
KITLG Promotes Glomerular Endothelial Cell Injury in Diabetic Nephropathy by an Autocrine Effect
 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
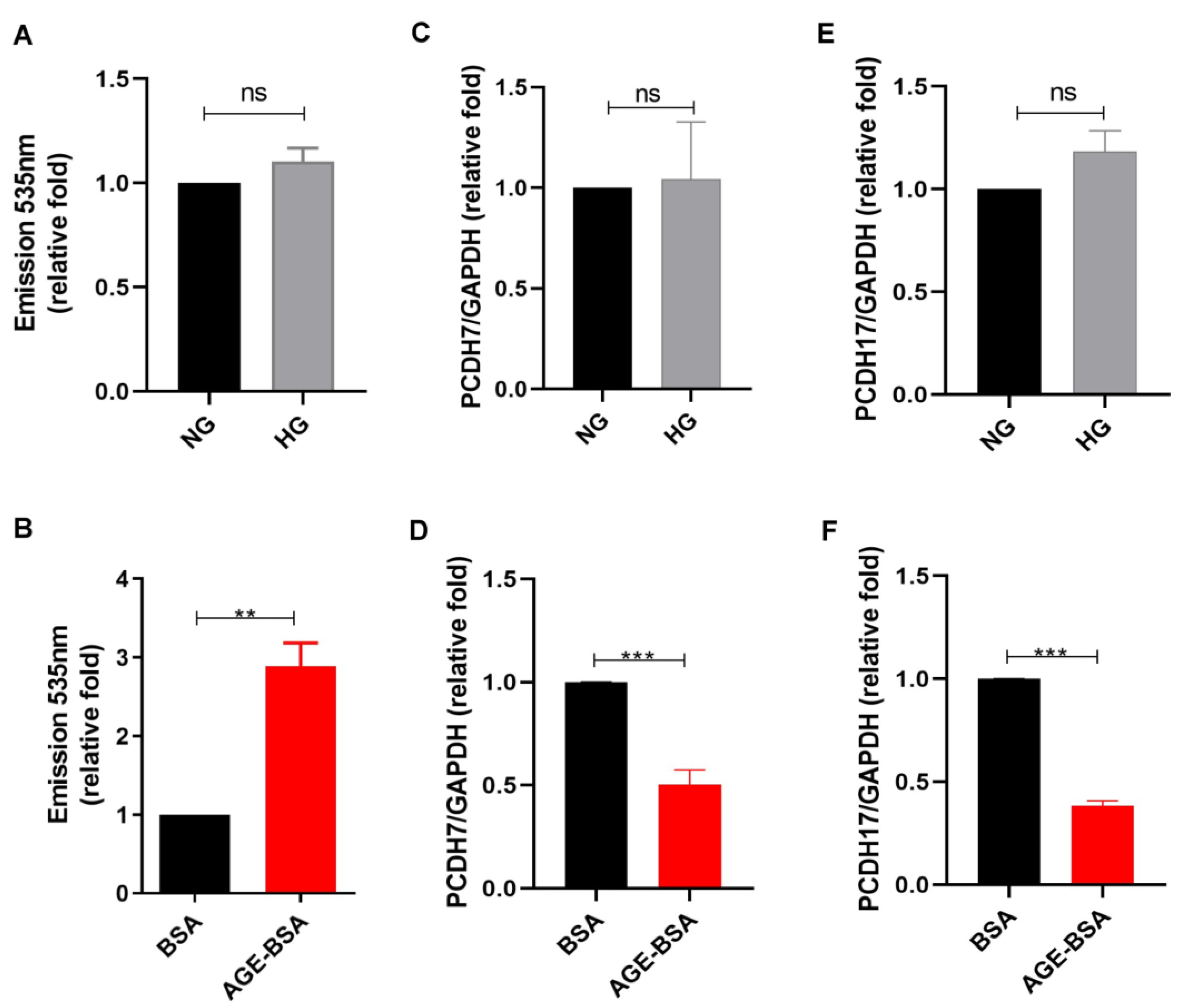
2.1. AGEs Increased Vascular Permeability of GECs
2.2. AGEs Increased KITLG Expression in GECs
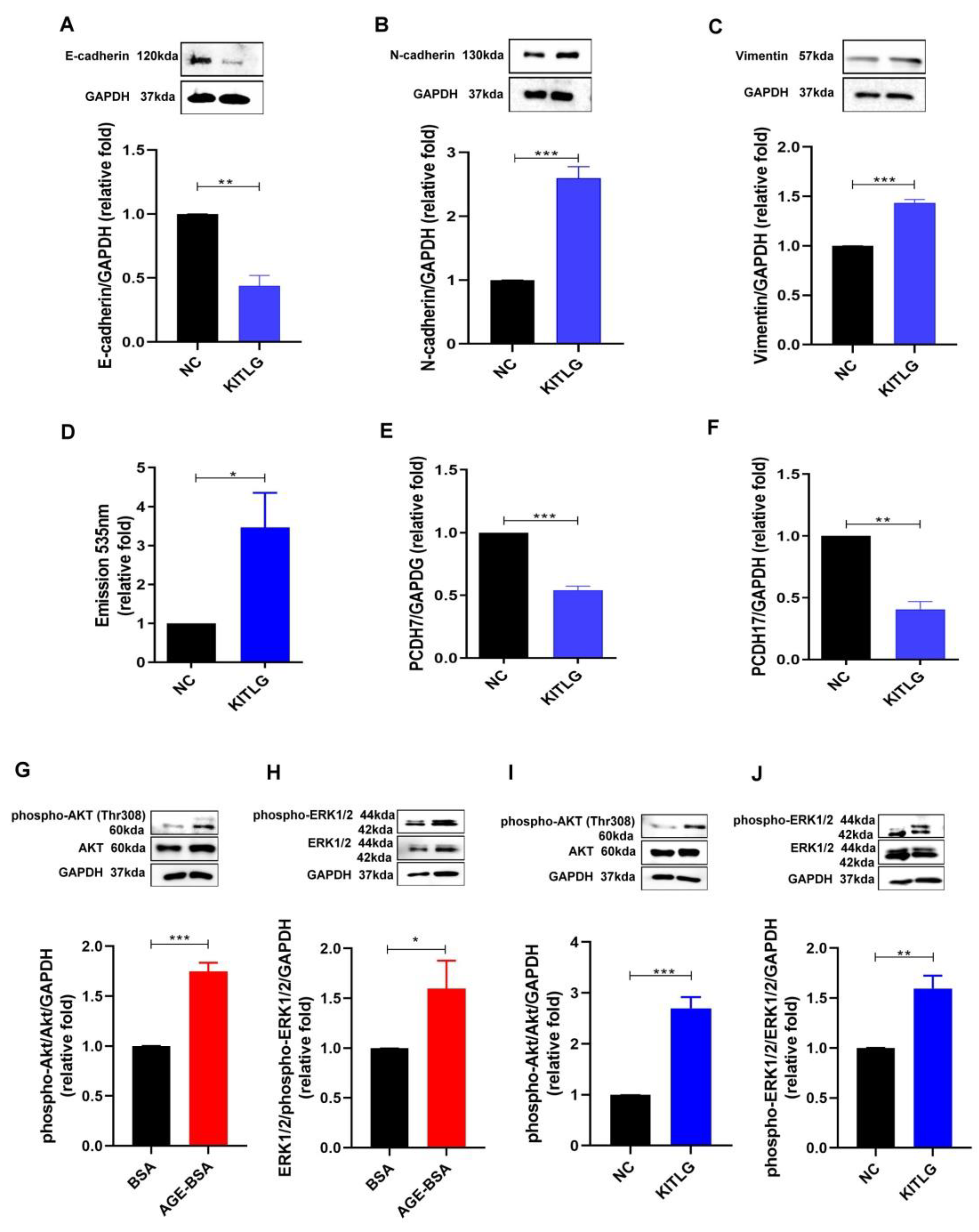
2.3. KITLG Promoted EndoMT and Furthered the Increase in Permeability of HGECs through AKT (Protein Kinase B)/Extracellular-Signal-Regulated Kinase (ERK) 1/2 Signaling Pathway
2.4. Imatinib, as a KITLG Inhibitor, Rescued the Glomerular Endothelial Injury Induced by AGEs
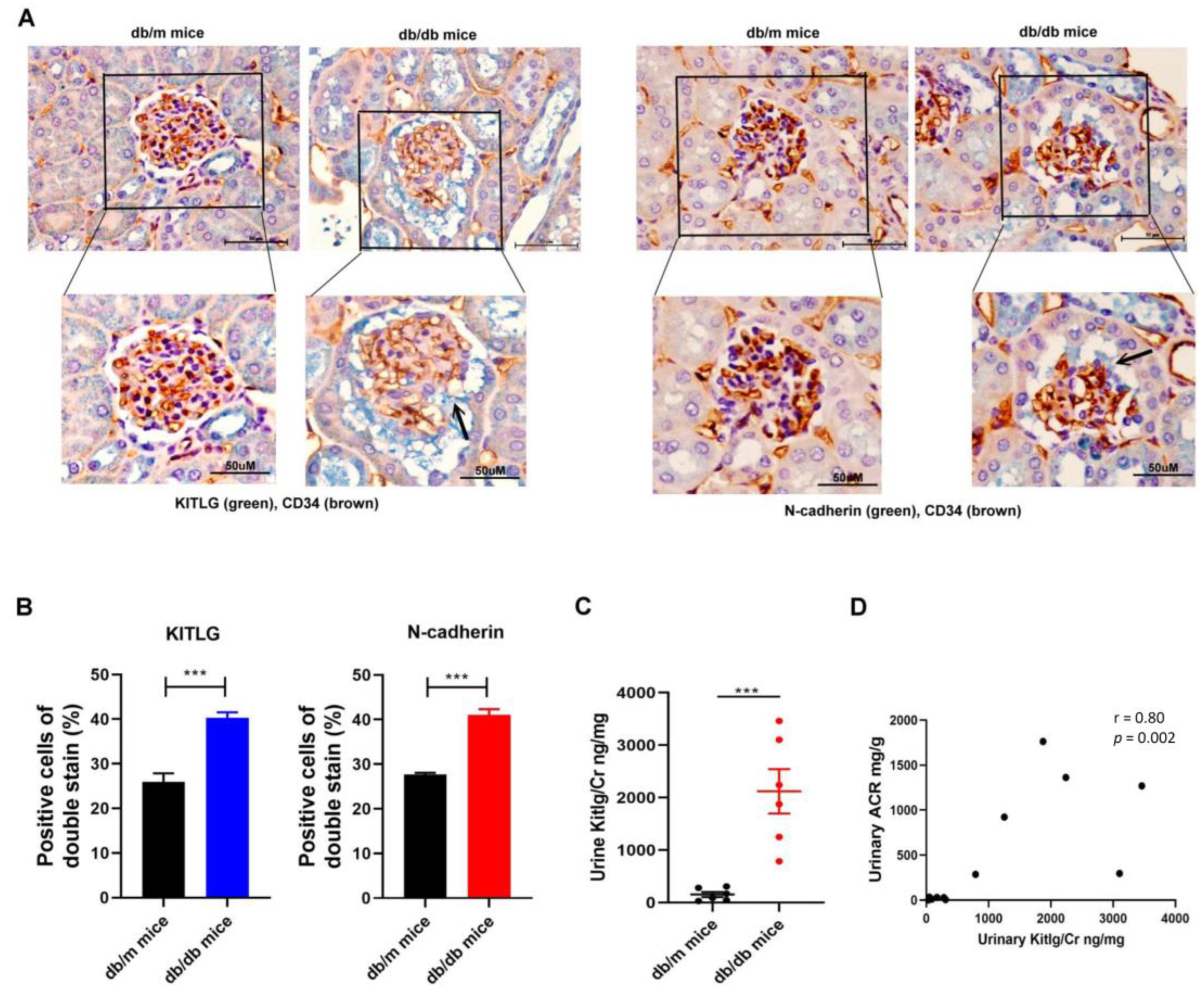
2.5. Positive Relationship between KITLG Expression and Kidney Dysfunction in Mice with DN
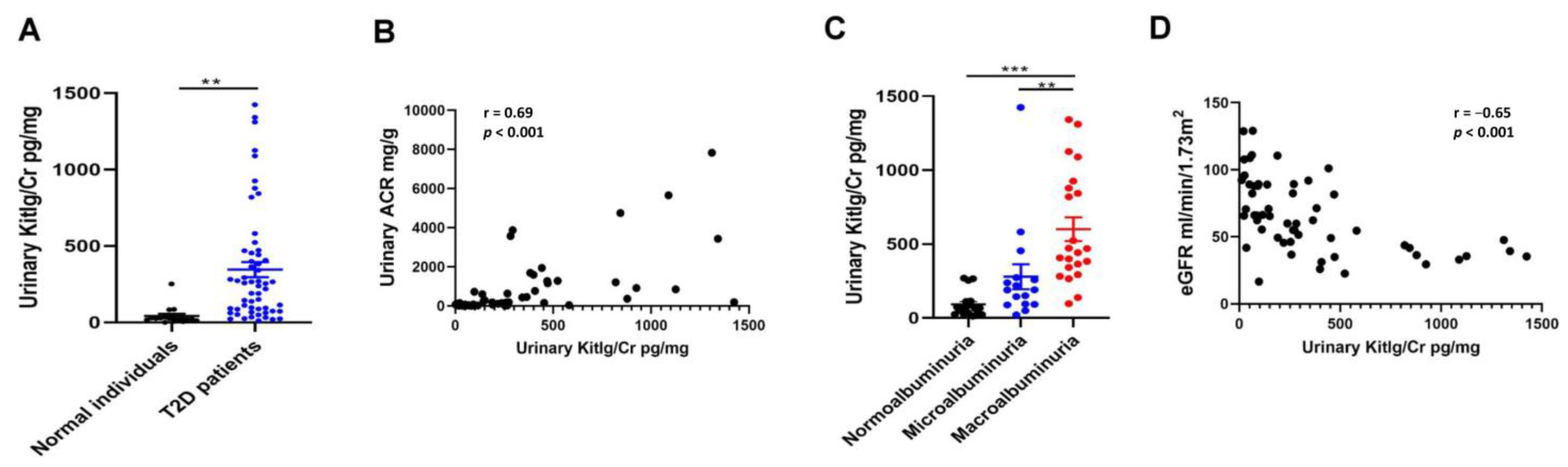
2.6. Positive Relationship between Urinary KITLG Levels and Kidney Dysfunction in Humans
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Cultures
4.2. Permeability Analysis of HGECs
4.3. Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.4. Quantification of KITLG Using ELISA
4.5. Western Blot Analysis
4.6. Experimental Animals
4.7. IHC Stain
4.8. Human Study Participants
4.9. Measurement of Urinary ACR Levels
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Johansen, K.L.; Chertow, G.M.; Gilbertson, D.T.; Herzog, C.A.; Ishani, A.; Israni, A.K.; Ku, E.; Li, S.; Li, S.; Liu, J.; et al. US Renal Data System 2021 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2022, 79, A8–A12. [Google Scholar] [CrossRef]
- Penno, G.; Solini, A.; Orsi, E.; Bonora, E.; Fondelli, C.; Trevisan, R.; Vedovato, M.; Cavalot, F.; Lamacchia, O.; Scardapane, M.; et al. Non-albuminuric renal impairment is a strong predictor of mortality in individuals with type 2 diabetes: The Renal Insufficiency And Cardiovascular Events (RIACE) Italian multicentre study. Diabetologia 2018, 61, 2277–2289. [Google Scholar] [CrossRef] [Green Version]
- Afkarian, M.; Sachs, M.C.; Kestenbaum, B.; Hirsch, I.B.; Tuttle, K.R.; Himmelfarb, J.; de Boer, I.H. Kidney disease and increased mortality risk in type 2 diabetes. J. Am. Soc. Nephrol. 2013, 24, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Sawaf, H.; Thomas, G.; Taliercio, J.J.; Nakhoul, G.; Vachharajani, T.J.; Mehdi, A. Therapeutic Advances in Diabetic Nephropathy. J. Clin. Med. 2022, 11, 378. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, H.; Lu, Y.; Wu, T.; Ji, G. New progress in drugs treatment of diabetic kidney disease. Biomed. Pharmacother. 2021, 141, 111918. [Google Scholar] [CrossRef]
- Tervaert, T.W.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; de Heer, E.; et al. Renal Pathology Society. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Hung, P.H.; Hsu, Y.C.; Chen, T.H.; Lin, C.L. Recent Advances in Diabetic Kidney Diseases: From Kidney Injury to Kidney Fibrosis. Int. J. Mol. Sci. 2021, 22, 11857. [Google Scholar] [CrossRef]
- Stehouwer, C.D.A. Microvascular Dysfunction and Hyperglycemia: A Vicious Cycle with Widespread Consequences. Diabetes 2018, 67, 1729–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassen, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Kim, Y.S. The Role of Advanced Glycation End Products in Diabetic Vascular Complications. Diabetes Metab. J. 2018, 42, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczuko, M.; Kaczkan, M.; Drozd, A.; Maciejewska, D.; Palma, J.; Owczarzak, A.; Marczuk, N.; Rutkowski, P.; Małgorzewicz, S. Comparison of Fatty Acid Profiles in a Group of Female Patients with Chronic Kidney Diseases (CKD) and Metabolic Syndrome (MetS)—Similar Trends of Changes, Different Pathophysiology. Int. J. Mol. Sci. 2019, 20, 1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opazo-Ríos, L.; Mas, S.; Marín-Royo, G.; Mezzano, S.; Gómez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sol, M.; Kamps, J.; van den Born, J.; van den Heuvel, M.C.; van der Vlag, J.; Krenning, G.; Hillebrands, J.L. Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases. Front. Pharmacol. 2020, 11, 573557. [Google Scholar] [CrossRef]
- Liang, X.; Duan, N.; Wang, Y.; Shu, S.; Xiang, X.; Guo, T.; Yang, L.; Zhang, S.; Tang, X.; Zhang, J. Advanced oxidation protein products induce endothelial-to-mesenchymal transition in human renal glomerular endothelial cells through induction of endoplasmic reticulum stress. J. Diabetes Complicat. 2016, 30, 573–579. [Google Scholar] [CrossRef]
- Shang, J.; Zhang, Y.; Jiang, Y.; Li, Z.; Duan, Y.; Wang, L.; Xiao, J.; Zhao, Z. NOD2 promotes endothelial-to-mesenchymal transition of glomerular endothelial cells via MEK/ERK signaling pathway in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2017, 484, 435–441. [Google Scholar] [CrossRef]
- Peng, H.; Li, Y.; Wang, C.; Zhang, J.; Chen, Y.; Chen, W.; Cao, J.; Wang, Y.; Hu, Z.; Lou, T. ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Sci. Rep. 2016, 6, 20304. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lu, L.; Hou, W.; Wang, F.; Huang, T.; Meng, Z.; Zhu, M. The SETD8/ELK1/bach1 complex regulates hyperglycaemia-mediated EndMT in diabetic nephropathy. J. Transl. Med. 2022, 20, 147. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zou, H.; Lu, H.; Xiang, H.; Chen, S. Research progress of endothelial-mesenchymal transition in diabetic kidney disease. J. Cell. Mol. Med. 2022, 26, 3313–3322. [Google Scholar] [CrossRef] [PubMed]
- Broudy, V.C. Stem cell factor and hematopoiesis. Blood 1997, 90, 1345–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, S.; Kitoh, T. Measurement of KIT ligand/stem cell factor: Clinical and biochemical significance. Curr. Opin. Hematol. 2000, 7, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Kong, X.; Liu, W.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. Phosphorylation and nuclear translocation of integrin beta4 induced by a chemical small molecule contribute to apoptosis in vascular endothelial cells. Apoptosis 2013, 18, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.; Wakabayashi, T.; Asada, M.; Yoshimatsu, K.; Okada, M. Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J. Biol. Chem. 2004, 279, 18600–18607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, E.P.; Mohan, M.; McClelland, A.; Tikellis, C.; Ziemann, M.; Kaspi, A.; Gray, S.P.; Pickering, R.; Tan, S.M.; Ali-Shah, S.T.; et al. Lipoxins Regulate the Early Growth Response-1 Network and Reverse Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1437–1448. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.; Reznichenko, A.; Witasp, A.; Liu, P.; Greasley, P.J.; Sorrentino, A.; Blondal, T.; Zambrano, S.; Nordstrom, J.; Bruchfeld, A.; et al. Novel insights into the disease transcriptome of human diabetic glomeruli and tubulointerstitium. Nephrol. Dial. Transplant. 2020, 35, 2059–2072. [Google Scholar] [CrossRef]
- Hsu, P.C.; Huang, J.C.; Tsai, W.C.; Hung, W.W.; Chang, W.A.; Wu, L.Y.; Chang, C.Y.; Tsai, Y.C.; Hsu, Y.L. Tumor Necrosis Factor Receptor Superfamily Member 21 Induces Endothelial-Mesenchymal Transition in Coronary Artery Endothelium of Type 2 Diabetes Mellitus. Biomedicines 2022, 10, 1282. [Google Scholar] [CrossRef]
- Yang, S.; Li, W.S.; Dong, F.; Sun, H.M.; Wu, B.; Tan, J.; Zou, W.J.; Zhou, D.S. KITLG is a novel target of miR-34c that is associated with the inhibition of growth and invasion in colorectal cancer cells. J. Cell. Mol. Med. 2014, 18, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Salomonsson, A.; Jonsson, M.; Isaksson, S.; Karlsson, A.; Jonsson, P.; Gaber, A.; Bendahl, P.O.; Johansson, L.; Brunnstrom, H.; Jirstrom, K.; et al. Histological specificity of alterations and expression of KIT and KITLG in non-small cell lung carcinoma. Genes Chromosomes Cancer 2013, 52, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Aslam, N.; Abusharieh, E.; Abuarqoub, D.; Ali, D.; Al-Hattab, D.; Wehaibi, S.; Al-Kurdi, B.; Jamali, F.; Alshaer, W.; Jafar, H.; et al. Anti-oncogenic activities exhibited by paracrine factors of MSCs can be mediated by modulation of KITLG and DKK1 genes in glioma SCs in vitro. Mol. Ther. Oncolytics 2021, 20, 147–165. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, S.; Wang, Y.; Chen, Y.; Zhang, P.; Liu, Y.; Zhang, H.; Zhang, P.; Tao, Z.; Xiong, K. High expression of KITLG is a new hallmark activating the MAPK pathway in type A and AB thymoma. Thorac. Cancer 2020, 11, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Veneri, D.; Franchini, M.; Bonora, E. Imatinib and regression of type 2 diabetes. N. Engl. J. Med. 2005, 352, 1049–1050. [Google Scholar] [CrossRef]
- Lassila, M.; Allen, T.J.; Cao, Z.; Thallas, V.; Jandeleit-Dahm, K.A.; Candido, R.; Cooper, M.E. Imatinib attenuates diabetes-associated atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 935–942. [Google Scholar] [CrossRef]
- Choi, S.S.; Kim, E.S.; Jung, J.E.; Marciano, D.P.; Jo, A.; Koo, J.Y.; Choi, S.Y.; Yang, Y.R.; Jang, H.J.; Kim, E.K.; et al. PPARgamma Antagonist Gleevec Improves Insulin Sensitivity and Promotes the Browning of White Adipose Tissue. Diabetes 2016, 65, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C.; et al. The role of PDGF-B/TGF-beta1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell. Signal. 2016, 28, 1489–1501. [Google Scholar] [CrossRef]
- Gitelman, S.E.; Bundy, B.N.; Ferrannini, E.; Lim, N.; Blanchfield, J.L.; DiMeglio, L.A.; Felner, E.I.; Gaglia, J.L.; Gottlieb, P.A.; Long, S.A.; et al. Gleevec Trial Study Group. Imatinib therapy for patients with recent-onset type 1 diabetes: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2021, 9, 502–514. [Google Scholar] [CrossRef]
- Zhou, L.; Sun, X.; Huang, Z.; Zhou, T.; Zhu, X.; Liu, Y.; Wang, J.; Cheng, B.; Li, M.; He, C.; et al. Imatinib Ameliorated Retinal Neovascularization by Suppressing PDGFR-α and PDGFR-β. Cell. Physiol. Biochem. 2018, 48, 263–273. [Google Scholar] [CrossRef]
- Lassila, M.; Jandeleit-Dahm, K.; Seah, K.K.; Smith, C.M.; Calkin, A.C.; Allen, T.J.; Cooper, M.E. Imatinib attenuates diabetic nephropathy in apolipoprotein E-knockout mice. J. Am. Soc. Nephrol. 2005, 16, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef]
- Winiarska, A.; Knysak, M.; Nabrdalik, K.; Gumprecht, J.; Stompór, T. Inflammation and Oxidative Stress in Diabetic Kidney Disease: The Targets for SGLT2 Inhibitors and GLP-1 Receptor Agonists. Int. J. Mol. Sci. 2021, 22, 10822. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Zoja, C.; Conti, S.; Cerullo, D.; Corna, D.; Rottoli, D.; Zanchi, C.; Tomasoni, S.; Remuzzi, G.; Benigni, A. Empagliflozin protects glomerular endothelial cell architecture in experimental diabetes through the VEGF-A/caveolin-1/PV-1 signaling pathway. J. Pathol. 2022, 256, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Bosch, J.P.; Lewis, J.B.; Greene, T.; Rogers, N.; Roth, D. A more accurate method to estimate glomerular filtration rate from serum creatinine: A new prediction equation. Modification of Diet in Renal Disease Study Group. Ann. Intern. Med. 1999, 130, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Benzing, T.; Salant, D. Insights into Glomerular Filtration and Albuminuria. N. Engl. J. Med. 2021, 384, 1437–1446. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Individuals n = 16 | Type 2 Diabetes n = 56 | p-Value | |
|---|---|---|---|
| Age, years | 61.8 ± 5.1 | 62.7 ± 12.4 | 0.77 |
| Sex (male), % | 65.5 | 50.0 | 0.37 |
| Fasting blood glucose, mg/dL | 98.3 ± 9.2 | 131.5 ± 32.5 | 0.001 |
| Blood urea nitrogen, mg/dL | 15.7 (12.7–17.7) | 17.5 (13.4–22.7) | 0.12 |
| Serum creatinine, mg/dL | 0.8 ± 0.2 | 1.2 ± 0.6 | <0.001 |
| Estimated glomerular filtration rate, mL/min/1.73 m2 | 96.5 ± 19.7 | 62.0 ± 27.1 | <0.001 |
| Urinary ACR, mg/g | 10.5 (4.9–66.2) | 157.1 (27.9–896.2) | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.-C.; Chen, S.-C.; Chang, W.-A.; Hung, W.-W.; Wu, P.-H.; Wu, L.-Y.; Chang, J.-M.; Hsu, Y.-L.; Tsai, Y.-C. KITLG Promotes Glomerular Endothelial Cell Injury in Diabetic Nephropathy by an Autocrine Effect. Int. J. Mol. Sci. 2022, 23, 11723. https://doi.org/10.3390/ijms231911723
Huang J-C, Chen S-C, Chang W-A, Hung W-W, Wu P-H, Wu L-Y, Chang J-M, Hsu Y-L, Tsai Y-C. KITLG Promotes Glomerular Endothelial Cell Injury in Diabetic Nephropathy by an Autocrine Effect. International Journal of Molecular Sciences. 2022; 23(19):11723. https://doi.org/10.3390/ijms231911723
Chicago/Turabian StyleHuang, Jiun-Chi, Szu-Chia Chen, Wei-An Chang, Wei-Wen Hung, Ping-Hsun Wu, Ling-Yu Wu, Jer-Ming Chang, Ya-Ling Hsu, and Yi-Chun Tsai. 2022. "KITLG Promotes Glomerular Endothelial Cell Injury in Diabetic Nephropathy by an Autocrine Effect" International Journal of Molecular Sciences 23, no. 19: 11723. https://doi.org/10.3390/ijms231911723