
Unique Structural Fold of LonBA Protease from Bacillus subtilis, a Member of a Newly Identified Subfamily of Lon Proteases
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Analysis of the Primary Structures of Lon Proteases Belonging to ID Group S16.005
2.2. Preparation, Limited Proteolysis, and Preliminary Characterization of the Recombinant BsLonBA
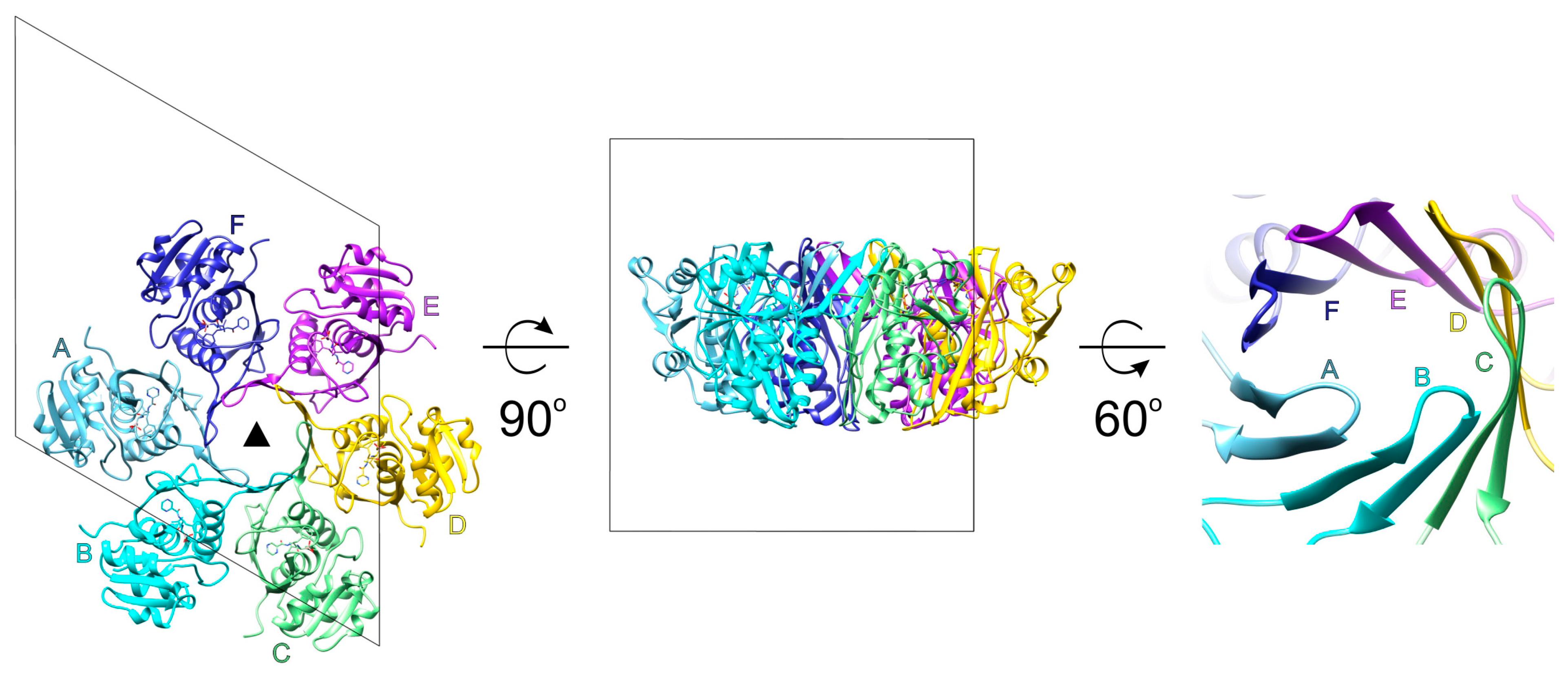
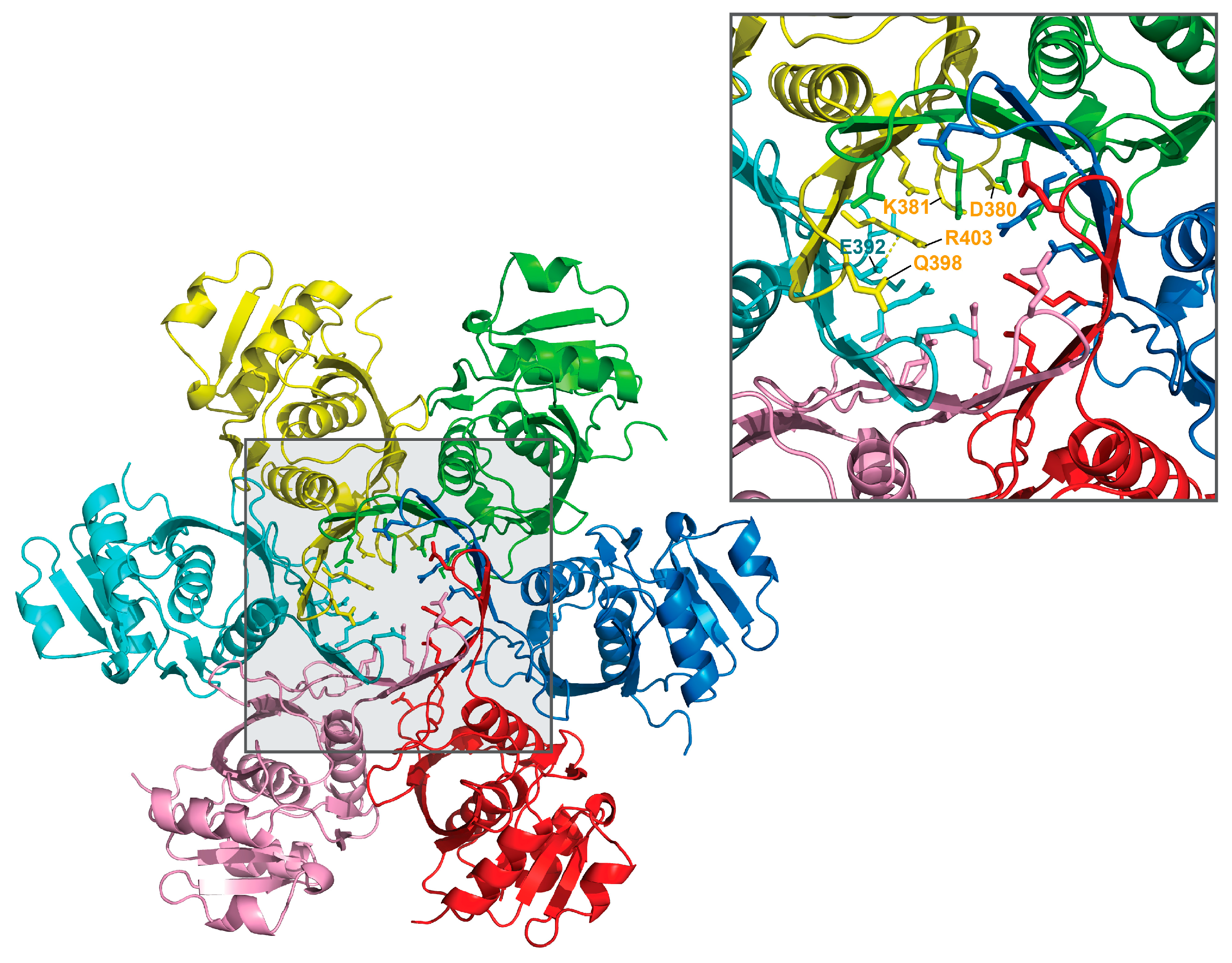
2.3. Crystal Structure of the P Domain of BsLonBA
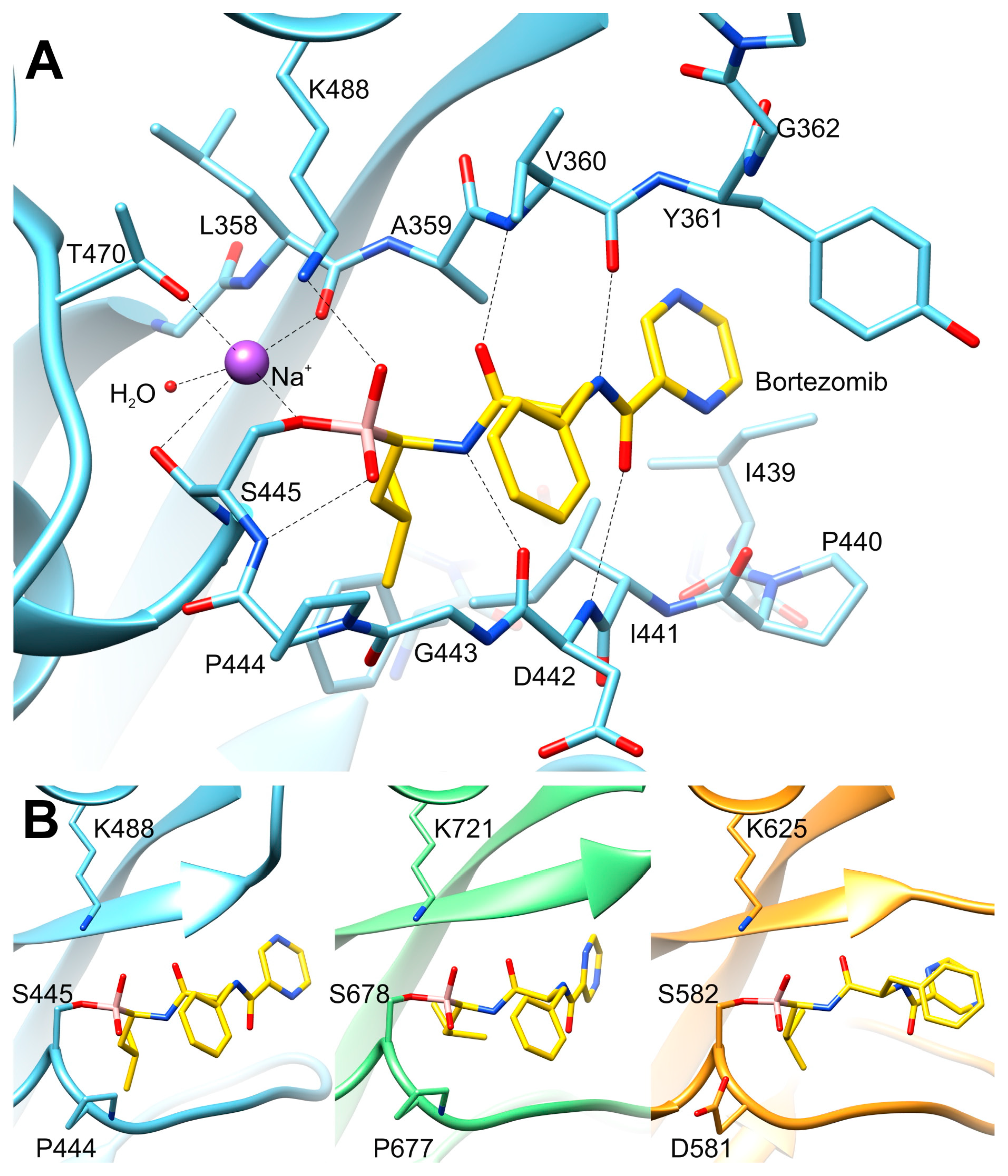
2.4. The Mode of Binding of Bortezomib to the P Domain of LonBA and Other Lon Proteases
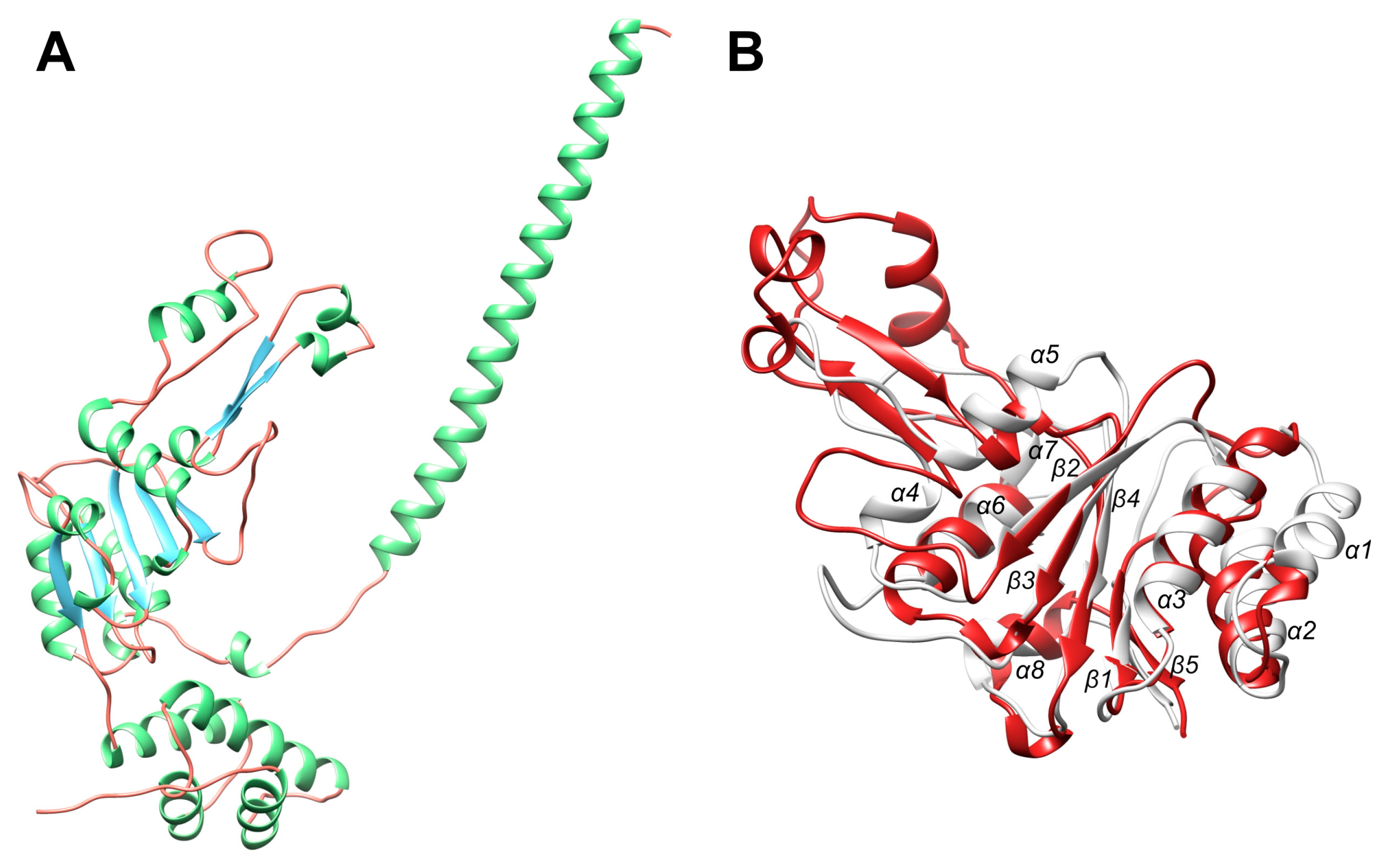
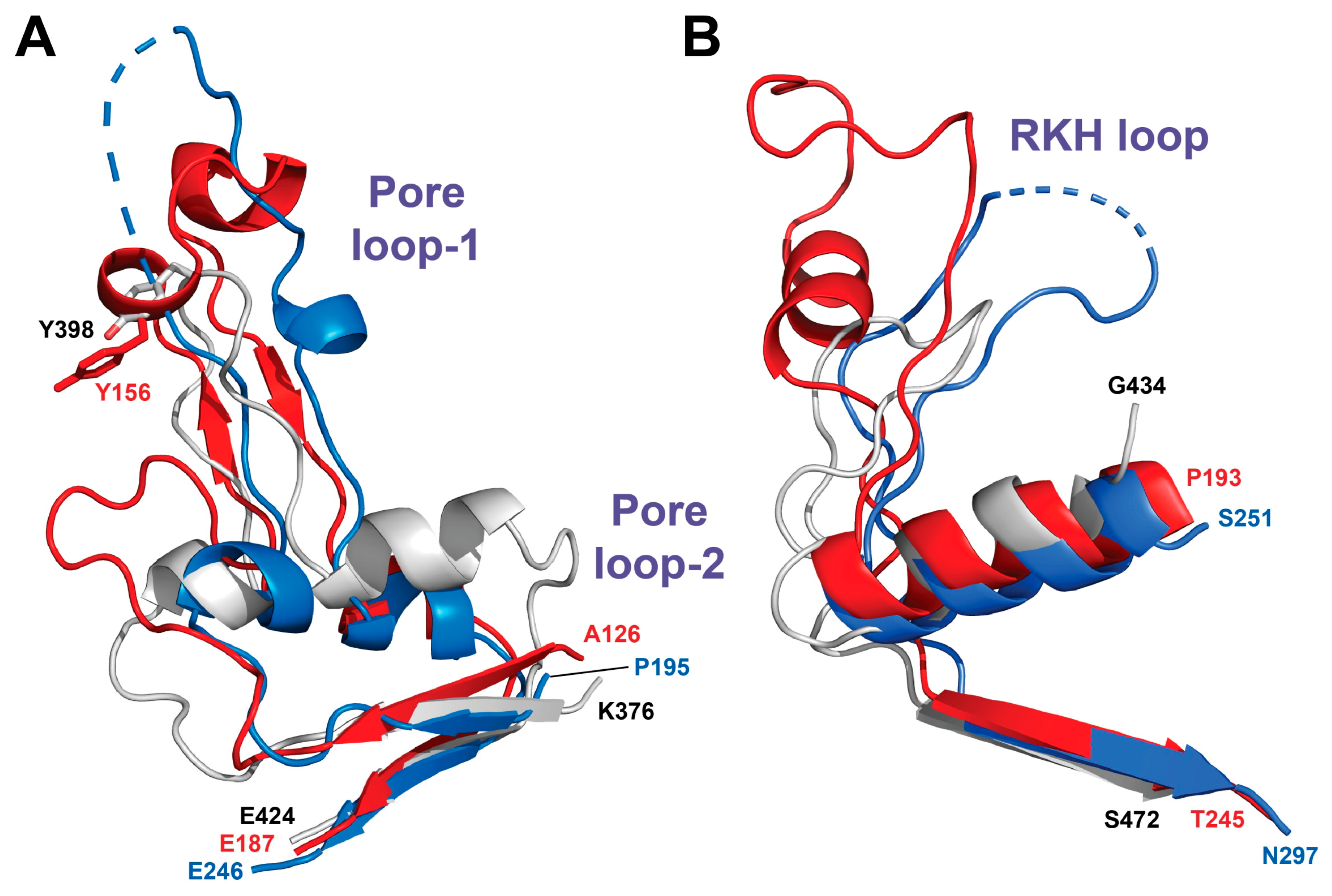
2.5. Comparison of the BsLonBA P Domain with Its Counterparts in Other Lon Proteases
2.6. Model Building of the ATPase Domain of BsLonBA
2.7. Comparison of the Functionally Important Elements in the AAA+ Module of BsLonBA with Their Counterparts in LonA and LonB
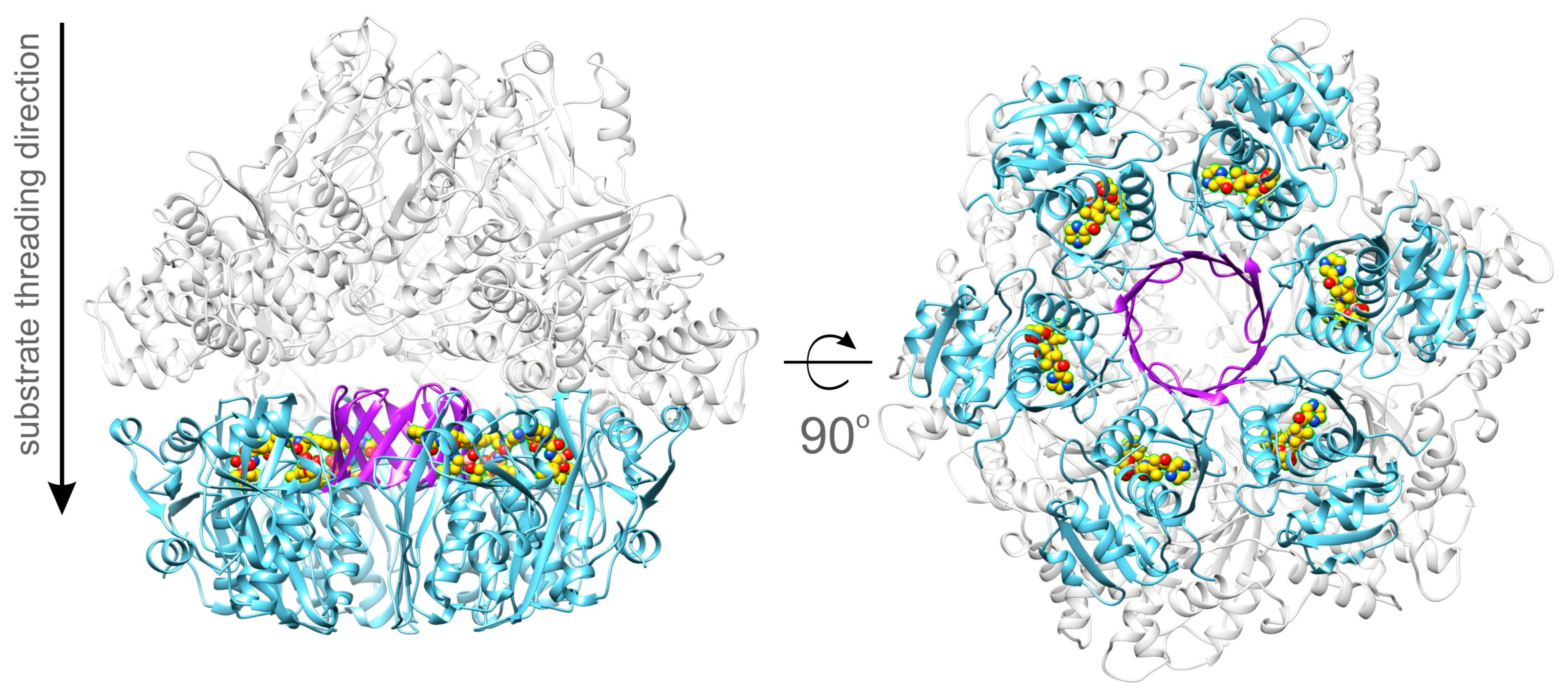
2.8. A Model of the Full-Length BsLonBA
3. Materials and Methods
3.1. Analysis of the Amino acid Sequences of Lon Proteases
3.2. Molecular Cloning, Purification, Characterization, and Limited Proteolysis of the Recombinant BsLonBA
3.3. Cloning, Protein Expression, and Purification of the Protease Domain of BsLonBA
3.4. Crystallographic Procedures
3.5. Modeling Procedures
4. Conclusions
A Proposal to Assign LonBA as a Separate Lon Subfamily
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gottesman, S.; Wickner, S.; Maurizi, M.R. Protein quality control: Triage by chaperones and proteases. Genes Dev. 1997, 11, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.; Bewley, M. Protein Quality Control in Bacterial Cells: Integrated Networks of Chaperones and ATP-Dependent Proteases. Gen. Eng. 2002, 24, 17–47. [Google Scholar] [CrossRef]
- Buchberger, A.; Bukau, B.; Sommer, T. Protein quality control in the cytosol and the endoplasmic reticulum: Brothers in arms. Mol. Cell 2010, 40, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, D.; Szabo, B.; Pancsa, R.; Tompa, P. Intrinsically disordered proteins undergo and assist folding transitions in the proteome. Arch. Biochem. Biophys. 2013, 531, 80–89. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef]
- Doyle, S.M.; Wickner, S. Hsp104 and ClpB: Protein disaggregating machines. Trends Biochem. Sci. 2009, 34, 40–48. [Google Scholar] [CrossRef]
- Clare, D.K.; Saibil, H.R. ATP-driven molecular chaperone machines. Biopolymers 2013, 99, 846–859. [Google Scholar] [CrossRef]
- Mattoo, R.U.; Goloubinoff, P. Molecular chaperones are nanomachines that catalytically unfold misfolded and alternatively folded proteins. Cell. Mol. Life Sci. 2014, 71, 3311–3325. [Google Scholar] [CrossRef]
- Sauer, R.T.; Bolon, D.N.; Burton, B.M.; Burton, R.E.; Flynn, J.M.; Grant, R.A.; Hersch, G.L.; Joshi, S.A.; Kenniston, J.A.; Levchenko, I.; et al. Sculpting the proteome with AAA+ proteases and disassembly machines. Cell 2004, 119, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Striebel, F.; Kress, W.; Weber-Ban, E. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr. Opin. Struct. Biol. 2009, 19, 209–217. [Google Scholar] [CrossRef]
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef]
- Mahmoud, S.A.; Chien, P. Regulated Proteolysis in Bacteria. Annu. Rev. Biochem. 2018, 87, 677–696. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Jee, H. Size dependent classification of heat shock proteins: A mini-review. J. Exerc. Rehabil. 2016, 12, 255–259. [Google Scholar] [CrossRef]
- Nishii, W.; Suzuki, T.; Nakada, M.; Kim, Y.T.; Muramatsu, T.; Takahashi, K. Cleavage mechanism of ATP-dependent Lon protease toward ribosomal S2 protein. FEBS Lett. 2005, 579, 6846–6850. [Google Scholar] [CrossRef]
- Ondrovicova, G.; Liu, T.; Singh, K.; Tian, B.; Li, H.; Gakh, O.; Perecko, D.; Janata, J.; Granot, Z.; Orly, J.; et al. Cleavage site selection within a folded substrate by the ATP-dependent lon protease. J. Biol. Chem. 2005, 280, 25103–25110. [Google Scholar] [CrossRef]
- Mikita, N.; Cheng, I.; Fishovitz, J.; Huang, J.; Lee, I. Processive Degradation of Unstructured Protein by Escherichia coli Lon Occurs via the Slow, Sequential Delivery of Multiple Scissile Sites Followed by Rapid and Synchronized Peptide Bond Cleavage Events. Biochemistry 2013, 52, 5629–5644. [Google Scholar] [CrossRef]
- Li, S.; Hsieh, K.Y.; Kuo, C.I.; Su, S.C.; Huang, K.F.; Zhang, K.; Chang, C.I. Processive cleavage of substrate at individual proteolytic active sites of the Lon protease complex. Sci. Adv. 2021, 7, eabj7537. [Google Scholar] [CrossRef]
- Lupas, A.; Flanagan, J.M.; Tamura, T.; Baumeister, W. Self-compartmentalizing proteases. Trends Biochem. Sci. 1997, 22, 399–404. [Google Scholar] [CrossRef]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar] [CrossRef]
- Rotanova, T.V.; Melnikov, E.E. ATP-dependent proteases and proteolytic complexes involved into intracellular protein degradation. Biochem. (Mosc.) Suppl. Ser. B Biomed. Chem. 2008, 54, 512–530. [Google Scholar] [CrossRef]
- Bittner, L.M.; Arends, J.; Narberhaus, F. Mini review: ATP-dependent proteases in bacteria. Biopolymers 2016, 105, 505–517. [Google Scholar] [CrossRef]
- Swamy, K.H.; Goldberg, A.L. E. coli contains eight soluble proteolytic activities, one being ATP dependent. Nature 1981, 292, 652–654. [Google Scholar] [CrossRef]
- Gur, E.; Sauer, R.T. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008, 22, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Udaka, S.; Yamagata, H. Cloning, characterization, and inactivation of the Bacillus brevis lon gene. J. Bacteriol. 1992, 174, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Riethdorf, S.; Völker, U.; Gerth, U.; Winkler, A.; Engelmann, S.; Hecker, M. Cloning, nucleotide sequence, and expression of the Bacillus subtilis lon gene. J. Bacteriol. 1994, 176, 6518–6527. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Antonov, V.K.; Gorbalenya, A.E.; Kotova, S.A.; Rotanova, T.V.; Shimbarevich, E.V. Site-directed mutagenesis of La protease. A catalytically active serine residue. FEBS Lett. 1991, 287, 211–214. [Google Scholar] [CrossRef]
- Rotanova, T.V.; Melnikov, E.E.; Tsirulnikov, K.B. Catalytic dyad Ser-Lys at the active site of Escherichia coli ATP-dependent Lon-proteinase. Bioorg. Khimiia 2003, 29, 97–99. [Google Scholar]
- Rotanova, T.V.; Melnikov, E.E.; Khalatova, A.G.; Makhovskaya, O.V.; Botos, I.; Wlodawer, A.; Gustchina, A. Classification of ATP-dependent proteases Lon and comparison of the active sites of their proteolytic domains. Eur. J. Biochem. 2004, 271, 4865–4871. [Google Scholar] [CrossRef]
- Rotanova, T.V.; Botos, I.; Melnikov, E.E.; Rasulova, F.; Gustchina, A.; Maurizi, M.R.; Wlodawer, A. Slicing a protease: Structural features of the ATP-dependent Lon proteases gleaned from investigations of isolated domains. Protein Sci. 2006, 15, 1815–1828. [Google Scholar] [CrossRef] [Green Version]
- Lupas, A.N.; Martin, J. AAA proteins. Curr. Opin. Struct. Biol. 2002, 12, 746–753. [Google Scholar] [CrossRef]
- Wlodawer, A.; Sekula, B.; Gustchina, A.; Rotanova, T.V. Structure and the mode of activity of Lon proteases from diverse organisms. J. Mol. Biol. 2022, 434, 167504. [Google Scholar] [CrossRef]
- Botos, I.; Melnikov, E.E.; Cherry, S.; Tropea, J.E.; Khalatova, A.G.; Rasulova, F.; Dauter, Z.; Maurizi, M.R.; Rotanova, T.V.; Wlodawer, A.; et al. The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J. Biol. Chem. 2004, 279, 8140–8148. [Google Scholar] [CrossRef]
- Botos, I.; Melnikov, E.E.; Cherry, S.; Kozlov, S.; Makhovskaya, O.V.; Tropea, J.E.; Gustchina, A.; Rotanova, T.V.; Wlodawer, A. Atomic-resolution crystal structure of the proteolytic domain of Archaeoglobus fulgidus Lon reveals the conformational variability in the active sites of Lon proteases. J. Mol. Biol. 2005, 351, 144–157. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2000, 28, 323–325. [Google Scholar] [CrossRef]
- Liao, J.H.; Kuo, C.I.; Huang, Y.Y.; Lin, Y.C.; Lin, Y.C.; Yang, C.Y.; Wu, W.L.; Chang, W.H.; Liaw, Y.C.; Lin, L.H.; et al. A Lon-Like Protease with No ATP-Powered Unfolding Activity. PLoS ONE 2012, 7, e40226. [Google Scholar]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Besche, H.; Zwickl, P. The Thermoplasma acidophilum Lon protease has a Ser-Lys dyad active site. Eur. J. Biochem. 2004, 271, 4361–4365. [Google Scholar] [CrossRef]
- Ward, D.E.; Shockley, K.R.; Chang, L.S.; Levy, R.D.; Michel, J.K.; Conners, S.B.; Kelly, R.M. Proteolysis in hyperthermophilic microorganisms. Archaea 2002, 1, 63–74. [Google Scholar] [CrossRef]
- Melnikov, E.E.; Andrianova, A.G.; Morozkin, A.D.; Stepnov, A.A.; Makhovskaya, O.V.; Botos, I.; Gustchina, A.; Wlodawer, A.; Rotanova, T.V. Limited proteolysis of E. coli ATP-dependent protease Lon—A unified view of the subunit architecture and characterization of isolated enzyme fragments. Acta Biochim. Pol. 2008, 55, 281–296. [Google Scholar] [CrossRef]
- Chung, C.H.; Goldberg, A.L. DNA stimulates ATP-dependent proteolysis and protein-dependent ATPase activity of protease La from Escherichia coli. Proc. Natl. Acad. Sci. USA 1982, 79, 795–799. [Google Scholar] [CrossRef] [Green Version]
- Andrianova, A.G.; Kudzhaev, A.M.; Serova, O.V.; Dergousova, N.I.; Rotanova, T.V. Role of alpha-Helical Domains in Functioning of ATP-Dependent Lon Protease of Escherichia coli. Russ. J. Bioorg. Chem. 2014, 40, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, J.; Schlothauer, T.; Dougan, D.A.; Lilie, H.; Tischendorf, G.; Mogk, A.; Bukau, B.; Turgay, K. Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J. 2006, 25, 1481–1491. [Google Scholar] [CrossRef]
- Wang, F.; Mei, Z.; Qi, Y.; Yan, C.; Hu, Q.; Wang, J.; Shi, Y. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature 2011, 471, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Cooper, D.R.; Porebski, P.J.; Shabalin, I.G.; Handling, K.B.; Minor, W. CheckMyMetal: A macromolecular metal-binding validation tool. Acta Crystallogr. 2017, D73, 223–233. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Simmons, L.A.; Grossman, A.D.; Walker, G.C. Clp and Lon proteases occupy distinct subcellular positions in Bacillus subtilis. J. Bacteriol. 2008, 190, 6758–6768. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Enemark, E.J. Fundamental Characteristics of AAA+ Protein Family Structure and Function. Archaea 2016, 2016, 9294307. [Google Scholar] [CrossRef] [PubMed]
- Podobnik, M.; Weitze, T.F.; O’Donnell, M.; Kuriyan, J. Nucleotide-induced conformational changes in an isolated Escherichia coli DNA polymerase III clamp loader subunit. Structure 2003, 11, 253–263. [Google Scholar] [CrossRef]
- Rotanova, T.V.; Andrianova, A.G.; Kudzhaev, A.M.; Li, M.; Botos, I.; Wlodawer, A.; Gustchina, A. New insights into structural and functional relationships between LonA proteases and ClpB chaperones. FEBS Open Bio 2019, 9, 1536–1551. [Google Scholar] [CrossRef] [Green Version]
- An, Y.J.; Na, J.H.; Kim, M.I.; Cha, S.S. Structural basis for the ATP-independent proteolytic activity of LonB proteases and reclassification of their AAA+ modules. J. Microbiol. 2015, 53, 711–717. [Google Scholar] [CrossRef]
- Kudzhaev, A.; Dubovtseva, E.; Serova, O.; Andrianova, A.; Rotanova, T. Effect of the Deletion of the (173–280) Fragment of the Inserted α-Helical Domain on the Functional Properties of ATP-Dependent Lon Protease from E. coli. Russ. J. Bioorg. Chem. 2018, 44, 518–527. [Google Scholar] [CrossRef]
- Bencini, D.A.; Wild, J.R.; O’Donovan, G.A. Linear one-step assay for the determination of orthophosphate. Anal. Biochem. 1983, 132, 254–258. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Castillo, M.J.; Nakajima, K.; Zimmerman, M.; Powers, J.C. Sensitive substrates for human leukocyte and porcine pancreatic elastase: A study of the merits of various chromophoric and fluorogenic leaving groups in assays for serine proteases. Anal. Biochem. 1979, 99, 53–64. [Google Scholar] [CrossRef]
- Kapust, R.B.; Tözsér, J.; Fox, J.D.; Anderson, D.E.; Cherry, S.; Copeland, T.D.; Waugh, D.S. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001, 14, 993–1000. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. 2010, D66, 133–144. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. 2013, D69, 1204–1214. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Adams, P.D.; Read, R.J.; Zwart, P.H.; Hung, L.W. Iterative-build OMIT maps: Map improvement by iterative model building and refinement without model bias. Acta Crystallogr. 2008, D64, 515–524. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. 2010, D66, 486–501. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. 2011, D67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Isupov, M.N.; Murshudov, G.N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. 2001, D57, 122–133. [Google Scholar] [CrossRef]
- Winn, M.D.; Murshudov, G.N.; Papiz, M.Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003, 374, 300–321. [Google Scholar] [CrossRef] [PubMed]
- Brünger, A.T. The free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–474. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: Program to check the stereochemical quality of protein structures. J. Appl. Cryst 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010, D66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.S.; An, Y.J.; Lee, C.R.; Lee, H.S.; Kim, Y.G.; Kim, S.J.; Kwon, K.K.; De Donatis, G.M.; Lee, J.H.; Maurizi, M.R.; et al. Crystal structure of Lon protease: Molecular architecture of gated entry to a sequestered degradation chamber. EMBO J. 2010, 29, 3520–3530. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Cohen, G.E. ALIGN: A program to superimpose protein coordinates, accounting for insertions and deletions. J. Appl. Crystallogr. 1997, 30, 1160–1161. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | LonBA |
|---|---|
| Space group | P63 |
| Unit cell parameters a, b, c (Å) α, β, γ (˚) | 89.9, 89.9, 83.95 90, 120, 90 |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.000 |
| Resolution (Å) | 84–1.9 (1.94–1.90) |
| Rmerge (%) | 4.4 (82.7) |
| Completeness (%) | 99.9 (99.0) |
| Mean I/σ(I) | 30.1 (1.8) |
| Total reflections | 294,057 (10,969) |
| Unique reflections | 30,425 (1942) |
| Redundancy | 9.7 |
| CC1/2 | 1.000 (0.797) |
| B factor from Wilson plot (Å2) | 39.6 |
| No. of molecules in a.u. | 2 |
| Refinement statistics | |
| Resolution (Å) | 30–1.9 (1.95–1.90) |
| Working set: no. of reflections | 28,500 |
| Rfactor (%) | 19.4 (31.6) |
| Test set: no. of reflections | 951 |
| Rfree (%) | 23.7 (34.2) |
| Protein atoms | 2831 |
| Solvent atoms | 159 |
| Ligand atoms | 58 |
| Geometry statistics | |
| R.m.s.d. bond distance (Å) | 0.01 |
| R.m.s.d. bond angle (°) | 1.65 |
| Isotropic average B-factor (Å2) | 47.0 |
| Ramachandran plot (%) | |
| Favored regions | 97 |
| Allowed regions | 3 |
| Disallowed regions | 0 |
| PDB ID | 8dvh |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustchina, A.; Li, M.; Andrianova, A.G.; Kudzhaev, A.M.; Lountos, G.T.; Sekula, B.; Cherry, S.; Tropea, J.E.; Smirnov, I.V.; Wlodawer, A.; et al. Unique Structural Fold of LonBA Protease from Bacillus subtilis, a Member of a Newly Identified Subfamily of Lon Proteases. Int. J. Mol. Sci. 2022, 23, 11425. https://doi.org/10.3390/ijms231911425
Gustchina A, Li M, Andrianova AG, Kudzhaev AM, Lountos GT, Sekula B, Cherry S, Tropea JE, Smirnov IV, Wlodawer A, et al. Unique Structural Fold of LonBA Protease from Bacillus subtilis, a Member of a Newly Identified Subfamily of Lon Proteases. International Journal of Molecular Sciences. 2022; 23(19):11425. https://doi.org/10.3390/ijms231911425
Chicago/Turabian StyleGustchina, Alla, Mi Li, Anna G. Andrianova, Arsen M. Kudzhaev, George T. Lountos, Bartosz Sekula, Scott Cherry, Joseph E. Tropea, Ivan V. Smirnov, Alexander Wlodawer, and et al. 2022. "Unique Structural Fold of LonBA Protease from Bacillus subtilis, a Member of a Newly Identified Subfamily of Lon Proteases" International Journal of Molecular Sciences 23, no. 19: 11425. https://doi.org/10.3390/ijms231911425