] [CrossRef]
- Gazina, E.V.; Leaw, B.T.; Richards, K.L.; Wimmer, V.C.; Kim, T.H.; Aumann, T.D.; Featherby, T.J.; Churilov, L.; Hammond, V.E.; Reid, C.A.; et al. ‘Neonatal’ Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Hum. Mol. Genet. 2015, 24, 1457–1468. [Google Scholar] [CrossRef] [Green Version]
- Jan, L.Y.; Jan, Y.N. Voltage-gated potassium channels and the diversity of electrical signalling. J. Physiol. 2012, 590, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, J.S. Regulation of ion channel expression by cytoplasmic subunits. Curr. Opin. Neurobiol. 1998, 8, 370–374. [Google Scholar] [CrossRef]
- Eunson, L.H.; Rea, R.; Zuberi, S.M.; Youroukos, S.; Panayiotopoulos, C.P.; Liguori, R.; Avoni, P.; McWilliam, R.C.; Stephenson, J.B.; Hanna, M.G.; et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann. Neurol. 2000, 48, 647–656. [Google Scholar] [CrossRef]
- Maljevic, S.; Lerche, H. Potassium channels: A review of broadening therapeutic possibilities for neurological diseases. J. Neurol. 2013, 260, 2201–2211. [Google Scholar] [CrossRef]
- Syrbe, S.; Hedrich, U.B.S.; Riesch, E.; Djemie, T.; Muller, S.; Moller, R.S.; Maher, B.; Hernandez-Hernandez, L.; Synofzik, M.; Caglayan, H.S.; et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 2015, 47, 393–399. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Noebels, J. Pathway-driven discovery of epilepsy genes. Nat. Neurosci. 2015, 18, 344–350. [Google Scholar] [CrossRef]
- Schroer, R.J.; Phelan, M.C.; Michaelis, R.C.; Crawford, E.C.; Skinner, S.A.; Cuccaro, M.; Simensen, R.J.; Bishop, J.; Skinner, C.; Fender, D.; et al. Autism and maternally derived aberrations of chromosome 15q. Am. J. Med. Genet. 1998, 76, 327–336. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Silverman, J.M.; Smith, C.J.; Greenberg, D.A.; Kilifarski, M.; Reichert, J.; Cook, E.H., Jr.; Fang, Y.; Song, C.Y.; Vitale, R. Association between a GABRB3 polymorphism and autism. Mol. Psychiatry 2002, 7, 311–316. [Google Scholar] [CrossRef]
- Shinohe, A.; Hashimoto, K.; Nakamura, K.; Tsujii, M.; Iwata, Y.; Tsuchiya, K.J.; Sekine, Y.; Suda, S.; Suzuki, K.; Sugihara, G.; et al. Increased serum levels of glutamate in adult patients with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; Al Mahmoud, R.A.; Mekki, M.; El-Hattab, A.W. Metabolic Seizures. Front. Neurol. 2021, 12, 640371. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Casanova, M.F.; Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Brown, G.L.; Edelson, S.M.; Slattery, J.C.; Adams, J.B. Neuropathological Mechanisms of Seizures in Autism Spectrum Disorder. Front. Neurosci. 2016, 10, 192. [Google Scholar] [CrossRef] [PubMed]
- Novarino, G.; El-Fishawy, P.; Kayserili, H.; Meguid, N.A.; Scott, E.M.; Schroth, J.; Silhavy, J.L.; Kara, M.; Khalil, R.O.; Ben-Omran, T.; et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 2012, 338, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E. Metabolic and mitochondrial disorders associated with epilepsy in children with autism spectrum disorder. Epilepsy Behav. 2015, 47, 147–157. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.A. Identification and Treatment of Pathophysiological Comorbidities of Autism Spectrum Disorder to Achieve Optimal Outcomes. Clin. Med. Insights Pediatr. 2016, 10, 43–56. [Google Scholar] [CrossRef]
- Singh, R.; Turner, R.C.; Nguyen, L.; Motwani, K.; Swatek, M.; Lucke-Wold, B.P. Pediatric Traumatic Brain Injury and Autism: Elucidating Shared Mechanisms. Behav. Neurol. 2016, 2016, 8781725. [Google Scholar] [CrossRef]
- Chess, S. Follow-up report on autism in congenital rubella. J. Autism Child. Schizophr. 1977, 7, 69–81. [Google Scholar] [CrossRef]
- Velez-Ruiz, N.J.; Meador, K.J. Neurodevelopmental effects of fetal antiepileptic drug exposure. Drug Saf. 2015, 38, 271–278. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Seizures beget seizures: The quest for GABA as a key player. Crit. Rev. Neurobiol. 2006, 18, 135–144. [Google Scholar] [CrossRef]
- Banerjee, T.K.; Das, S.K. Refractory epilepsy. J. Assoc. Physicians India 2013, 61, 52–54. [Google Scholar]
- Strasser, L.; Downes, M.; Kung, J.; Cross, J.H.; De Haan, M. Prevalence and risk factors for autism spectrum disorder in epilepsy: A systematic review and meta-analysis. Dev. Med. Child Neurol. 2018, 60, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, H. Autism and epilepsy: A retrospective follow-up study. Brain Dev. 2007, 29, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, Y.; Provenzano, G.; Casarosa, S. Neurobiological bases of autism-epilepsy comorbidity: A focus on excitation/inhibition imbalance. Eur. J. Neurosci. 2018, 47, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Staba, R.J.; Stead, M.; Worrell, G.A. Electrophysiological biomarkers of epilepsy. Neurotherapeutics 2014, 11, 334–346. [Google Scholar] [CrossRef]
- Bragin, A.; Engel, J., Jr.; Wilson, C.L.; Fried, I.; Mathern, G.W. Hippocampal and entorhinal cortex high-frequency oscillations (100–500 Hz) in human epileptic brain and in kainic acid—Treated rats with chronic seizures. Epilepsia 1999, 40, 127–137. [Google Scholar] [CrossRef]
- Schevon, C.A.; Goodman, R.R.; McKhann, G., Jr.; Emerson, R.G. Propagation of epileptiform activity on a submillimeter scale. J. Clin. Neurophysiol. 2010, 27, 406–411. [Google Scholar] [CrossRef]
- Warren, C.P.; Hu, S.; Stead, M.; Brinkmann, B.H.; Bower, M.R.; Worrell, G.A. Synchrony in normal and focal epileptic brain: The seizure onset zone is functionally disconnected. J. Neurophysiol. 2010, 104, 3530–3539. [Google Scholar] [CrossRef]
- van Diessen, E.; Senders, J.; Jansen, F.E.; Boersma, M.; Bruining, H. Increased power of resting-state gamma oscillations in autism spectrum disorder detected by routine electroencephalography. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 537–540. [Google Scholar] [CrossRef]
- Operto, F.F.; Mazza, R.; Pastorino, G.M.G.; Verrotti, A.; Coppola, G. Epilepsy and genetic in Rett syndrome: A review. Brain Behav. 2019, 9, e01250. [Google Scholar] [CrossRef]
- Krajnc, N. Management of epilepsy in patients with Rett syndrome: Perspectives and considerations. Ther. Clin. Risk Manag. 2015, 11, 925–932. [Google Scholar] [CrossRef]
- Reiss, A.L.; Faruque, F.; Naidu, S.; Abrams, M.; Beaty, T.; Bryan, R.N.; Moser, H. Neuroanatomy of Rett syndrome: A volumetric imaging study. Ann. Neurol. 1993, 34, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Vlaskamp, D.R.M.; Shaw, B.J.; Burgess, R.; Mei, D.; Montomoli, M.; Xie, H.; Myers, C.T.; Bennett, M.F.; XiangWei, W.; Williams, D.; et al. SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy. Neurology 2019, 92, e96–e107. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E. Epilepsy in fragile X syndrome. Dev. Med. Child Neurol. 2002, 44, 724–728. [Google Scholar] [CrossRef]
- Davidson, M.; Sebastian, S.A.; Benitez, Y.; Desai, S.; Quinonez, J.; Ruxmohan, S.; Stein, J.D.; Cueva, W. Behavioral Problems in Fragile X Syndrome: A Review of Clinical Management. Cureus 2022, 14, e21840. [Google Scholar] [CrossRef]
- Sandoval, G.M.; Shim, S.; Hong, D.S.; Garrett, A.S.; Quintin, E.M.; Marzelli, M.J.; Patnaik, S.; Lightbody, A.A.; Reiss, A.L. Neuroanatomical abnormalities in fragile X syndrome during the adolescent and young adult years. J. Psychiatr. Res. 2018, 107, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Holder, J.L., Jr.; Quach, M.M. The spectrum of epilepsy and electroencephalographic abnormalities due to SHANK3 loss-of-function mutations. Epilepsia 2016, 57, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Paprocka, J.; Ziętkiewicz, S.; Kosińska, J.; Kaczorowska, E.; Płoski, R. Case Report: Lennox-Gastaut Epileptic Encephalopathy Responsive to Cannabidiol Treatment Associated With a Novel de novo Mosaic SHANK1 Variant. Front. Genet. 2021, 12, 735292. [Google Scholar] [CrossRef]
- Holmes, G.L.; Stafstrom, C.E. Tuberous sclerosis complex and epilepsy: Recent developments and future challenges. Epilepsia 2007, 48, 617–630. [Google Scholar] [CrossRef]
- Henriksen, M.W.; Breck, H.; von Tetzchner, S.; Paus, B.; Skjeldal, O.H.; Brodtkorb, E. Epilepsy in classic Rett syndrome: Course and characteristics in adult age. Epilepsy Res. 2018, 145, 134–139. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Neul, J.L.; Lane, J.B.; Lee, H.S.; Geerts, S.; Barrish, J.O.; Annese, F.; Baggett, L.M.; Barnes, K.; Skinner, S.A.; Motil, K.J.; et al. Developmental delay in Rett syndrome: Data from the natural history study. J. Neurodev. Disord. 2014, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renieri, A.; Mari, F.; Mencarelli, M.A.; Scala, E.; Ariani, F.; Longo, I.; Meloni, I.; Cevenini, G.; Pini, G.; Hayek, G.; et al. Diagnostic criteria for the Zappella variant of Rett syndrome (the preserved speech variant). Brain. Dev. 2009, 31, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Halbach, N.; Smeets, E.E.; Julu, P.; Witt-Engerstrom, I.; Pini, G.; Bigoni, S.; Hansen, S.; Apartopoulos, F.; Delamont, R.; van Roozendaal, K.; et al. Neurophysiology versus clinical genetics in Rett syndrome: A multicenter study. Am. J. Med. Genet. A 2016, 170, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.; Wong, K.; Jacoby, P.; Downs, J.; Leonard, H. Twenty years of surveillance in Rett syndrome: What does this tell us? Orphanet. J. Rare Dis. 2014, 9, 87. [Google Scholar] [CrossRef]
- Vignoli, A.; La Briola, F.; Peron, A.; Turner, K.; Savini, M.; Cogliati, F.; Russo, S.; Canevini, M.P. Medical care of adolescents and women with Rett syndrome: An Italian study. Am. J. Med. Genet. A 2012, 158A, 13–18. [Google Scholar] [CrossRef]
- Tarquinio, D.C.; Hou, W.; Berg, A.; Kaufmann, W.E.; Lane, J.B.; Skinner, S.A.; Motil, K.J.; Neul, J.L.; Percy, A.K.; Glaze, D.G. Longitudinal course of epilepsy in Rett syndrome and related disorders. Brain 2017, 140, 306–318. [Google Scholar] [CrossRef]
- Mari, F.; Azimonti, S.; Bertani, I.; Bolognese, F.; Colombo, E.; Caselli, R.; Scala, E.; Longo, I.; Grosso, S.; Pescucci, C.; et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005, 14, 1935–1946. [Google Scholar] [CrossRef]
- Florian, C.; Bahi-Buisson, N.; Bienvenu, T. FOXG1-Related Disorders: From Clinical Description to Molecular Genetics. Mol. Syndromol. 2012, 2, 153–163. [Google Scholar] [CrossRef]
- Kortum, F.; Das, S.; Flindt, M.; Morris-Rosendahl, D.J.; Stefanova, I.; Goldstein, A.; Horn, D.; Klopocki, E.; Kluger, G.; Martin, P.; et al. The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J. Med. Genet. 2011, 48, 396–406. [Google Scholar] [CrossRef]
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273. [Google Scholar] [CrossRef]
- Guerrini, R.; Parrini, E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia 2012, 53, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, L.E.; Ma, M.; Ahmed, S.; Bertrand, M.; Dobyns, W.B.; Wheless, J.; Paciorkowski, A.R. Epilepsy and outcome in FOXG1-related disorders. Epilepsia 2014, 55, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.E.; Tambunan, D.; LaCoursiere, C.; Goldenberg, M.; Pinsky, R.; Martin, E.; Ho, E.; Khwaja, O.; Kaufmann, W.E.; Poduri, A. Mutations in epilepsy and intellectual disability genes in patients with features of Rett syndrome. Am. J. Med. Genet. A 2015, 167A, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Liao, D.; Lau, L.F.; Huganir, R.L. SynGAP: A synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 1998, 20, 683–691. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Gauthier, J.; Spiegelman, D.; Noreau, A.; Yang, Y.; Pellerin, S.; Dobrzeniecka, S.; Cote, M.; Perreau-Linck, E.; Carmant, L.; et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 2009, 360, 599–605. [Google Scholar] [CrossRef]
- Verma, V.; Kumar, M.J.V.; Sharma, K.; Rajaram, S.; Muddashetty, R.; Manjithaya, R.; Behnisch, T.; Clement, J.P. Pharmacological intervention in young adolescents rescues synaptic physiology and behavioural deficits in Syngap1(+/−) mice. Exp. Brain Res. 2022, 240, 289–309. [Google Scholar] [CrossRef]
- Jeyabalan, N.; Clement, J.P. SYNGAP1: Mind the Gap. Front. Cell Neurosci. 2016, 10, 32. [Google Scholar] [CrossRef]
- Verma, V.; Paul, A.; Amrapali Vishwanath, A.; Vaidya, B.; Clement, J.P. Understanding intellectual disability and autism spectrum disorders from common mouse models: Synapses to behaviour. Open Biol. 2019, 9, 180265. [Google Scholar] [CrossRef]
- Zollino, M.; Gurrieri, F.; Orteschi, D.; Marangi, G.; Leuzzi, V.; Neri, G. Integrated analysis of clinical signs and literature data for the diagnosis and therapy of a previously undescribed 6p21.3 deletion syndrome. Eur. J. Hum. Genet. 2011, 19, 239–242. [Google Scholar] [CrossRef]
- Berryer, M.H.; Hamdan, F.F.; Klitten, L.L.; Moller, R.S.; Carmant, L.; Schwartzentruber, J.; Patry, L.; Dobrzeniecka, S.; Rochefort, D.; Neugnot-Cerioli, M.; et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum. Mutat. 2013, 34, 385–394. [Google Scholar] [CrossRef]
- Lo Barco, T.; Kaminska, A.; Solazzi, R.; Cances, C.; Barcia, G.; Chemaly, N.; Fontana, E.; Desguerre, I.; Canafoglia, L.; Hachon Le Camus, C.; et al. SYNGAP1-DEE: A visual sensitive epilepsy. Clin. Neurophysiol. 2021, 132, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Katsanevaki, D. Cortical Circuit and Behavioural Pathophysiology in Rodent Models of SYNGAP1 Haploinsufficiency. Ph.D. Thesis, University of Edinburgh, Edinburgh, UK, 2018. [Google Scholar]
- Niu, M.; Han, Y.; Dy, A.B.C.; Du, J.; Jin, H.; Qin, J.; Zhang, J.; Li, Q.; Hagerman, R.J. Autism Symptoms in Fragile X Syndrome. J. Child. Neurol. 2017, 32, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Filipink, R.A.; Frye, R.E.; Golla, S.; Morris, S.M.; Andrews, H.; Choo, T.H.; Kaufmann, W.E.; Consortium, F. Seizures in Fragile X Syndrome: Associations and Longitudinal Analysis of a Large Clinic-Based Cohort. Front. Pediatr. 2021, 9, 736255. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P.J.; Stafstrom, C.E. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 2009, 9, 108–112. [Google Scholar] [CrossRef]
- Wong, R.K.; Bianchi, R.; Chuang, S.C.; Merlin, L.R. Group I mGluR-induced epileptogenesis: Distinct and overlapping roles of mGluR1 and mGluR5 and implications for antiepileptic drug design. Epilepsy Curr. 2005, 5, 63–68. [Google Scholar] [CrossRef]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Asadi-Pooya, A.A. Lennox-Gastaut syndrome: A comprehensive review. Neurol. Sci. 2018, 39, 403–414. [Google Scholar] [CrossRef]
- Figura, M.G.; Coppola, A.; Bottitta, M.; Calabrese, G.; Grillo, L.; Luciano, D.; Del Gaudio, L.; Torniero, C.; Striano, S.; Elia, M. Seizures and EEG pattern in the 22q13.3 deletion syndrome: Clinical report of six Italian cases. Seizure 2014, 23, 774–779. [Google Scholar] [CrossRef]
- Smalley, S.L.; Tanguay, P.E.; Smith, M.; Gutierrez, G. Autism and tuberous sclerosis. J. Autism Dev. Disord. 1992, 22, 339–355. [Google Scholar] [CrossRef]
- Wiznitzer, M. Autism and tuberous sclerosis. J. Child. Neurol. 2004, 19, 675–679. [Google Scholar] [CrossRef]
- Chu-Shore, C.J.; Major, P.; Camposano, S.; Muzykewicz, D.; Thiele, E.A. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010, 51, 1236–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, E.; Juhasz, C.; Shah, A.; Sood, S.; Chugani, H.T. Role of subdural electrocorticography in prediction of long-term seizure outcome in epilepsy surgery. Brain 2009, 132, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.E.; Sadleir, L.G.; Harkin, L.A.; Vadlamudi, L.; McMahon, J.M.; Mulley, J.C.; Scheffer, I.E.; Berkovic, S.F. Severe myoclonic epilepsy of infancy (Dravet syndrome): Recognition and diagnosis in adults. Neurology 2006, 67, 2224–2226. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Aronica, E.; Jansen, A.; Jansen, F.; Kotulska, K.; Lagae, L.; Moavero, R.; Jozwiak, S. Early onset epileptic encephalopathy or genetically determined encephalopathy with early onset epilepsy? Lessons learned from TSC. Eur. J. Paediatr. Neurol 2016, 20, 203–211. [Google Scholar] [CrossRef]
- Peters, J.M.; Taquet, M.; Vega, C.; Jeste, S.S.; Fernandez, I.S.; Tan, J.; Nelson, C.A., 3rd; Sahin, M.; Warfield, S.K. Brain functional networks in syndromic and non-syndromic autism: A graph theoretical study of EEG connectivity. BMC Med. 2013, 11, 54. [Google Scholar] [CrossRef]
- Bryda, E.C. The Mighty Mouse: The impact of rodents on advances in biomedical research. Mo. Med. 2013, 110, 207–211. [Google Scholar]
- Thomson, H.H. A Case of Hystero-Epilepsy Simulating “Status Epilepticus”. Br. Med. J. 1897, 2, 464. [Google Scholar] [CrossRef]
- Cherian, A.; Thomas, S.V. Status epilepticus. Ann. Indian Acad. Neurol. 2009, 12, 140–153. [Google Scholar] [CrossRef]
- Löscher, W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 2011, 20, 359–368. [Google Scholar] [CrossRef]
- Lévesque, M.; Avoli, M.; Bernard, C. Animal models of temporal lobe epilepsy following systemic chemoconvulsant administration. J. Neurosci. Methods 2016, 260, 45–52. [Google Scholar] [CrossRef]
- Grone, B.P.; Baraban, S.C. Animal models in epilepsy research: Legacies and new directions. Nat. Neurosci. 2015, 18, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Bertoglio, D.; Amhaoul, H.; Van Eetveldt, A.; Houbrechts, R.; Van De Vijver, S.; Ali, I.; Dedeurwaerdere, S. Kainic Acid-Induced Post-Status Epilepticus Models of Temporal Lobe Epilepsy with Diverging Seizure Phenotype and Neuropathology. Front. Neurol. 2017, 8, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado Mde, A.; Braga, G.K.; Oliveira, J.A.; Del Vecchio, F.; Garcia-Cairasco, N. Behavioral, morphologic, and electroencephalographic evaluation of seizures induced by intrahippocampal microinjection of pilocarpine. Epilepsia 2002, 43 (Suppl. S5), 37–39. [Google Scholar] [CrossRef] [PubMed]
- Turski, W.A.; Cavalheiro, E.A.; Schwarz, M.; Czuczwar, S.J.; Kleinrok, Z.; Turski, L. Limbic seizures produced by pilocarpine in rats: Behavioural, electroencephalographic and neuropathological study. Behav. Brain Res. 1983, 9, 315–335. [Google Scholar] [CrossRef]
- Leite, J.P.; Nakamura, E.M.; Lemos, T.; Masur, J.; Cavalheiro, E.A. Learning impairment in chronic epileptic rats following pilocarpine-induced status epilepticus. Braz. J. Med. Biol. Res. 1990, 23, 681–683. [Google Scholar]
- Honchar, M.P.; Olney, J.W.; Sherman, W.R. Systemic cholinergic agents induce seizures and brain damage in lithium-treated rats. Science 1983, 220, 323–325. [Google Scholar] [CrossRef]
- Cavalheiro, E.A.; Leite, J.P.; Bortolotto, Z.A.; Turski, W.A.; Ikonomidou, C.; Turski, L. Long-term effects of pilocarpine in rats: Structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia 1991, 32, 778–782. [Google Scholar] [CrossRef]
- Glien, M.; Brandt, C.; Potschka, H.; Voigt, H.; Ebert, U.; Löscher, W. Repeated low-dose treatment of rats with pilocarpine: Low mortality but high proportion of rats developing epilepsy. Epilepsy Res. 2001, 46, 111–119. [Google Scholar] [CrossRef]
- Turski, W.A.; Cavalheiro, E.A.; Bortolotto, Z.A.; Mello, L.M.; Schwarz, M.; Turski, L. Seizures produced by pilocarpine in mice: A behavioral, electroencephalographic and morphological analysis. Brain Res. 1984, 321, 237–253. [Google Scholar] [CrossRef]
- Avoli, M.; de Curtis, M.; Gnatkovsky, V.; Gotman, J.; Köhling, R.; Lévesque, M.; Manseau, F.; Shiri, Z.; Williams, S. Specific imbalance of excitatory/inhibitory signaling establishes seizure onset pattern in temporal lobe epilepsy. J. Neurophysiol. 2016, 115, 3229–3237. [Google Scholar] [CrossRef]
- Lévesque, M.; Salami, P.; Gotman, J.; Avoli, M. Two seizure-onset types reveal specific patterns of high-frequency oscillations in a model of temporal lobe epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 13264–13272. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, M.; Biagini, G.; de Curtis, M.; Gnatkovsky, V.; Pitsch, J.; Wang, S.; Avoli, M. The pilocarpine model of mesial temporal lobe epilepsy: Over one decade later, with more rodent species and new investigative approaches. Neurosci. Biobehav. Rev. 2021, 130, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Velasco, A.L.; Wilson, C.L.; Babb, T.L.; Engel, J., Jr. Functional and anatomic correlates of two frequently observed temporal lobe seizure-onset patterns. Neural Plast. 2000, 7, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memarian, N.; Madsen, S.K.; Macey, P.M.; Fried, I.; Engel, J., Jr.; Thompson, P.M.; Staba, R.J. Ictal depth EEG and MRI structural evidence for two different epileptogenic networks in mesial temporal lobe epilepsy. PLoS ONE 2015, 10, e0123588. [Google Scholar] [CrossRef]
- Behr, C.; Lévesque, M.; Stroh, T.; Avoli, M. Time-dependent evolution of seizures in a model of mesial temporal lobe epilepsy. Neurobiol. Dis. 2017, 106, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Nadler, J.V.; Perry, B.W.; Gentry, C.; Cotman, C.W. Degeneration of hippocampal CA3 pyramidal cells induced by intraventricular kainic acid. J. Comp. Neurol. 1980, 192, 333–359. [Google Scholar] [CrossRef]
- Cavalheiro, E.A.; Riche, D.A.; Le Gal La Salle, G. Long-term effects of intrahippocampal kainic acid injection in rats: A method for inducing spontaneous recurrent seizures. Electroencephalogr. Clin. Neurophysiol. 1982, 53, 581–589. [Google Scholar] [CrossRef]
- Hellier, J.L.; Patrylo, P.R.; Buckmaster, P.S.; Dudek, F.E. Recurrent spontaneous motor seizures after repeated low-dose systemic treatment with kainate: Assessment of a rat model of temporal lobe epilepsy. Epilepsy Res. 1998, 31, 73–84. [Google Scholar] [CrossRef]
- Lothman, E.W.; Bertram, E.H., 3rd; Stringer, J.L. Functional anatomy of hippocampal seizures. Prog. Neurobiol. 1991, 37, 1–82. [Google Scholar] [CrossRef]
- Khalilov, I.; Dzhala, V.; Medina, I.; Leinekugel, X.; Melyan, Z.; Lamsa, K.; Khazipov, R.; Ben-Ari, Y. Maturation of kainate-induced epileptiform activities in interconnected intact neonatal limbic structures in vitro. Eur. J. Neurosci. 1999, 11, 3468–3480. [Google Scholar] [CrossRef]
- Vincent, P.; Mulle, C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience 2009, 158, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.M.; Polygalov, D.; Wintzer, M.E.; Chiang, M.C.; McHugh, T.J. CA3 Synaptic Silencing Attenuates Kainic Acid-Induced Seizures and Hippocampal Network Oscillations. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.P.; Garcia-Cairasco, N.; Cavalheiro, E.A. New insights from the use of pilocarpine and kainate models. Epilepsy Res. 2002, 50, 93–103. [Google Scholar] [CrossRef]
- Kandratavicius, L.; Balista, P.A.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, E.H.; Garcia-Cairasco, N.; Bueno-Junior, L.S.; Leite, J.P. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Mishra, A.; Goel, R.K. PTZ kindling model for epileptogenesis, refractory epilepsy, and associated comorbidities: Relevance and reliability. Metab. Brain Dis. 2021, 36, 1573–1590. [Google Scholar] [CrossRef]
- Weller, A.; Mostofsky, D.I. Ontogenetic development and pentylenetetrazol seizure thresholds in rats. Physiol. Behav. 1995, 57, 629–631. [Google Scholar] [CrossRef]
- Holmes, G.L.; Sarkisian, M.; Ben-Ari, Y.; Chevassus-Au-Louis, N. Mossy fiber sprouting after recurrent seizures during early development in rats. J. Comp. Neurol. 1999, 404, 537–553. [Google Scholar] [CrossRef]
- Pitkänen, A.; Buckmaster, P.; Galanopoulou, A.S.; Moshé, S.L. Models of Seizures and Epilepsy; Academic Press: Cambridge, MA, USA, 2005; pp. 1–14. [Google Scholar]
- Mason, C.R.; Cooper, R.M. A permanent change in convulsive threshold in normal and brain-damaged rats with repeated small doses of pentylenetetrazol. Epilepsia 1972, 13, 663–674. [Google Scholar] [CrossRef]
- Karler, R.; Murphy, V.; Calder, L.D.; Turkanis, S.A. Pentylenetetrazol kindling in mice. Neuropharmacology 1989, 28, 775–780. [Google Scholar] [CrossRef]
- Bertram, E. The relevance of kindling for human epilepsy. Epilepsia 2007, 48 (Suppl. S2), 65–74. [Google Scholar] [CrossRef]
- Welzel, L.; Schidlitzki, A.; Twele, F.; Anjum, M.; Löscher, W. A face-to-face comparison of the intra-amygdala and intrahippocampal kainate mouse models of mesial temporal lobe epilepsy and their utility for testing novel therapies. Epilepsia 2020, 61, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Poh, M.Z.; Loddenkemper, T.; Reinsberger, C.; Swenson, N.C.; Goyal, S.; Madsen, J.R.; Picard, R.W. Autonomic changes with seizures correlate with postictal EEG suppression. Neurology 2012, 78, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Van Erum, J.; Van Dam, D.; De Deyn, P.P. PTZ-induced seizures in mice require a revised Racine scale. Epilepsy Behav. 2019, 95, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Reddy, D.S.; Kuruba, R. Experimental models of status epilepticus and neuronal injury for evaluation of therapeutic interventions. Int. J. Mol. Sci. 2013, 14, 18284–18318. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, D.H.; Simon, R.P.; Sharp, F.R. The pattern of 72-kDa heat shock protein-like immunoreactivity in the rat brain following flurothyl-induced status epilepticus. Brain Res. 1990, 531, 173–182. [Google Scholar] [CrossRef]
- Holmes, G.L.; Gairsa, J.L.; Chevassus-Au-Louis, N.; Ben-Ari, Y. Consequences of neonatal seizures in the rat: Morphological and behavioral effects. Ann. Neurol. 1998, 44, 845–857. [Google Scholar] [CrossRef]
- Huang, L.; Cilio, M.R.; Silveira, D.C.; McCabe, B.K.; Sogawa, Y.; Stafstrom, C.E.; Holmes, G.L. Long-term effects of neonatal seizures: A behavioral, electrophysiological, and histological study. Brain Res. Dev. Brain Res. 1999, 118, 99–107. [Google Scholar] [CrossRef]
- Cooper, K.; Fink, M. The chemical induction of seizures in psychiatric therapy: Were flurothyl (indoklon) and pentylenetetrazol (metrazol) abandoned prematurely? J. Clin. Psychopharmacol. 2014, 34, 602–607. [Google Scholar] [CrossRef]
- Kadiyala, S.B.; Yannix, J.Q.; Nalwalk, J.W.; Papandrea, D.; Beyer, B.S.; Herron, B.J.; Ferland, R.J. Eight Flurothyl-Induced Generalized Seizures Lead to the Rapid Evolution of Spontaneous Seizures in Mice: A Model of Epileptogenesis with Seizure Remission. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 7485–7496. [Google Scholar] [CrossRef]
- Löscher, W.; Schmidt, D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res 1988, 2, 145–181. [Google Scholar] [CrossRef]
- Browning, R.A.; Nelson, D.K. Variation in threshold and pattern of electroshock-induced seizures in rats depending on site of stimulation. Life Sci. 1985, 37, 2205–2211. [Google Scholar] [CrossRef]
- Velísek, L.; Mares, P. Hippocampal afterdischarges in rats. I. Effects of antiepileptics. Physiol. Res. 2004, 53, 453–461. [Google Scholar] [PubMed]
- Sutula, T.P. Mechanisms of epilepsy progression: Current theories and perspectives from neuroplasticity in adulthood and development. Epilepsy Res. 2004, 60, 161–171. [Google Scholar] [CrossRef] [PubMed]
- He, X.P.; Wen, R.; McNamara, J.O. Impairment of kindling development in phospholipase Cγ1 heterozygous mice. Epilepsia 2014, 55, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Goddard, G.V.; McIntyre, D.C.; Leech, C.K. A permanent change in brain function resulting from daily electrical stimulation. Exp. Neurol. 1969, 25, 295–330. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, L. Clustering of Spontaneous Recurrent Seizures in a Mouse Model of Extended Hippocampal Kindling. Front. Neurol. 2021, 12, 738986. [Google Scholar] [CrossRef]
- Lowenstein, D.H. Epilepsy after head injury: An overview. Epilepsia 2009, 50 (Suppl. S2), 4–9. [Google Scholar] [CrossRef]
- Pitkänen, A.; Immonen, R. Epilepsy related to traumatic brain injury. Neurotherapeutics 2014, 11, 286–296. [Google Scholar] [CrossRef]
- Keith, K.A.; Huang, J.H. Animal Models of Post-Traumatic Epilepsy. Diagnostics 2019, 10, 4. [Google Scholar] [CrossRef]
- Mukherjee, S.; Zeitouni, S.; Cavarsan, C.F.; Shapiro, L.A. Increased seizure susceptibility in mice 30 days after fluid percussion injury. Front. Neurol. 2013, 4, 28. [Google Scholar] [CrossRef]
- D’Ambrosio, R.; Fairbanks, J.P.; Fender, J.S.; Born, D.E.; Doyle, D.L.; Miller, J.W. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain 2004, 127, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.; Fujioka, W.; Lifshitz, J.; Crockett, D.P.; Thakker-Varia, S. Lateral fluid percussion: Model of traumatic brain injury in mice. J. Vis. Exp. 2011, 54, e3063. [Google Scholar] [CrossRef] [PubMed]
- Frey, L.C.; Hellier, J.; Unkart, C.; Lepkin, A.; Howard, A.; Hasebroock, K.; Serkova, N.; Liang, L.; Patel, M.; Soltesz, I.; et al. A novel apparatus for lateral fluid percussion injury in the rat. J. Neurosci. Methods 2009, 177, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Dixon, C.E.; Clifton, G.L.; Lighthall, J.W.; Yaghmai, A.A.; Hayes, R.L. A controlled cortical impact model of traumatic brain injury in the rat. J. Neurosci. Methods 1991, 39, 253–262. [Google Scholar] [CrossRef]
- Reddy, D.S.; Golub, V.M.; Ramakrishnan, S.; Abeygunaratne, H.; Dowell, S.; Wu, X. A Comprehensive and Advanced Mouse Model of Post-Traumatic Epilepsy with Robust Spontaneous Recurrent Seizures. Curr. Protoc. 2022, 2, e447. [Google Scholar] [CrossRef] [PubMed]
- Bakalkin, G.; Nosova, O.; Sarkisyan, D.; Hallberg, M.; Zhang, M.; Schouenborg, J.; Marklund, N.; Watanabe, H. Unilateral traumatic brain injury of the left and right hemisphere produces the left hindlimb response in rats. Exp. Brain Res. 2021, 239, 2221–2232. [Google Scholar] [CrossRef]
- Singh, T.; Joshi, S.; Williamson, J.M.; Kapur, J. Neocortical injury-induced status epilepticus. Epilepsia 2020, 61, 2811–2824. [Google Scholar] [CrossRef]
- Singh, T.; Batabyal, T.; Kapur, J. Neuronal circuits sustaining neocortical-injury-induced status epilepticus. Neurobiol. Dis. 2022, 165, 105633. [Google Scholar] [CrossRef]
- Berkovic, S.F.; Mulley, J.C.; Scheffer, I.E.; Petrou, S. Human epilepsies: Interaction of genetic and acquired factors. Trends Neurosci. 2006, 29, 391–397. [Google Scholar] [CrossRef]
- Cela, E.; McFarlan, A.R.; Chung, A.J.; Wang, T.; Chierzi, S.; Murai, K.K.; Sjöström, P.J. An Optogenetic Kindling Model of Neocortical Epilepsy. Sci. Rep. 2019, 9, 5236. [Google Scholar] [CrossRef]
- Krushinsky, L.; Molodkina, L.; Fless, D.; Dobrokhotova, L.; Steshenko, A.; Semiokhina, A.; Zorina, Z.; Romanova, L. The functional state of the brain during sonic stimulation. In Physiological Effects of Noise; Springer: Berlin/Heidelberg, Germany, 1970; pp. 159–183. [Google Scholar]
- Garcia-Cairasco, N. A critical review on the participation of inferior colliculus in acoustic-motor and acoustic-limbic networks involved in the expression of acute and kindled audiogenic seizures. Hear. Res. 2002, 168, 208–222. [Google Scholar] [CrossRef]
- Garcia-Cairasco, N.; Terra, V.C.; Doretto, M.C. Midbrain substrates of audiogenic seizures in rats. Behav. Brain Res. 1993, 58, 57–67. [Google Scholar] [CrossRef]
- Löscher, W. Genetic animal models of epilepsy. In Genetically Defined Animal Models of Neurobehavioral Dysfunctions; Springer: Berlin/Heidelberg, Germany, 1992; pp. 111–135. [Google Scholar]
- Roberts, R.C.; Ribak, C.E. Anatomical changes of the GABAergic system in the inferior colliculus of the genetically epilepsy-prone rat. Life Sci. 1986, 39, 789–798. [Google Scholar] [CrossRef]
- Jobe, P.C.; Picchioni, A.L.; Chin, L. Role of brain norepinephrine in audiogenic seizure in the rat. J. Pharmacol. Exp. Ther. 1973, 184, 1–10. [Google Scholar]
- Zhao, D.Y.; Wu, X.R.; Pei, Y.Q.; Zuo, Q.H. Long-term effects of febrile convulsion on seizure susceptibility in P77PMC rat--resistant to acoustic stimuli but susceptible to kainate-induced seizures. Exp. Neurol. 1985, 88, 688–695. [Google Scholar] [CrossRef]
- Van Luijtelaar, E.L.; Coenen, A.M. Two types of electrocortical paroxysms in an inbred strain of rats. Neurosci. Lett. 1986, 70, 393–397. [Google Scholar] [CrossRef]
- Marescaux, C.; Vergnes, M.; Kiesmann, M.; Depaulis, A.; Micheletti, G.; Warter, J. Kindling of audiogenic seizures in Wistar rats: An EEG study. Exp. Neurol. 1987, 97, 160–168. [Google Scholar] [CrossRef]
- Doretto, M.; Fonseca, C.; Lobo, R.; Terra, V.; Oliveira, J.; Garcia-Cairasco, N. Quantitative study of the response to genetic selection of the Wistar audiogenic rat strain (WAR). Behav. Genet. 2003, 33, 33–42. [Google Scholar] [CrossRef]
- Consroe, P.; Picchioni, A.; Chin, L. Audiogenic seizure susceptible rats. Fed. Proc. 1979, 38, 2411–2416. [Google Scholar]
- Ross, K.C.; Coleman, J.R. Developmental and genetic audiogenic seizure models: Behavior and biological substrates. Neurosci. Biobehav. Rev. 2000, 24, 639–653. [Google Scholar] [CrossRef]
- Garcia-Cairasco, N.; Wakamatsu, H.; Oliveira, J.A.C.; Gomes, E.L.T.; Del Bel, E.A.; Mello, L.E.A.M. Neuroethological and morphological (Neo-Timm staining) correlates of limbic recruitment during the development of audiogenic kindling in seizure susceptible Wistar rats. Epilepsy Res. 1996, 26, 177–192. [Google Scholar] [CrossRef]
- Naritoku, D.K.; Mecozzi, L.B.; Aiello, M.T.; Faingold, C.L. Repetition of audiogenic seizures in genetically epilepsy-prone rats induces cortical epileptiform activity and additional seizure behaviors. Exp. Neurol. 1992, 115, 317–324. [Google Scholar] [CrossRef]
- Akman, O.; Demiralp, T.; Ates, N.; Onat, F.Y. Electroencephalographic differences between WAG/Rij and GAERS rat models of absence epilepsy. Epilepsy Res. 2010, 89, 185–193. [Google Scholar] [CrossRef]
- Micheletti, G.; Vergnes, M.; Marescaux, C.; Reis, J.; Depaulis, A.; Rumbach, L.; Warter, J.M. Antiepileptic drug evaluation in a new animal model: Spontaneous petit mal epilepsy in the rat. Arzneim. Forsch. 1985, 35, 483–485. [Google Scholar]
- Vergnes, M.; Marescaux, C.; Micheletti, G.; Reis, J.; Depaulis, A.; Rumbach, L.; Warter, J.M. Spontaneous paroxysmal electroclinical patterns in rat: A model of generalized non-convulsive epilepsy. Neurosci. Lett. 1982, 33, 97–101. [Google Scholar] [CrossRef]
- Fletcher, C.F.; Lutz, C.M.; O’Sullivan, T.N.; Shaughnessy, J.D., Jr.; Hawkes, R.; Frankel, W.N.; Copeland, N.G.; Jenkins, N.A. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell 1996, 87, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Felix, R. Insights from mouse models of absence epilepsy into Ca2+ channel physiology and disease etiology. Cell. Mol. Neurobiol. 2002, 22, 103–120. [Google Scholar] [CrossRef]
- Green, M.C.; Sidman, R.L. Tottering--a neuromusclar mutation in the mouse. And its linkage with oligosyndacylism. J. Hered. 1962, 53, 233–237. [Google Scholar] [CrossRef]
- Doyle, J.; Ren, X.; Lennon, G. Mutations in the Cacnl1a4 calcium channel gene are associated with seizures, cerebellar degeneration, and ataxia in tottering and leaner mutant mice. Mamm. Genome 1997, 8, 113–120. [Google Scholar] [CrossRef]
- Vicini, S.; Ferguson, C.; Prybylowski, K.; Kralic, J.; Morrow, A.L.; Homanics, G.E. GABA(A) receptor alpha1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 3009–3016. [Google Scholar] [CrossRef]
- Mulcahey, P.J.; Tang, S.; Takano, H.; White, A.; Davila Portillo, D.R.; Kane, O.M.; Marsh, E.D.; Zhou, Z.; Coulter, D.A. Aged heterozygous Cdkl5 mutant mice exhibit spontaneous epileptic spasms. Exp. Neurol. 2020, 332, 113388. [Google Scholar] [CrossRef]
- Chen, C.; Westenbroek, R.E.; Xu, X.; Edwards, C.A.; Sorenson, D.R.; Chen, Y.; McEwen, D.P.; O’Malley, H.A.; Bharucha, V.; Meadows, L.S.; et al. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 4030–4042. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, H.A.; Hull, J.M.; Clawson, B.C.; Chen, C.; Owens-Fiestan, G.; Jameson, M.B.; Aton, S.J.; Parent, J.M.; Isom, L.L. Scn1b deletion in adult mice results in seizures and SUDEP. Ann. Clin. Transl. Neurol. 2019, 6, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Nabbout, R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia 2019, 60 (Suppl. S3), S17–S24. [Google Scholar] [CrossRef]
- Escayg, A.; MacDonald, B.T.; Meisler, M.H.; Baulac, S.; Huberfeld, G.; An-Gourfinkel, I.; Brice, A.; LeGuern, E.; Moulard, B.; Chaigne, D.; et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 2000, 24, 343–345. [Google Scholar] [CrossRef]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; de la Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, L.A.; Escayg, A.; Kearney, J.A.; Trudeau, M.; MacDonald, B.T.; Mori, M.; Reichert, J.; Buxbaum, J.D.; Meisler, M.H. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 2003, 8, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef]
- Oakley, J.C.; Kalume, F.; Yu, F.H.; Scheuer, T.; Catterall, W.A. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. USA 2009, 106, 3994–3999. [Google Scholar] [CrossRef]
- Kalume, F.; Yu, F.H.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: Implications for ataxia in severe myoclonic epilepsy in infancy. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 11065–11074. [Google Scholar] [CrossRef]
- Mistry, A.M.; Thompson, C.H.; Miller, A.R.; Vanoye, C.G.; George, A.L., Jr.; Kearney, J.A. Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol. Dis. 2014, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fadila, S.; Quinn, S.; Turchetti Maia, A.; Yakubovich, D.; Ovadia, M.; Anderson, K.L.; Giladi, M.; Rubinstein, M. Convulsive seizures and some behavioral comorbidities are uncoupled in the Scn1a(A1783V) Dravet syndrome mouse model. Epilepsia 2020, 61, 2289–2300. [Google Scholar] [CrossRef] [PubMed]
- Mattis, J.H.; Somarowthu, A.; Goff, K.M.; Jiang, E.; Yom, J.; Sotuyo, N.P.; McGarry, L.M.; Feng, H.; Kaneko, K.; Goldberg, E.M. Corticohippocampal circuit dysfunction in a mouse model of Dravet syndrome. eLife 2022, 11, e69293. [Google Scholar] [CrossRef]
- Tran, C.H.; Vaiana, M.; Nakuci, J.; Somarowthu, A.; Goff, K.M.; Goldstein, N.; Murthy, P.; Muldoon, S.F.; Goldberg, E.M. Interneuron Desynchronization Precedes Seizures in a Mouse Model of Dravet Syndrome. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 2764–2775. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Zhu, B.; Xie, Y.; Zeng, L.; Pham, A.T.; Neumann, J.C.; Safrina, O.; Benavides, D.R.; MacGregor, G.R.; Schutte, S.S.; et al. Interneuron Dysfunction in a New Mouse Model of SCN1A GEFS. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Ogiwara, I.; Miyamoto, H.; Morita, N.; Atapour, N.; Mazaki, E.; Inoue, I.; Takeuchi, T.; Itohara, S.; Yanagawa, Y.; Obata, K.; et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 5903–5914. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.S.; Stella, N.; Catterall, W.A.; Westenbroek, R.E. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 11229–11234. [Google Scholar] [CrossRef]
- Gamache, T.R.; Araki, Y.; Huganir, R.L. Twenty Years of SynGAP Research: From Synapses to Cognition. J. Neurosci. 2020, 40, 1596–1605. [Google Scholar] [CrossRef]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Clement, J.P.; Aceti, M.; Creson, T.K.; Ozkan, E.D.; Shi, Y.; Reish, N.J.; Almonte, A.G.; Miller, B.H.; Wiltgen, B.J.; Miller, C.A.; et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 2012, 151, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, E.D.; Creson, T.K.; Kramár, E.A.; Rojas, C.; Seese, R.R.; Babyan, A.H.; Shi, Y.; Lucero, R.; Xu, X.; Noebels, J.L.; et al. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 2014, 82, 1317–1333. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.J.; Ammanuel, S.; Kipnis, P.A.; Araki, Y.; Huganir, R.L.; Kadam, S.D. Low-Dose Perampanel Rescues Cortical Gamma Dysregulation Associated With Parvalbumin Interneuron GluA2 Upregulation in Epileptic Syngap1(+/−) Mice. Biol. Psychiatry 2020, 87, 829–842. [Google Scholar] [CrossRef]
- Striano, P.; Huppke, P. A synaptic protein defect associated with reflex seizure disorder. Neurology 2019, 92, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Creson, T.K.; Rojas, C.; Hwaun, E.; Vaissiere, T.; Kilinc, M.; Jimenez-Gomez, A.; Holder, J.L., Jr.; Tang, J.; Colgin, L.L.; Miller, C.A.; et al. Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. eLife 2019, 8, e46752. [Google Scholar] [CrossRef]
- Kilinc, M.; Creson, T.; Rojas, C.; Aceti, M.; Ellegood, J.; Vaissiere, T.; Lerch, J.P.; Rumbaugh, G. Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders. Mol. Cell. Neurosci. 2018, 91, 140–150. [Google Scholar] [CrossRef]
- Kooy, R.F. Of mice and the fragile X syndrome. Trends Genet. 2003, 19, 148–154. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Kaufmann, W.E.; Ono, M.Y.; Tartaglia, N.; Lachiewicz, A.; Kronk, R.; Delahunty, C.; Hessl, D.; Visootsak, J.; et al. Advances in the treatment of fragile X syndrome. Pediatrics 2009, 123, 378–390. [Google Scholar] [CrossRef]
- Chen, L.; Toth, M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 2001, 103, 1043–1050. [Google Scholar] [CrossRef]
- Musumeci, S.A.; Bosco, P.; Calabrese, G.; Bakker, C.; De Sarro, G.B.; Elia, M.; Ferri, R.; Oostra, B.A. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 2000, 41, 19–23. [Google Scholar] [CrossRef]
- The Dutch-Belgian Fragile X Consorthium; Bakker, C.E.; Verheij, C.; Willemsen, R.; van der Helm, R.; Oerlemans, F.; Vermey, M.; Bygrave, A.; Hoogeveen, A.; Oostra, B.A.; et al. Fmr1 knockout mice: A model to study fragile X mental retardation. Cell 1994, 78, 23–33. [Google Scholar]
- Jessica, L.A.; Tanishka, S.S.; Omkar, B.; Clinton, E.C. Spontaneous seizures in adult Fmr1 knockout mice: FVB.129P2-Pde6b+ Tyrc-ch Fmr1tm1Cgr/J. Epilepsy Res. 2022, 182, 106891. [Google Scholar]
- Sinclair, D.; Featherstone, R.; Naschek, M.; Nam, J.; Du, A.; Wright, S.; Pance, K.; Melnychenko, O.; Weger, R.; Akuzawa, S.; et al. GABA-B Agonist Baclofen Normalizes Auditory-Evoked Neural Oscillations and Behavioral Deficits in the Fmr1 Knockout Mouse Model of Fragile X Syndrome. eNeuro 2017, 4. [Google Scholar] [CrossRef]
- Paluszkiewicz, S.M.; Olmos-Serrano, J.L.; Corbin, J.G.; Huntsman, M.M. Impaired inhibitory control of cortical synchronization in fragile X syndrome. J. Neurophysiol. 2011, 106, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.T.; Anstey, J.E.; Golshani, P.; Portera-Cailliau, C. Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci. 2013, 16, 903–909. [Google Scholar] [CrossRef]
- Yan, Q.J.; Rammal, M.; Tranfaglia, M.; Bauchwitz, R.P. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 2005, 49, 1053–1066. [Google Scholar] [CrossRef]
- Nimchinsky, E.A.; Oberlander, A.M.; Svoboda, K. Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 5139–5146. [Google Scholar] [CrossRef]
- Rais, M.; Binder, D.K.; Razak, K.A.; Ethell, I.M. Sensory Processing Phenotypes in Fragile X Syndrome. ASN Neuro 2018, 10, 1759091418801092. [Google Scholar] [CrossRef] [Green Version]
- Roussignol, G.; Ango, F.; Romorini, S.; Tu, J.C.; Sala, C.; Worley, P.F.; Bockaert, J.; Fagni, L. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 3560–3570. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [PubMed]
- Dhamne, S.C.; Silverman, J.L.; Super, C.E.; Lammers, S.H.T.; Hameed, M.Q.; Modi, M.E.; Copping, N.A.; Pride, M.C.; Smith, D.G.; Rotenberg, A.; et al. Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism 2017, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.E.; Yoo, T.; Lee, S.; Lee, J.; Kim, D.; Han, H.M.; Bae, Y.C.; Kim, E. Shank3 Mice Carrying the Human Q321R Mutation Display Enhanced Self-Grooming, Abnormal Electroencephalogram Patterns, and Suppressed Neuronal Excitability and Seizure Susceptibility. Front. Mol. Neurosci. 2019, 12, 155. [Google Scholar] [CrossRef]
- Han, K.; Holder, J.L., Jr.; Schaaf, C.P.; Lu, H.; Chen, H.; Kang, H.; Tang, J.; Wu, Z.; Hao, S.; Cheung, S.W.; et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 2013, 503, 72–77. [Google Scholar] [CrossRef]
- McCormick, D.A.; Contreras, D. On the cellular and network bases of epileptic seizures. Annu. Rev. Physiol. 2001, 63, 815–846. [Google Scholar] [CrossRef]
- Jin, C.; Zhang, Y.; Kim, S.; Kim, Y.; Lee, Y.; Han, K. Spontaneous seizure and partial lethality of juvenile Shank3-overexpressing mice in C57BL/6 J background. Mol. Brain 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S.; Rogawski, M.A. Molecular targets for antiepileptic drug development. Neurotherapeutics 2007, 4, 18–61. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Cerri, C.; Nencetti, S.; Orlandini, E. Carbonic Anhydrase Inhibitors and Epilepsy: State of the Art and Future Perspectives. Molecules 2021, 26, 6380. [Google Scholar] [CrossRef]
- Riva, A.; Golda, A.; Balagura, G.; Amadori, E.; Vari, M.S.; Piccolo, G.; Iacomino, M.; Lattanzi, S.; Salpietro, V.; Minetti, C. New trends and most promising therapeutic strategies for epilepsy treatment. Front. Neurol. 2021, 2150. [Google Scholar] [CrossRef]
- Fattorusso, A.; Matricardi, S.; Mencaroni, E.; Dell’Isola, G.B.; Di Cara, G.; Striano, P.; Verrotti, A. The Pharmacoresistant Epilepsy: An overview on existant and new emerging therapies. Front. Neurol. 2021, 12, 1030. [Google Scholar] [CrossRef]
- Mukhtar, I. Inflammatory and immune mechanisms underlying epileptogenesis and epilepsy: From pathogenesis to treatment target. Seizure 2020, 82, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Vitaliti, G.; Pavone, P.; Marino, S.; Saporito, M.A.N.; Corsello, G.; Falsaperla, R. Molecular mechanism involved in the pathogenesis of early-onset epileptic encephalopathy. Front. Mol. Neurosci. 2019, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Brueggeman, L.; Sturgeon, M.L.; Martin, R.M.; Grossbach, A.J.; Nagahama, Y.; Zhang, A.; Howard III, M.A.; Kawasaki, H.; Wu, S.; Cornell, R.A. Drug repositioning in epilepsy reveals novel antiseizure candidates. Ann. Clin. Transl. Neurol. 2019, 6, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, P.R.; Amorim, H.A.; Scorza, C.A.; Arida, R.M.; Cavalheiro, E.A.; Scorza, F.A. Animal study results suggest that an antifungal drug works against neuronal loss in epilepsy. Epilepsy Behav. 2012, 23, 174–175. [Google Scholar] [CrossRef] [PubMed]
- Marshall, G.F.; Gonzalez-Sulser, A.; Abbott, C.M. Modelling epilepsy in the mouse: Challenges and solutions. Dis. Models Mech. 2021, 14, dmm047449. [Google Scholar] [CrossRef]
- Galindo-Mendez, B.; Mayor, L.C.; Velandia-Hurtado, F.; Calderon-Ospina, C. Failure of antiepileptic drugs in controlling seizures in epilepsy: What do we do next? Epilepsy Behav. Case Rep. 2015, 4, 6–8. [Google Scholar] [CrossRef]
- Bonnett, L.J.; Smith, C.T.; Donegan, S.; Marson, A.G. Treatment outcome after failure of a first antiepileptic drug. Neurology 2014, 83, 552–560. [Google Scholar] [CrossRef]
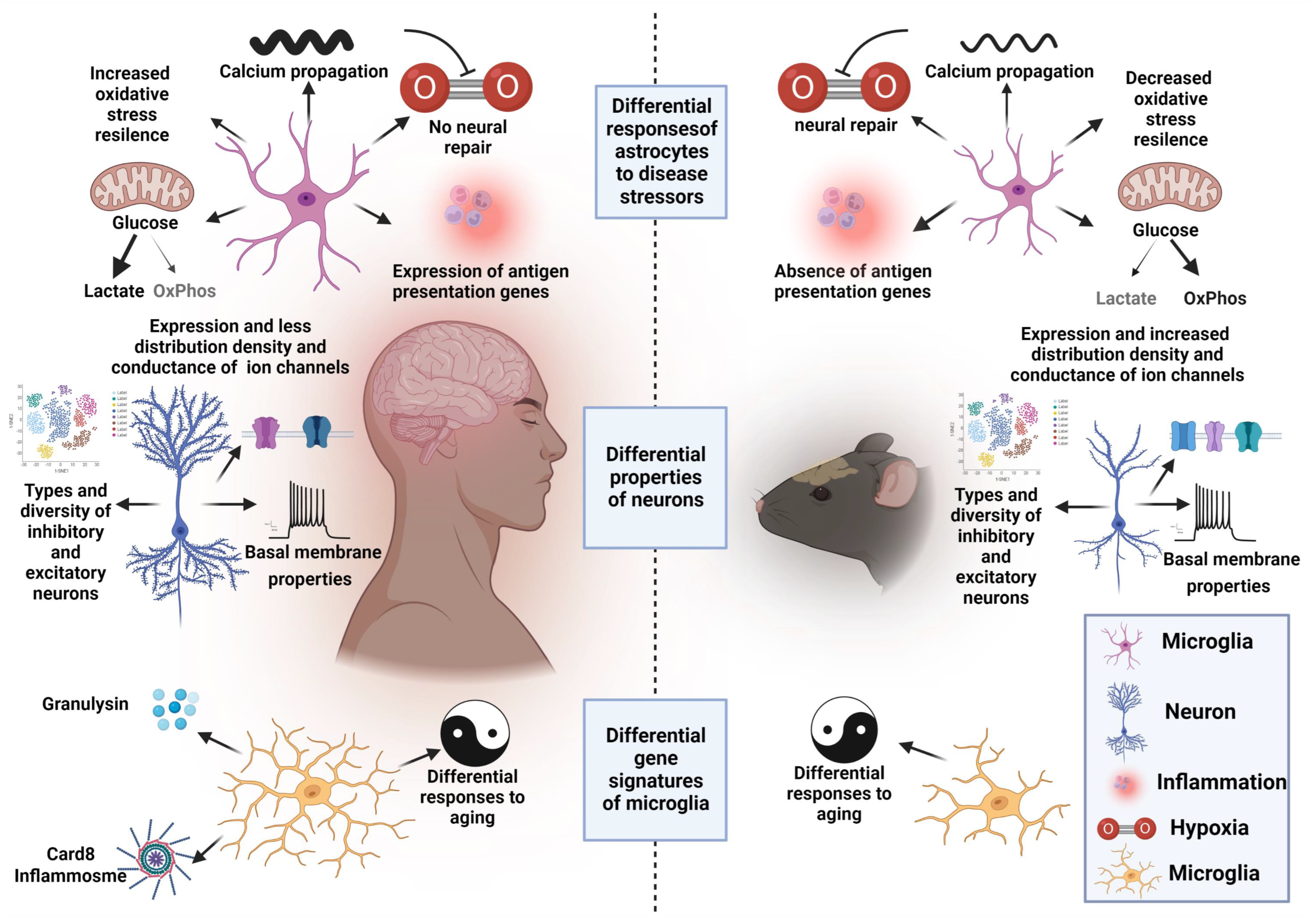
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Li, J.; Pan, L.; Pembroke, W.G.; Rexach, J.E.; Godoy, M.I.; Condro, M.C.; Alvarado, A.G.; Harteni, M.; Chen, Y.-W.; Stiles, L. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat. Commun. 2021, 12, 3958. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Johansen, N.; Penn, O. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019, 573, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Eyal, G.; Verhoog, M.B.; Testa-Silva, G.; Deitcher, Y.; Lodder, J.C.; Benavides-Piccione, R.; Morales, J.; DeFelipe, J.; de Kock, C.P.; Mansvelder, H.D. Unique membrane properties and enhanced signal processing in human neocortical neurons. eLife 2016, 5, e16553. [Google Scholar] [CrossRef] [PubMed]
- Testa-Silva, G.; Verhoog, M.B.; Linaro, D.; De Kock, C.P.; Baayen, J.C.; Meredith, R.M.; De Zeeuw, C.I.; Giugliano, M.; Mansvelder, H.D. High bandwidth synaptic communication and frequency tracking in human neocortex. PLoS Biol. 2014, 12, e1002007. [Google Scholar] [CrossRef] [PubMed]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.; Möller, T. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef]
- Simkin, D.; Kiskinis, E. Modeling pediatric epilepsy through iPSC-Based technologies. Epilepsy Curr. 2018, 18, 240–245. [Google Scholar] [CrossRef]
- Niu, W.; Parent, J.M. Modeling genetic epilepsies in a dish. Dev. Dyn. 2020, 249, 56–75. [Google Scholar] [CrossRef]
- Grainger, A.I.; King, M.C.; Nagel, D.A.; Parri, H.R.; Coleman, M.D.; Hill, E.J. In vitro models for seizure-liability testing using induced pluripotent stem cells. Front. Neurosci. 2018, 590. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Estévez, V.; Hsieh, J. Human brain organoid models of developmental epilepsies. Epilepsy Curr. 2020, 20, 282–290. [Google Scholar] [CrossRef]
- Sun, A.X.; Yuan, Q.; Fukuda, M.; Yu, W.; Yan, H.; Lim, G.G.Y.; Nai, M.H.; D’agostino, G.A.; Tran, H.-D.; Itahana, Y. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 2019, 366, 1486–1492. [Google Scholar] [CrossRef]
- Samarasinghe, R.A.; Miranda, O.A.; Buth, J.E.; Mitchell, S.; Ferando, I.; Watanabe, M.; Allison, T.F.; Kurdian, A.; Fotion, N.N.; Gandal, M.J. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 2021, 24, 1488–1500. [Google Scholar] [CrossRef] [PubMed]
- Shiri, Z.; Simorgh, S.; Naderi, S.; Baharvand, H. Optogenetics in the era of cerebral organoids. Trends Biotechnol. 2019, 37, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.P.; Stice, S.L. Electrophysiological analysis of brain organoids: Current approaches and advancements. Front. Neurosci. 2021, 14, 1405. [Google Scholar] [CrossRef] [PubMed]
- Wang, H. Modeling neurological diseases with human brain organoids. Front. Synaptic Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering stem cell organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sá, R.; Van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.-J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.-S. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Fair, S.R.; Julian, D.; Hartlaub, A.M.; Pusuluri, S.T.; Malik, G.; Summerfied, T.L.; Zhao, G.; Hester, A.B.; Ackerman IV, W.E.; Hollingsworth, E.W. Electrophysiological maturation of cerebral organoids correlates with dynamic morphological and cellular development. Stem Cell Rep. 2020, 15, 855–868. [Google Scholar] [CrossRef]
- Kebir, H.; Carmant, L.; Fontaine, F.; Béland, K.; Bosoi, C.M.; Sanon, N.T.; Alvarez, J.I.; Desgent, S.; Pittet, C.L.; Hébert, D. Humanized mouse model of Rasmussen’s encephalitis supports the immune-mediated hypothesis. J. Clin. Investig. 2018, 128, 2000–2009. [Google Scholar] [CrossRef]
- Li, T.; Ren, G.; Kaplan, D.L.; Boison, D. Human mesenchymal stem cell grafts engineered to release adenosine reduce chronic seizures in a mouse model of CA3-selective epileptogenesis. Epilepsy Res. 2009, 84, 238–241. [Google Scholar] [CrossRef]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Espuny-Camacho, I.; Michelsen, K.A.; Gall, D.; Linaro, D.; Hasche, A.; Bonnefont, J.; Bali, C.; Orduz, D.; Bilheu, A.; Herpoel, A. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron 2013, 77, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H.; Björklund, A. Reconstruction of brain circuitry by neural transplants generated from pluripotent stem cells. Neurobiol. Dis. 2015, 79, 28–40. [Google Scholar] [CrossRef]
- Cunningham, M.; Cho, J.-H.; Leung, A.; Savvidis, G.; Ahn, S.; Moon, M.; Lee, P.K.; Han, J.J.; Azimi, N.; Kim, K.-S. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell 2014, 15, 559–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Genetic Mutation | Disease | Epilepsy Characteristics | Behavioral and Structural Deficits | References |
|---|---|---|---|---|
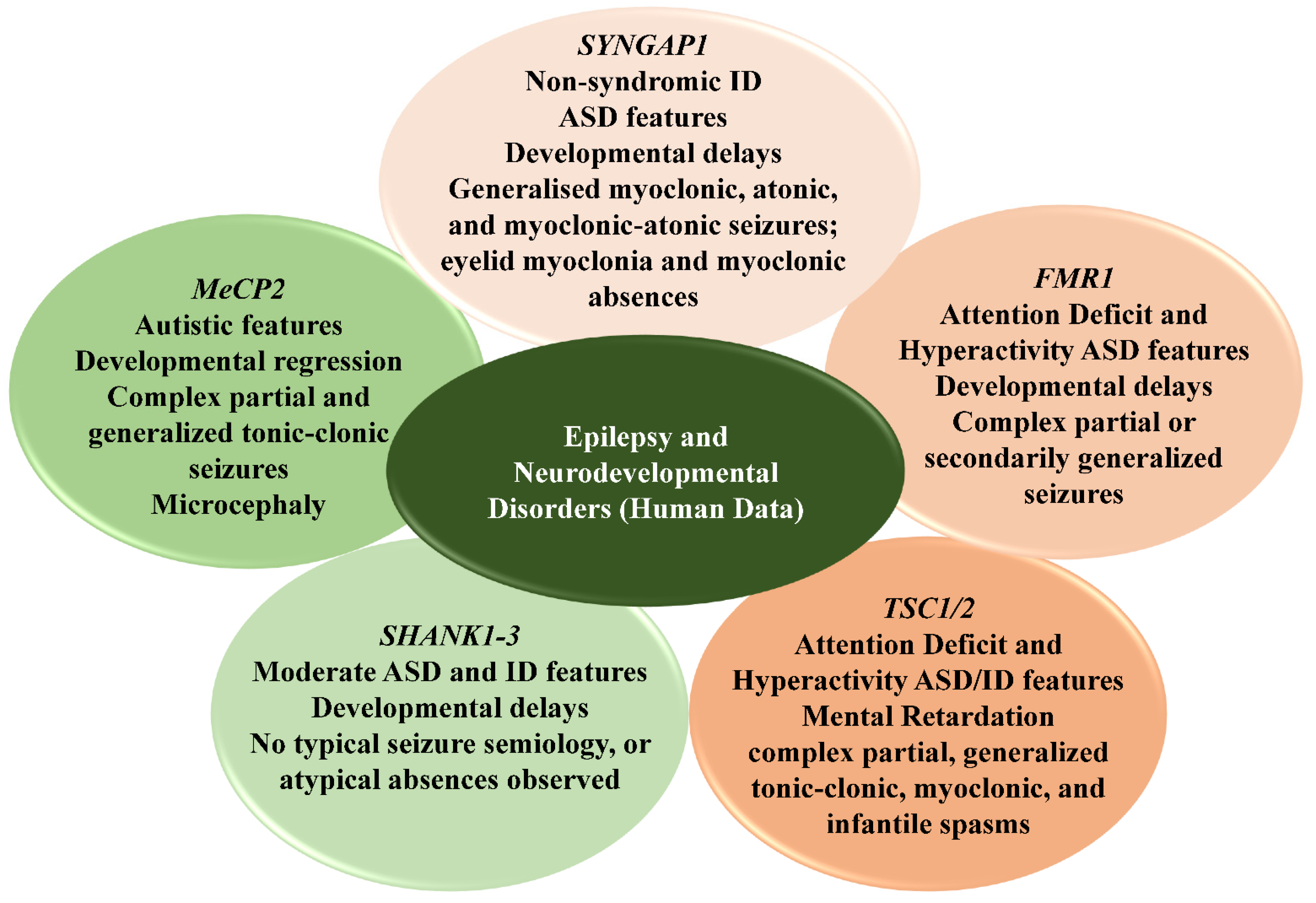
| MeCP2 | Rett syndrome (typical and atypical) | Complex partial and generalized tonic-clonic seizures. Stage I RTT has normal EEG features; Stage II RTT EEG shows loss of non-REM sleep characteristics and focal spikes or sharp waves; Stage III RTT EEG shows bilaterally synchronous bursts of pseudoperiodic delta activity and generalized rhythmic spike discharges characterizing a high seizure burden; Stage IV RTT EEG shows significant slowing of the background activity with delta rhythms, multifocal epileptiform activity in the awake state, and generalized slow spike-wave activity in sleep. | Developmental regression and delays, partial or complete loss of motor functions, gait abnormalities, abnormal sleep patterns, hand stereotypies, reduced cerebral volume and cortical grey matter mainly in frontal regions, acquired microcephaly | [59,60,61] |
| SYNGAP1 | SYNGAP1-related NSID | Psychomotor delays precede epilepsy onset, seizures are mostly generalized: myoclonic, atonic, and myoclonic-atonic seizures; atypical absences; eyelid myoclonia and myoclonic absences. Ictal EEG shows generalized spike-wave discharges coinciding with the eyelid myoclonia, followed by a spike-wave complex correlating with a myoclonic (spike) and an atonic (slow-wave) component. Focal or multifocal epileptiform discharges are often observed along with generalized spike-wave discharges. | Developmental delays, language impairments, high pain and low seizure thresholds, sleeping and eating abnormalities, nonspecific MRI findings with enlarged ventricles or subarachnoid spaces, discrete hippocampal tissue loss; astrocytosis and cerebellar Purkinje neuron losses are also seen | [62] |
| FMR1 | Fragile X Mental retardation syndrome | Common form of epilepsy in FXS resembles benign focal epilepsy with centrotemporal spikes (benign focal epilepsy of childhood (BFEC), benign Rolandic epilepsy). Centrotemporal spikes are the most common epileptiform feature on EEG; focal spikes or sharp discharges are seen in some patients; seizures can be partial complex and generalized tonic-clonic. | Attention-deficit/hyperactivity disorders, ASD features, aggression and self-injurious behaviors, anxiety, hand stereotypies, language deficits, regional variation in grey matter volume, linear increase in white matter volume, enlarged caudate nucleus, microcephaly | [63,64,65] |
| SHANK1-3 | Phelan-McDermid syndrome | Moderate ASD/ID and refractory epilepsy of the Lennox-Gastaut type; electroencephalographic abnormalities are heterogenous: from slowing or absence of the dominant occipital rhythm to focal spike and slow-wave discharges to generalized spike and slow-wave discharges; generalised tonic-clonic, myoclonic, and tonic seizures have been reported. SHANK3 duplications can cause episodes of status epilepticus. | Developmental delay, ASD, and schizophrenia, progressive loss of skills, attention-deficit/hyperactivity disorder, dysmorphisms of corpus callosum, severe white matter alterations | [66,67] |
| TSC1/2 | Tuberous sclerosis | Complex partial, generalized tonic-clonic, myoclonic, and infantile spasms characterized by multifocal EEG abnormalities | ASD/ID features, mental retardation, attention deficit, hyperactivity, aggression, anxiety, sleep disturbances, depression, altered neuronal network topology, and excitation/inhibition balance | [68] |
| S.No. | Intervention | Mechanism of Action | Type of Epilepsy | Clinical Phase | Clinicaltrials.gov |
|---|---|---|---|---|---|
| 1 | XEN1101 | Potassium channel modulator | Focal onset | 2 | NCT03796962 |
| 2 | Clobazam | Potentiation of GABAergic transmission | Refractory focal | 4 | NCT02726919 |
| 3 | EQU-001 | NA | All | 2 | NCT05063877 |
| 4 | Cenobamate | Positive allosteric modulation of GABAA ion channels | Primary generalized tonic-clonic | 3 | NCT03678753 |
| 5 | CX-998 | T-type calcium channels | Idiopathic generalized epilepsy with absence seizures | 2 | NCT03406702 |
| 6 | Brivaracetam | Synaptic vesicle 2A | Childhood absence | 3 | NCT05109234 |
| Rolandic benign | |||||
| 4 | NCT00181116 | ||||
| 7 | YKP3089 | Positive modulation of GABAA receptors and voltage-gated sodium channels | Photosensitive | 2 | NCT00616148 |
| 8 | Ganaxolone | Allosteric to GABAA receptor | Photosensitive, drug-resistant, partial onset | 2 | NCT01963208 |
| 9 | Prednisolone | Immunotherapy | Cryptogenic | 4 | NCT02695797 |
| 10 | Topiramate | Inhibits carbonic anhydrase enzyme | Childhood absence | 2 | NCT00210574 |
| 11 | BGG492 | Antagonism of AMPAR | Photosensitive | 2 | NCT00784212 |
| Partial | 2 | NCT00887861 | |||
| 12 | BM430C | Inhibition of voltage gated sodium channels | All | 3 | NCT00395694 |
| 13 | Lu AG06466 | Inhibits monoacylglycerol lipase (MGLL)-serine hydrolase | Focal | 1 | NCT05081518 |
| 14 | ABI-009 (Nab-rapamycin) | Inhibition of mTOR | Surgically refractory | 1 | NCT03646240 |
| 15 | Vorinostat | Inhibition of histone deacetylases (HDAC) | Drug-resistant | 2 | NCT03894826 |
| 16 | ACT-709478 | Inhibition of T-type Ca2+ channels | Photosensitive | 2 | NCT03239691 |
| 17 | UCB0942 | Antagonism of GBAA receptors | Drug-resistant focal | 2 | NCT02495844 |
| 18 | PF-06372865 | Agonism of GBAA receptors | Photosensitive | 2 | NCT02564029 |
| 19 | VX-765 | Inhibition of caspase 1 | Drug-resistant partial epilepsy | 2 | NCT01048255 |
| 20 | TAVT-18 (sirolimus) | Inhibition of mTOR | Pediatric drug-resistant | 1/2 | NCT04595513 |
| 21 | MGCND00EP1 | Modulation of 5HT1a receptors | Adolescent drug-resistant | 2 | NCT04406948 |
| 22 | RWJ-333369 | Neuromodulator | Complex partial, focal | 3 | NCT00433667 |
| 23 | Soticlestat | Inhibition of cholesterol 24-hydroxylase | Dravet syndrome (DS) Lennox-Gastaut syndrome (LGS) | 2 | NCT03635073 |
| 3 | NCT04940624 | ||||
| 24 | OPC-214870 | Not known | Drug-resistant | 1 | NCT04241965 |
| 25 | TAK-935 | Conversion of cholesterol to 24HC | Epileptic encephalopathies | 1/2 | NCT03166215 |
| 26 | NBI-921352 | Inhibition of Nav 1.6 | SCN8A developmental and epileptic encephalopathy syndrome | 2 | NCT04873869 |
| 27 | NBI-827104 | Triple T-type calcium channel blocker | Epileptic encephalopathy | 2 | NCT04625101 |
| 28 | LP352 | 5-HT2c receptor super agonist | Epileptic encephalopathy | 1/2 | NCT05364021 |
| 29 | GWP42003-P | Cannabidiol oral solution | Dravet syndrome | 3 | NCT02091375 |
| 30 | Ropinirole | Agonist of dopamine | Myoclonic | 2 | NCT00639119 |
| 31 | Rufinamide | Stabilizes inactivation state of voltage-gated sodium channel | Drug-resistant | 3 | NCT00334958 |
| 32 | Allopregnanolone injection | Positively modulates GABAA receptors | Post-traumatic | 2 | NCT01673828 |
| 33 | Ezogabine | Positive allosteric modulation of (K(v) 7.2–7.5) channels | KCNQ2 developmental and epileptic encephalopathy | 3 | NCT04639310 |
| 34 | Aspirin | Inhibition of mTOR | Tuberous sclerosis complex | 2 | NCT03356769 |
| 35 | Phenylbutyrate | Removal of ammonia | STXBP1 encephalopathy | 1 | NCT04937062 |
| 36 | STK-001 | Antisense oligonucleotide to SCN1A mRNA | Dravet syndrome | 2 | NCT04740476 |
| 37 | Triheptanoin | Medium-chain triglyceride | Rett syndrome | 2 | NCT02696044 |
| 38 | Carisbamate | Moderate inhibition of high-voltage-activated calcium channels | Lennox-Gastaut syndrome | 1 | NCT04062981 |
| NCT03731715 | |||||
| 39 | CVL-865 | GABAA modulation | Focal onset drug-resistant | 2 | NCT04244175 |
| 40 | Cysteamine bitartrate (RP103) | Lysosomal metabolism of cysteine | Mitochondrial diseases, including Leigh syndrome | 2 | NCT02023866 |
| 41 | Telampanel | Antagonism of AMPA receptors | Drug-resistant | 2 | NCT00057460 |
| S.No. | Intervention | Mechanism of Action | Type of Epilepsy | Clinical Phase | Clinicaltrials.gov |
|---|---|---|---|---|---|
| 1 | Pulvinar deep stimulation | Stimulation of pulvinar thalamic nucleus | Drug-resistant | NA | NCT04692701 |
| 2 | Trans auricular vagus nerve stimulation | Experience-dependent neural plasticity | Pediatric | NA | NCT02004340 |
| 3 | MRI-guided laser interstitial thermal therapy (MgLiTT) | Sinovation Laser Ablation System | Drug-resistant | NA | NCT04569071 |
| 4 | Green light exposure | Engagement of thalamocortical inhibitory circuits | Drug-resistant | NA | NCT03857074 |
| 5 | Fecal microbiota suspension | Modulation of gut–brain axis | Drug-resistant | 3 | NCT02889627 |
| 6 | Bilateral thalamic central lateral nuclei stimulation | Restoration of conscious awareness | Temporal lobe | NA | NCT04897776 |
| 7 | Cerebellar continuous θ burst stimulation (cTBS) | Inhibition of cortical and motor evoked potentials | Drug-resistant | NA | NCT05042726 |
| 8 | Transcranial deep brain stimulation | Modulation of cortical excitability | Drug-resistant | NA | NCT04325360 |
| 9 | Stereotactic laser ablation | Necrosis of epileptic foci | Temporal lobe | 3 | NCT02844465 |
| 10 | Vagus nerve stimulation | Experience-dependent neural plasticity | Drug-resistant | 1 | NCT02378792 |
| 11 | Lentiviral engineered potassium (K+) channel (EKC) | Gene therapy for hyperpolarization | Drug-resistant | 0 | NCT04601974 |
| 12 | Autologous bone marrow stem cell transplantation | Tissue repair | Temporal lobe | 1 | NCT00916266 |
| 13 | Transplantation of adipose-derived regenerative cells (ADRCs) | Tissue repair | Autoimmune drug-resistant | 1 | NCT03676569 |
| 14 | Modified Atkins diet | Metabolism | Drug-resistant | NA | NCT01311440 |
| 15 | Physical exercise program | Life-style improvement | Pediatric drug-resistant | NA | NCT05323682 |
| 16 | Focused ultrasound | Modulate neuronal firing | Drug-resistant | NA | NCT03868293 |
| 17 | External trigeminal nerve stimulation | Alternative to neurostimulation | Drug-resistant | 2 | NCT01159431 |
| 18 | Vitamin D supplementation | Metabolic stimulation | Drug-resistant | 3 | NCT03475225 |
| 19 | Betashot (a medium chain triglyceride- based (MCT) food) | Metabolic stimulation | All | NA | NCT02825745 |
| 20 | Music periodicity | Musical stimulation | Benign childhood with centrotemporal spikes (BCECTS) or Rolandic | NA | NCT01515436 |
| 22 | Polyunsaturated fatty acids | Anti-inflammation | Drug-resistant | 3 | NCT00299533 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, S.; Parayil, R.; Mishra, S.; Nongthomba, U.; Clement, J.P. Epilepsy Characteristics in Neurodevelopmental Disorders: Research from Patient Cohorts and Animal Models Focusing on Autism Spectrum Disorder. Int. J. Mol. Sci. 2022, 23, 10807. https://doi.org/10.3390/ijms231810807
Chakraborty S, Parayil R, Mishra S, Nongthomba U, Clement JP. Epilepsy Characteristics in Neurodevelopmental Disorders: Research from Patient Cohorts and Animal Models Focusing on Autism Spectrum Disorder. International Journal of Molecular Sciences. 2022; 23(18):10807. https://doi.org/10.3390/ijms231810807
Chicago/Turabian StyleChakraborty, Sukanya, Rrejusha Parayil, Shefali Mishra, Upendra Nongthomba, and James P. Clement. 2022. "Epilepsy Characteristics in Neurodevelopmental Disorders: Research from Patient Cohorts and Animal Models Focusing on Autism Spectrum Disorder" International Journal of Molecular Sciences 23, no. 18: 10807. https://doi.org/10.3390/ijms231810807