
The Role of Small Heat Shock Proteins in Protein Misfolding Associated Motoneuron Diseases
 , , , , , , , ,
, , , , , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
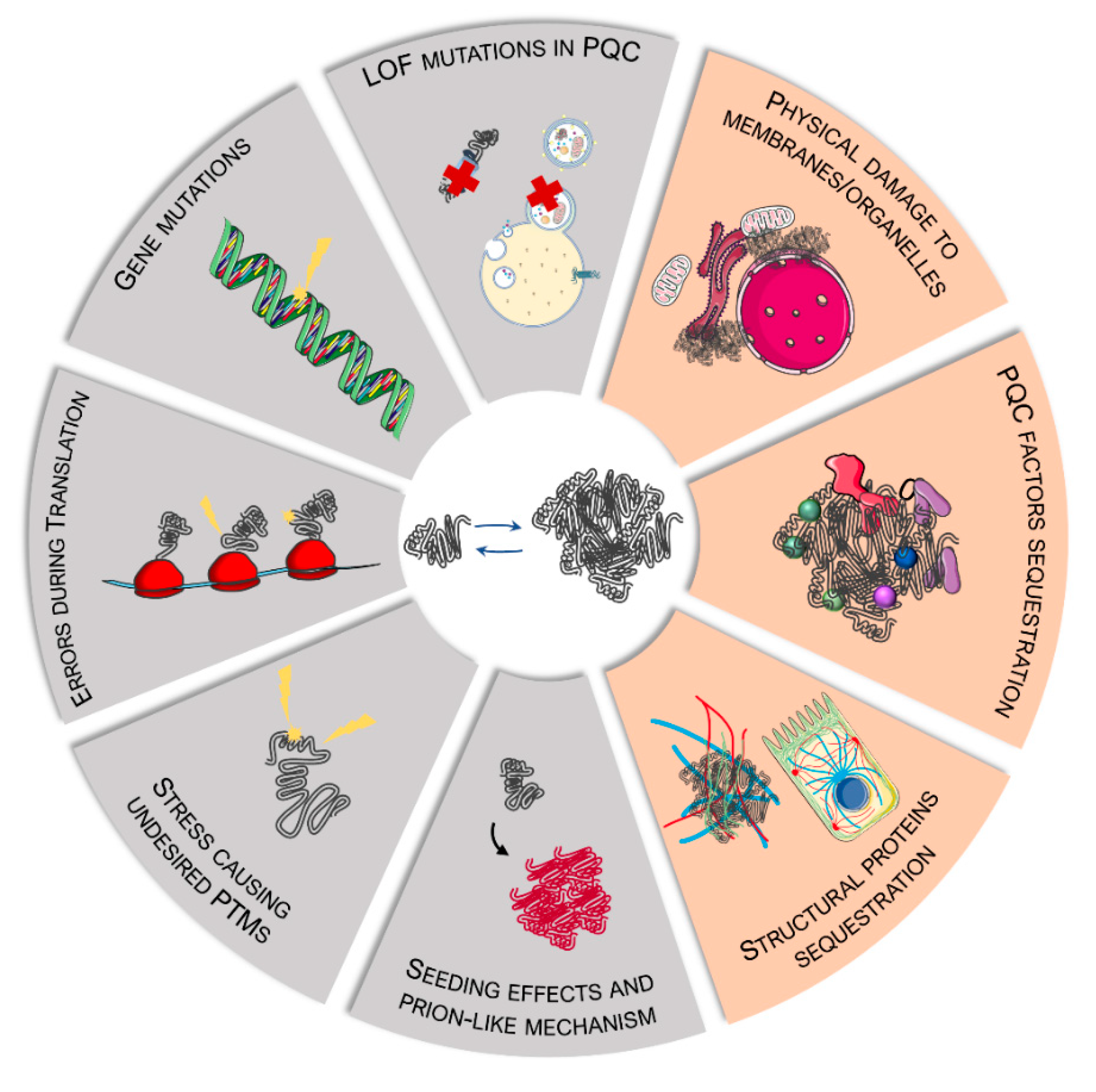
2. Sources and Mechanisms of Protein Misfolding in MNDs
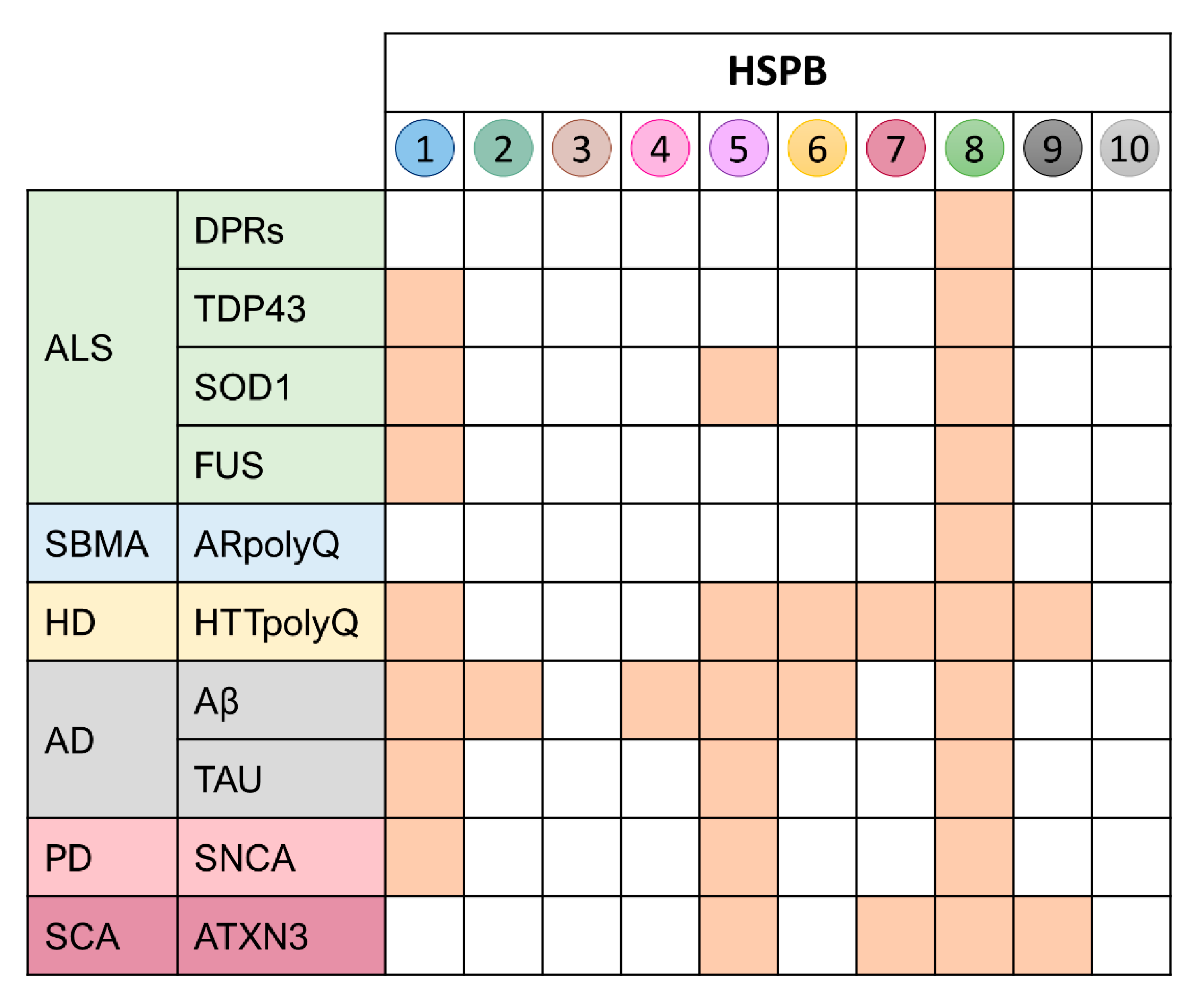
3. The Family of the Small HSPs (HSPBs) and Their Role in Protein Misfolding in MNDs
3.1. HSPB1
3.2. HSPB2 and HSPB3
3.3. HSPB4 and HSPB5
3.4. HSPB6
3.5. HSPB7
3.6. HSPB8
3.7. HSPB9 and HSPB10
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Figlewicz, D.A.; Orrell, R.W. The genetics of motor neuron diseases. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2003, 4, 225–231. [Google Scholar] [CrossRef] [PubMed]
- James, P.A.; Talbot, K. The molecular genetics of non-ALS motor neuron diseases. Biochim. Biophys. Acta 2006, 1762, 986–1000. [Google Scholar] [CrossRef] [Green Version]
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet 2009, 10, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Casterton, R.L.; Hunt, R.J.; Fanto, M. Pathomechanism Heterogeneity in the Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Disease Spectrum: Providing Focus Through the Lens of Autophagy. J. Mol. Biol. 2020, 432, 2692–2713. [Google Scholar] [CrossRef]
- Pasquini, J.; Trogu, F.; Morelli, C.; Poletti, B.; Girotti, F.; Peverelli, S.; Brusati, A.; Ratti, A.; Ciammola, A.; Silani, V.; et al. Parkinsonian Syndromes in Motor Neuron Disease: A Clinical Study. Front. Aging Neurosci. 2022, 14, 917706. [Google Scholar] [CrossRef]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef] [Green Version]
- Cykowski, M.D.; Powell, S.Z.; Appel, J.W.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Phosphorylated TDP-43 (pTDP-43) aggregates in the axial skeletal muscle of patients with sporadic and familial amyotrophic lateral sclerosis. Acta Neuropathol. Comm. 2018, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Onesto, E.; Rusmini, P.; Crippa, V.; Ferri, N.; Zito, A.; Galbiati, M.; Poletti, A. Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J. Neurochem. 2011, 118, 266–280. [Google Scholar] [CrossRef] [Green Version]
- Crippa, V.; Galbiati, M.; Boncoraglio, A.; Rusmini, P.; Onesto, E.; Giorgetti, E.; Cristofani, R.; Zito, A.; Poletti, A. Motoneuronal and muscle-selective removal of ALS-related misfolded proteins. Biochem. Soc. Trans. 2013, 41, 1598–1604. [Google Scholar] [CrossRef]
- Musaro, A. Understanding ALS: New therapeutic approaches. FEBS J. 2013, 280, 4315–4322. [Google Scholar] [CrossRef]
- Rusmini, P.; Polanco, M.J.; Cristofani, R.; Cicardi, M.E.; Meroni, M.; Galbiati, M.; Piccolella, M.; Messi, E.; Giorgetti, E.; Lieberman, A.P.; et al. Aberrant Autophagic Response in The Muscle of A Knock-in Mouse Model of Spinal and Bulbar Muscular Atrophy. Sci. Rep. 2015, 5, 15174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Halievski, K.; Henley, C.; Atchison, W.D.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Jordan, C.L. Defects in Neuromuscular Transmission May Underlie Motor Dysfunction in Spinal and Bulbar Muscular Atrophy. J. Neurosci. 2016, 36, 5094–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malacarne, C.; Galbiati, M.; Giagnorio, E.; Cavalcante, P.; Salerno, F.; Andreetta, F.; Cagnoli, C.; Taiana, M.; Nizzardo, M.; Corti, S.; et al. Dysregulation of Muscle-Specific MicroRNAs as Common Pathogenic Feature Associated with Muscle Atrophy in ALS, SMA and SBMA: Evidence from Animal Models and Human Patients. Int. J. Mol. Sci. 2021, 22, 5673. [Google Scholar] [CrossRef] [PubMed]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslesh, T.; Yokota, T. Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy. Cells 2022, 11, 417. [Google Scholar] [CrossRef]
- Chilcott, E.M.; Muiruri, E.W.; Hirst, T.C.; Yanez-Munoz, R.J. Systematic review and meta-analysis determining the benefits of in vivo genetic therapy in spinal muscular atrophy rodent models. Gene Ther. 2022, 29, 498–512. [Google Scholar] [CrossRef]
- Krokidis, M.G. Transcriptomics and Metabolomics in Amyotrophic Lateral Sclerosis. Adv. Exp. Med. Biol. 2020, 1195, 205–212. [Google Scholar]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.S., Jr.; Fraley, G.S.; Damian, V.; Woodke, L.B.; Zapata, F.; Sopher, B.L.; Plymate, S.R.; La Spada, A.R. Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2006, 15, 2225–2238. [Google Scholar] [CrossRef] [Green Version]
- Hauser, S.; Schuster, S.; Heuten, E.; Hoflinger, P.; Admard, J.; Schelling, Y.; Velic, A.; Macek, B.; Ossowski, S.; Schols, L. Comparative Transcriptional Profiling of Motor Neuron Disorder-Associated Genes in Various Human Cell Culture Models. Front. Cell Dev. Biol. 2020, 8, 544043. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Crippa, V.; Rusmini, P.; Boncoraglio, A.; Minoia, M.; Giorgetti, E.; Kampinga, H.H.; Poletti, A. Alteration of protein folding and degradation in motor neuron diseases: Implications and protective functions of small heat shock proteins. Progr. Neurobiol. 2012, 97, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Kanning, K.C.; Kaplan, A.; Henderson, C.E. Motor neuron diversity in development and disease. Annu. Rev. Neurosci. 2010, 33, 409–440. [Google Scholar] [CrossRef] [PubMed]
- Vegeto, E.; Villa, A.; Della Torre, S.; Crippa, V.; Rusmini, P.; Cristofani, R.; Galbiati, M.; Maggi, A.; Poletti, A. The Role of Sex and Sex Hormones in Neurodegenerative Diseases. Endocr. Rev. 2020, 41, 273–319. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Carra, S.; Rusmini, P.; Crippa, V.; Giorgetti, E.; Boncoraglio, A.; Cristofani, R.; Naujock, M.; Meister, M.; Minoia, M.; Kampinga, H.H.; et al. Different anti-aggregation and pro-degradative functions of the members of the mammalian sHSP family in neurological disorders. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20110409. [Google Scholar] [CrossRef] [Green Version]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Carra, S.; Alberti, S.; Benesch, J.L.P.; Boelens, W.; Buchner, J.; Carver, J.A.; Cecconi, C.; Ecroyd, H.; Gusev, N.; Hightower, L.E.; et al. Small heat shock proteins: Multifaceted proteins with important implications for life. Cell Stress Chaperones 2019, 24, 295–308. [Google Scholar] [CrossRef]
- Tedesco, B.; Ferrari, V.; Cozzi, M.; Chierichetti, M.; Casarotto, E.; Pramaggiore, P.; Mina, F.; Piccolella, M.; Cristofani, R.; Crippa, V.; et al. The role of autophagy-lysosomal pathway in motor neuron diseases. Biochem. Soc. Trans. 2022. [Google Scholar] [CrossRef]
- Tedesco, B.; Cristofani, R.; Ferrari, V.; Cozzi, M.; Rusmini, P.; Casarotto, E.; Chierichetti, M.; Mina, F.; Galbiati, M.; Piccolella, M.; et al. Insights on Human Small Heat Shock Proteins and Their Alterations in Diseases. Front. Mol. Biosci. 2022, 9, 842149. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Kirola, L.; Mukherjee, A.; Mutsuddi, M. Recent Updates on the Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Mol. Neurobiol. 2022, 59, 5673–5694. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Cicardi, M.E.; Marrone, L.; Azzouz, M.; Trotti, D. Proteostatic imbalance and protein spreading in amyotrophic lateral sclerosis. EMBO J. 2021, 40, e106389. [Google Scholar] [CrossRef]
- Mori, S.; Honda, H.; Hamasaki, H.; Sasagasako, N.; Suzuki, S.O.; Furuya, H.; Taniwaki, T.; Iwaki, T. Transactivation response DNA-binding protein of 43 kDa proteinopathy and lysosomal abnormalities in spastic paraplegia type 11. Neuropathology 2021, 41, 253–265. [Google Scholar] [CrossRef]
- Toupenet Marchesi, L.; Leblanc, M.; Stevanin, G. Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia. Cells 2021, 10, 1678. [Google Scholar] [CrossRef]
- Boutry, M.; Morais, S.; Stevanin, G. Update on the Genetics of Spastic Paraplegias. Curr. Neurol. Neurosci. Rep. 2019, 19, 18. [Google Scholar] [CrossRef]
- Panza, E.; Meyyazhagan, A.; Orlacchio, A. Hereditary spastic paraplegia: Genetic heterogeneity and common pathways. Exp. Neurol. 2022, 357, 114203. [Google Scholar] [CrossRef]
- Murphy, N.A.; Arthur, K.C.; Tienari, P.J.; Houlden, H.; Chio, A.; Traynor, B.J. Age-related penetrance of the C9orf72 repeat expansion. Sci. Rep. 2017, 7, 2116. [Google Scholar] [CrossRef] [Green Version]
- Dharmadasa, T.; Scaber, J.; Edmond, E.; Marsden, R.; Thompson, A.; Talbot, K.; Turner, M.R. Genetic testing in motor neurone disease. Pract. Neurol. 2022, 22, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Corrado, L.; Castellotti, B.; Del Bo, R.; Fogh, I.; Cereda, C.; Tiloca, C.; D’Ascenzo, C.; Bagarotti, A.; Pensato, V.; et al. C9ORF72 repeat expansion in a large Italian ALS cohort: Evidence of a founder effect. Neurobiol. Aging 2012, 33, 2528.e7–2528.e14. [Google Scholar] [CrossRef] [PubMed]
- Al Khleifat, A.; Iacoangeli, A.; van Vugt, J.; Bowles, H.; Moisse, M.; Zwamborn, R.A.J.; van der Spek, R.A.A.; Shatunov, A.; Cooper-Knock, J.; Topp, S.; et al. Structural variation analysis of 6,500 whole genome sequences in amyotrophic lateral sclerosis. NPJ Genom. Med. 2022, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Soto, C.; Estrada, L.; Castilla, J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 2006, 31, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Tanaka, F.; Robitschek, J.; Sandoval, C.M.; Taye, A.; Markovic-Plese, S.; Fischbeck, K.H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003, 12, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2021, 22, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H.; et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell 2017, 65, 1044–1055.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Simeoni, S.; Mancini, M.A.; Stenoien, D.L.; Marcelli, M.; Weigel, N.L.; Zanisi, M.; Martini, L.; Poletti, A. Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum. Mol. Genet. 2000, 9, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Amzallag, E.; Hornstein, E. Crosstalk between Biomolecular Condensates and Proteostasis. Cells 2022, 11, 2415. [Google Scholar] [CrossRef]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappe, G.; Franck, E.; Verschuure, P.; Boelens, W.C.; Leunissen, J.A.; de Jong, W.W. The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones 2003, 8, 53–61. [Google Scholar] [CrossRef]
- Lambert, H.; Charette, S.J.; Bernier, A.F.; Guimond, A.; Landry, J. HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J. Biol. Chem. 1999, 274, 9378–9385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobb, B.A.; Petrash, J.M. Characterization of alpha-crystallin-plasma membrane binding. J. Biol. Chem. 2000, 275, 6664–6672. [Google Scholar] [CrossRef] [Green Version]
- van Montfort, R.L.; Basha, E.; Friedrich, K.L.; Slingsby, C.; Vierling, E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 2001, 8, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Zavialov, A.; Benndorf, R.; Ehrnsperger, M.; Zav’yalov, V.; Dudich, I.; Buchner, J.; Gaestel, M. The effect of the intersubunit disulfide bond on the structural and functional properties of the small heat shock protein Hsp25. Int. J. Biol. Macromol. 1998, 22, 163–173. [Google Scholar] [CrossRef]
- Zavialov, A.V.; Gaestel, M.; Korpela, T.; Zav’yalov, V.P. Thiol/disulfide exchange between small heat shock protein 25 and glutathione. Biochim. Biophys. Acta 1998, 1388, 123–132. [Google Scholar] [CrossRef]
- Diaz-Latoud, C.; Buache, E.; Javouhey, E.; Arrigo, A.P. Substitution of the unique cysteine residue of murine Hsp25 interferes with the protective activity of this stress protein through inhibition of dimer formation. Antiox. Redox Signal 2005, 7, 436–445. [Google Scholar] [CrossRef]
- Zhang, H.; Rajasekaran, N.S.; Orosz, A.; Xiao, X.; Rechsteiner, M.; Benjamin, I.J. Selective degradation of aggregate-prone CryAB mutants by HSPB1 is mediated by ubiquitin-proteasome pathways. J. Mol. Cell Cardiol. 2010, 49, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Haidar, M.; Asselbergh, B.; Adriaenssens, E.; De Winter, V.; Timmermans, J.P.; Auer-Grumbach, M.; Juneja, M.; Timmerman, V. Neuropathy-causing mutations in HSPB1 impair autophagy by disturbing the formation of SQSTM1/p62 bodies. Autophagy 2019, 15, 1051–1068. [Google Scholar] [CrossRef] [PubMed]
- Bolhuis, S.; Richter-Landsberg, C. Effect of proteasome inhibition by MG-132 on HSP27 oligomerization, phosphorylation, and aggresome formation in the OLN-93 oligodendroglia cell line. J. Neurochem. 2010, 114, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Bryantsev, A.L.; Kurchashova, S.Y.; Golyshev, S.A.; Polyakov, V.Y.; Wunderink, H.F.; Kanon, B.; Budagova, K.R.; Kabakov, A.E.; Kampinga, H.H. Regulation of stress-induced intracellular sorting and chaperone function of Hsp27 (HspB1) in mammalian cells. Biochem J. 2007, 407, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2000, 2, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Paul, C.; Fromentin, A.; Hilpert, S.; Arrigo, A.P.; Solary, E.; Garrido, C. Differential regulation of HSP27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene 2000, 19, 4855–4863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, P.; Saleh, A.; Nakazawa, A.; Kumar, S.; Srinivasula, S.M.; Kumar, V.; Weichselbaum, R.; Nalin, C.; Alnemri, E.S.; Kufe, D.; et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000, 19, 4310–4322. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.; Simon, S.; Gibert, B.; Virot, S.; Manero, F.; Arrigo, A.P. Dynamic processes that reflect anti-apoptotic strategies set up by HspB1 (Hsp27). Exp. Cell Res. 2010, 316, 1535–1552. [Google Scholar] [CrossRef]
- Perng, M.D.; Cairns, L.; Van Den IJssel, P.; Prescott, A.; Hutcheson, A.M.; Quinlan, R.A. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J. Cell Sci. 1999, 112 Pt 13, 2099–2112. [Google Scholar] [CrossRef]
- Miron, T.; Vancompernolle, K.; Vandekerckhove, J.; Wilchek, M.; Geiger, B. A 25-kD inhibitor of actin polymerization is a low molecular mass heat shock protein. J. Cell Biol. 1991, 114, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Perng, M.D.; Muchowski, P.J.; van Den, I.P.; Wu, G.J.; Hutcheson, A.M.; Clark, J.I.; Quinlan, R.A. The cardiomyopathy and lens cataract mutation in alphaB-crystallin alters its protein structure, chaperone activity, and interaction with intermediate filaments in vitro. J. Biol. Chem. 1999, 274, 33235–33243. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, J.N.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J. Biol. Chem. 1993, 268, 24210–24214. [Google Scholar] [CrossRef]
- Lavoie, J.N.; Lambert, H.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol. Cell Biol. 1995, 15, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryantsev, A.L.; Loktionova, S.A.; Ilyinskaya, O.P.; Tararak, E.M.; Kampinga, H.H.; Kabakov, A.E. Distribution, phosphorylation, and activities of Hsp25 in heat-stressed H9c2 myoblasts: A functional link to cytoprotection. Cell Stress Chaperones 2002, 7, 146–155. [Google Scholar] [CrossRef]
- Katz, M.; Davis, M.; Garton, F.C.; Henderson, R.; Bharti, V.; Wray, N.; McCombe, P. Mutations in heat shock protein beta-1 (HSPB1) are associated with a range of clinical phenotypes related to different patterns of motor neuron dysfunction: A case series. J. Neurol. Sci. 2020, 413, 116809. [Google Scholar] [CrossRef]
- Capponi, S.; Geuens, T.; Geroldi, A.; Origone, P.; Verdiani, S.; Cichero, E.; Adriaenssens, E.; De Winter, V.; Bandettini di Poggio, M.; Barberis, M.; et al. Molecular Chaperones in the Pathogenesis of Amyotrophic Lateral Sclerosis: The Role of HSPB1. Hum. Mutat. 2016, 37, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.P.; Rubinsztein, D.C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 2002, 11, 1137–1151. [Google Scholar] [CrossRef]
- Zourlidou, A.; Payne Smith, M.D.; Latchman, D.S. HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J. Neurochem. 2004, 88, 1439–1448. [Google Scholar] [CrossRef]
- An, J.J.; Lee, Y.P.; Kim, D.W.; Sohn, E.J.; Jeong, H.J.; Kang, H.W.; Shin, M.J.; Kim, M.J.; Ahn, E.H.; Jang, S.H.; et al. Transduced HSP27 protein protects neuronal cell death by enhancing FALS-associated SOD1 mutant activity. BMB Rep. 2009, 42, 136–141. [Google Scholar] [CrossRef]
- Krishnan, J.; Lemmens, R.; Robberecht, W.; Van Den Bosch, L. Role of heat shock response and Hsp27 in mutant SOD1-dependent cell death. Exp. Neurol. 2006, 200, 301–310. [Google Scholar] [CrossRef]
- Krishnan, J.; Vannuvel, K.; Andries, M.; Waelkens, E.; Robberecht, W.; Van Den Bosch, L. Over-expression of Hsp27 does not influence disease in the mutant SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2008, 106, 2170–2183. [Google Scholar]
- Vleminckx, V.; Van Damme, P.; Goffin, K.; Delye, H.; Van Den Bosch, L.; Robberecht, W. Upregulation of HSP27 in a transgenic model of ALS. J. Neuropathol. Exp. Neurol. 2002, 61, 968–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilman, P.L.; Song, S.; Miranda, C.J.; Meyer, K.; Srivastava, A.K.; Knapp, A.; Wier, C.G.; Kaspar, B.K.; Kolb, S.J. HSPB1 mutations causing hereditary neuropathy in humans disrupt non-cell autonomous protection of motor neurons. Exp. Neurol. 2017, 297, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Hu, J.; Arogundade, O.A.; Goginashvili, A.; Vazquez-Sanchez, S.; Diedrich, J.K.; Gu, J.; Blum, J.; Oung, S.; Ye, Q.; et al. Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat. Cell Biol. 2022, 24, 1378–1393. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, S.; Gu, J.; Tong, Y.; Li, Y.; Gui, X.; Long, H.; Wang, C.; Zhao, C.; Lu, J.; et al. Hsp27 chaperones FUS phase separation under the modulation of stress-induced phosphorylation. Nat. Struct. Mol. Biol. 2020, 27, 363–372. [Google Scholar] [CrossRef]
- Mo, K.; Razak, Z.; Rao, P.; Yu, Z.; Adachi, H.; Katsuno, M.; Sobue, G.; Lieberman, A.P.; Westwood, J.T.; Monks, D.A. Microarray analysis of gene expression by skeletal muscle of three mouse models of Kennedy disease/spinal bulbar muscular atrophy. PLoS ONE 2010, 5, e12922. [Google Scholar] [CrossRef]
- Chen, C.M.; Chen, W.L.; Hung, C.T.; Lin, T.H.; Chao, C.Y.; Lin, C.H.; Wu, Y.R.; Chang, K.H.; Yao, C.F.; Lee-Chen, G.J.; et al. The indole compound NC009-1 inhibits aggregation and promotes neurite outgrowth through enhancement of HSPB1 in SCA17 cells and ameliorates the behavioral deficits in SCA17 mice. Neurotoxicology 2018, 67, 259–269. [Google Scholar] [CrossRef]
- Chang, K.H.; Lin, C.H.; Chen, H.C.; Huang, H.Y.; Chen, S.L.; Lin, T.H.; Ramesh, C.; Huang, C.C.; Fung, H.C.; Wu, Y.R.; et al. The Potential of Indole/Indolylquinoline Compounds in Tau Misfolding Reduction by Enhancement of HSPB1. CNS Neurosci. Ther. 2017, 23, 45–56. [Google Scholar] [CrossRef]
- Iwaki, A.; Nagano, T.; Nakagawa, M.; Iwaki, T.; Fukumaki, Y. Identification and characterization of the gene encoding a new member of the alpha-crystallin/small hsp family, closely linked to the alphaB-crystallin gene in a head-to-head manner. Genomics 1997, 45, 386–394. [Google Scholar] [CrossRef]
- Suzuki, A.; Sugiyama, Y.; Hayashi, Y.; Nyu-i, N.; Yoshida, M.; Nonaka, I.; Ishiura, S.; Arahata, K.; Ohno, S. MKBP, a novel member of the small heat shock protein family, binds and activates the myotonic dystrophy protein kinase. J. Cell Biol. 1998, 140, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, M.; Tsujimoto, N.; Nakagawa, H.; Iwaki, T.; Fukumaki, Y.; Iwaki, A. Association of HSPB2, a member of the small heat shock protein family, with mitochondria. Exp. Cell Res. 2001, 271, 161–168. [Google Scholar] [CrossRef]
- Shama, K.M.; Suzuki, A.; Harada, K.; Fujitani, N.; Kimura, H.; Ohno, S.; Yoshida, K. Transient up-regulation of myotonic dystrophy protein kinase-binding protein, MKBP, and HSP27 in the neonatal myocardium. Cell Struct. Funct. 1999, 24, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschuure, P.; Croes, Y.; van den IJssel, P.R.; Quinlan, R.A.; de Jong, W.W.; Boelens, W.C. Translocation of small heat shock proteins to the actin cytoskeleton upon proteasomal inhibition. J. Mol. Cell Cardiol. 2002, 34, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Morelli, F.F.; Mediani, L.; Heldens, L.; Bertacchini, J.; Bigi, I.; Carra, A.D.; Vinet, J.; Carra, S. An interaction study in mammalian cells demonstrates weak binding of HSPB2 to BAG3, which is regulated by HSPB3 and abrogated by HSPB8. Cell Stress Chaperones 2017, 22, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, Y.; Suzuki, A.; Kishikawa, M.; Akutsu, R.; Hirose, T.; Waye, M.M.; Tsui, S.K.; Yoshida, S.; Ohno, S. Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J. Biol. Chem. 2000, 275, 1095–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Engelsman, J.; Boros, S.; Dankers, P.Y.; Kamps, B.; Vree Egberts, W.T.; Bode, C.S.; Lane, L.A.; Aquilina, J.A.; Benesch, J.L.; Robinson, C.V.; et al. The small heat-shock proteins HSPB2 and HSPB3 form well-defined heterooligomers in a unique 3 to 1 subunit ratio. J. Mol. Biol. 2009, 393, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, M.M.; Boelens, W.C.; Otte-Holler, I.; Kamps, B.; de Waal, R.M.; Verbeek, M.M. Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity. Brain Res. 2006, 1089, 67–78. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Otte-Holler, I.; Wesseling, P.; de Waal, R.M.; Boelens, W.C.; Verbeek, M.M. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 2006, 32, 119–130. [Google Scholar] [CrossRef]
- Kim, N.; Yu, L.; Dawe, R.; Petyuk, V.A.; Gaiteri, C.; De Jager, P.L.; Schneider, J.A.; Arfanakis, K.; Bennett, D.A. Microstructural changes in the brain mediate the association of AK4, IGFBP5, HSPB2, and ITPK1 with cognitive decline. Neurobiol. Aging 2019, 84, 17–25. [Google Scholar] [CrossRef]
- Lam, W.Y.; Wing Tsui, S.K.; Law, P.T.; Luk, S.C.; Fung, K.P.; Lee, C.Y.; Waye, M.M. Isolation and characterization of a human heart cDNA encoding a new member of the small heat shock protein family--HSPL27. Biochim. Biophys. Acta 1996, 1314, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Boelens, W.C.; Croes, Y.; de Ruwe, M.; de Reu, L.; de Jong, W.W. Negative charges in the C-terminal domain stabilize the alphaB-crystallin complex. J. Biol. Chem. 1998, 273, 28085–28090. [Google Scholar] [CrossRef] [Green Version]
- Boelens, W.C.; Van Boekel, M.A.; De Jong, W.W. HspB3, the most deviating of the six known human small heat shock proteins. Biochim. Biophys. Acta 1998, 1388, 513–516. [Google Scholar] [CrossRef]
- Asthana, A.; Raman, B.; Ramakrishna, T.; Rao Ch, M. Structural aspects and chaperone activity of human HspB3: Role of the “C-terminal extension”. Cell Biochem. Biophys. 2012, 64, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Morelli, F.F.; Verbeek, D.S.; Bertacchini, J.; Vinet, J.; Mediani, L.; Marmiroli, S.; Cenacchi, G.; Nasi, M.; De Biasi, S.; Brunsting, J.F.; et al. Aberrant Compartment Formation by HSPB2 Mislocalizes Lamin A and Compromises Nuclear Integrity and Function. Cell Rep. 2017, 20, 2100–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Padula, V.; Staszewski, O.; Nestel, S.; Busch, H.; Boerries, M.; Roussa, E.; Prinz, M.; Krieglstein, K. HSPB3 protein is expressed in motoneurons and induces their survival after lesion-induced degeneration. Exp. Neurol. 2016, 286, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Tiago, T.; Hummel, B.; Morelli, F.F.; Basile, V.; Vinet, J.; Galli, V.; Mediani, L.; Antoniani, F.; Pomella, S.; Cassandri, M.; et al. Small heat-shock protein HSPB3 promotes myogenesis by regulating the lamin B receptor. Cell Death Dis. 2021, 12, 452. [Google Scholar] [CrossRef]
- Kolb, S.J.; Snyder, P.J.; Poi, E.J.; Renard, E.A.; Bartlett, A.; Gu, S.; Sutton, S.; Arnold, W.D.; Freimer, M.L.; Lawson, V.H.; et al. Mutant small heat shock protein B3 causes motor neuropathy: Utility of a candidate gene approach. Neurology 2010, 74, 502–506. [Google Scholar] [CrossRef]
- Lassuthova, P.; Safka Brozkova, D.; Krutova, M.; Mazanec, R.; Zuchner, S.; Gonzalez, M.A.; Seeman, P. Severe axonal Charcot-Marie-Tooth disease with proximal weakness caused by de novo mutation in the MORC2 gene. Brain 2016, 139 Pt 4, e26. [Google Scholar] [CrossRef] [Green Version]
- Bassnett, S. On the mechanism of organelle degradation in the vertebrate lens. Exp. Eye Res. 2009, 88, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Bassnett, S.; Wilmarth, P.A.; David, L.L. The membrane proteome of the mouse lens fiber cell. Mol. Vis. 2009, 15, 2448–2463. [Google Scholar]
- Horwitz, J. Alpha-crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. USA 1992, 89, 10449–10453. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andley, U.P. Crystallins in the eye: Function and pathology. Prog. Retin. Eye Res. 2007, 26, 78–98. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, P.J.; MacElroy, K.S.; Rosser, M.P.; Church, R.L. Association of alpha-crystallin with actin in cultured lens cells. Curr. Eye Res. 1984, 3, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Maddala, R.; Rao, V.P. alpha-Crystallin localizes to the leading edges of migrating lens epithelial cells. Exp. Cell Res. 2005, 306, 203–215. [Google Scholar] [CrossRef]
- Andley, U.P.; Song, Z.; Wawrousek, E.F.; Bassnett, S. The molecular chaperone alphaA-crystallin enhances lens epithelial cell growth and resistance to UVA stress. J. Biol. Chem. 1998, 273, 31252–31261. [Google Scholar] [CrossRef] [Green Version]
- Golenhofen, N.; Htun, P.; Ness, W.; Koob, R.; Schaper, W.; Drenckhahn, D. Binding of the stress protein alpha B-crystallin to cardiac myofibrils correlates with the degree of myocardial damage during ischemia/reperfusion in vivo. J. Mol. Cell Cardiol. 1999, 31, 569–580. [Google Scholar] [CrossRef]
- Bullard, B.; Ferguson, C.; Minajeva, A.; Leake, M.C.; Gautel, M.; Labeit, D.; Ding, L.; Labeit, S.; Horwitz, J.; Leonard, K.R.; et al. Association of the chaperone alphaB-crystallin with titin in heart muscle. J. Biol. Chem. 2004, 279, 7917–7924. [Google Scholar] [CrossRef] [Green Version]
- Doran, P.; Gannon, J.; O’Connell, K.; Ohlendieck, K. Aging skeletal muscle shows a drastic increase in the small heat shock proteins alphaB-crystallin/HspB5 and cvHsp/HspB7. Eur. J. Cell Biol. 2007, 86, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Bajramovic, J.J.; Bsibsi, M.; Geutskens, S.B.; Hassankhan, R.; Verhulst, K.C.; Stege, G.J.; de Groot, C.J.; van Noort, J.M. Differential expression of stress proteins in human adult astrocytes in response to cytokines. J. Neuroimmunol. 2000, 106, 14–22. [Google Scholar] [CrossRef]
- Bajramovic, J.J.; Plomp, A.C.; Goes, A.; Koevoets, C.; Newcombe, J.; Cuzner, M.L.; van Noort, J.M. Presentation of alpha B-crystallin to T cells in active multiple sclerosis lesions: An early event following inflammatory demyelination. J. Immunol. 2000, 164, 4359–4366. [Google Scholar] [CrossRef] [Green Version]
- Ousman, S.S.; Tomooka, B.H.; van Noort, J.M.; Wawrousek, E.F.; O’Connor, K.C.; Hafler, D.A.; Sobel, R.A.; Robinson, W.H.; Steinman, L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 2007, 448, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L.; Boelens, W.C.; Wawrousek, E.F.; Messing, A. Suppression of GFAP toxicity by alphaB-crystallin in mouse models of Alexander disease. Hum. Mol. Genet. 2009, 18, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.O.; Osmand, A.; Outeiro, T.F.; Muchowski, P.J.; Finkbeiner, S. alphaB-Crystallin overexpression in astrocytes modulates the phenotype of the BACHD mouse model of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Gorter, R.P.; Stephenson, J.; Nutma, E.; Anink, J.; de Jonge, J.C.; Baron, W.; Jahreibeta, M.C.; Belien, J.A.M.; van Noort, J.M.; Mijnsbergen, C.; et al. Rapidly progressive amyotrophic lateral sclerosis is associated with microglial reactivity and small heat shock protein expression in reactive astrocytes. Neuropathol. Appl. Neurobiol. 2019, 45, 459–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, D.W.; Amor, S.; Story, D.; Torvell, M.; Bsibsi, M.; van Noort, J.M.; Chandran, S. HspB5 Activates a Neuroprotective Glial Cell Response in Experimental Tauopathy. Front. Neurosci. 2020, 14, 574. [Google Scholar] [CrossRef] [PubMed]
- Pras, E.; Frydman, M.; Levy-Nissenbaum, E.; Bakhan, T.; Raz, J.; Assia, E.I.; Goldman, B.; Pras, E. A nonsense mutation (W9X) in CRYAA causes autosomal recessive cataract in an inbred Jewish Persian family. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3511–3515. [Google Scholar] [PubMed]
- Mackay, D.S.; Andley, U.P.; Shiels, A. Cell death triggered by a novel mutation in the alphaA-crystallin gene underlies autosomal dominant cataract linked to chromosome 21q. Eur. J. Hum. Genet. 2003, 11, 784–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhiya, S.T.; Soker, T.; Klopp, N.; Illig, T.; Prakash, M.V.; Selvaraj, B.; Gopinath, P.M.; Graw, J. Identification of a novel, putative cataract-causing allele in CRYAA (G98R) in an Indian family. Mol. Vis. 2006, 12, 768–773. [Google Scholar]
- Andley, U.P. The lens epithelium: Focus on the expression and function of the alpha-crystallin chaperones. Int. J. Biochem. Cell Biol. 2008, 40, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.L.; Chen, J.; Wang, Y.P.; Zheng, H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011, 7, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Khoshaman, K.; Yousefi, R.; Tamaddon, A.M.; Abolmaali, S.S.; Oryan, A.; Moosavi-Movahedi, A.A.; Kurganov, B.I. The impact of different mutations at Arg54 on structure, chaperone-like activity and oligomerization state of human alphaA-crystallin: The pathomechanism underlying congenital cataract-causing mutations R54L, R54P and R54C. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 604–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoshaman, K.; Yousefi, R.; Moosavi-Movahedi, A.A. Protective role of antioxidant compounds against peroxynitrite-mediated modification of R54C mutant alphaA-crystallin. Arch Biochem. Biophys. 2017, 629, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Raju, I.; Abraham, E.C. Congenital cataract causing mutants of alphaA-crystallin/sHSP form aggregates and aggresomes degraded through ubiquitin-proteasome pathway. PLoS ONE 2011, 6, e28085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raju, I.; Abraham, E.C. Mutants of human alphaB-crystallin cause enhanced protein aggregation and apoptosis in mammalian cells: Influence of co-expression of HspB1. Biochem. Biophys. Res. Comm. 2013, 430, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Ghahramani, M.; Yousefi, R.; Krivandin, A.; Muranov, K.; Kurganov, B.; Moosavi-Movahedi, A.A. Structural and functional characterization of D109H and R69C mutant versions of human alphaB-crystallin: The biochemical pathomechanism underlying cataract and myopathy development. Int. J. Biol. Macromol. 2020, 146, 1142–1160. [Google Scholar] [CrossRef]
- Waudby, C.A.; Knowles, T.P.; Devlin, G.L.; Skepper, J.N.; Ecroyd, H.; Carver, J.A.; Welland, M.E.; Christodoulou, J.; Dobson, C.M.; Meehan, S. The interaction of alphaB-crystallin with mature alpha-synuclein amyloid fibrils inhibits their elongation. Biophys J. 2010, 98, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Shammas, S.L.; Waudby, C.A.; Wang, S.; Buell, A.K.; Knowles, T.P.; Ecroyd, H.; Welland, M.E.; Carver, J.A.; Dobson, C.M.; Meehan, S. Binding of the molecular chaperone alphaB-crystallin to Abeta amyloid fibrils inhibits fibril elongation. Biophys J. 2011, 101, 1681–1689. [Google Scholar] [CrossRef] [Green Version]
- Yerbury, J.J.; Gower, D.; Vanags, L.; Roberts, K.; Lee, J.A.; Ecroyd, H. The small heat shock proteins alphaB-crystallin and Hsp27 suppress SOD1 aggregation in vitro. Cell Stress Chaperones 2013, 18, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Ecroyd, H. The small heat shock proteins alphaB-crystallin (HSPB5) and Hsp27 (HSPB1) inhibit the intracellular aggregation of alpha-synuclein. Cell Stress Chaperones 2017, 22, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu, G.; Li, H.; Gonzales, V.; Fromholt, D.; Karch, C.; Copeland, N.G.; Jenkins, N.A.; Borchelt, D.R. Somatodendritic accumulation of misfolded SOD1-L126Z in motor neurons mediates degeneration: AlphaB-crystallin modulates aggregation. Hum. Mol. Genet. 2005, 14, 2335–2347. [Google Scholar] [CrossRef] [Green Version]
- Karch, C.M.; Borchelt, D.R. An examination of alpha B-crystallin as a modifier of SOD1 aggregate pathology and toxicity in models of familial amyotrophic lateral sclerosis. J. Neurochem. 2010, 113, 1092–1100. [Google Scholar] [PubMed] [Green Version]
- Xu, G.; Fromholt, S.; Ayers, J.I.; Brown, H.; Siemienski, Z.; Crosby, K.W.; Mayer, C.A.; Janus, C.; Borchelt, D.R. Substantially elevating the levels of alphaB-crystallin in spinal motor neurons of mutant SOD1 mice does not significantly delay paralysis or attenuate mutant protein aggregation. J. Neurochem. 2015, 133, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Goto, S.; Inaguma, Y.; Hasegawa, K.; Morishita, R.; Asano, T. Purification and characterization of a 20-kDa protein that is highly homologous to alpha B crystallin. J. Biol. Chem. 1994, 269, 15302–15309. [Google Scholar] [CrossRef]
- Brophy, C.M.; Dickinson, M.; Woodrum, D. Phosphorylation of the small heat shock-related protein, HSP20, in vascular smooth muscles is associated with changes in the macromolecular associations of HSP20. J. Biol. Chem. 1999, 274, 6324–6329. [Google Scholar] [CrossRef]
- van de Klundert, F.A.; Gijsen, M.L.; van den IJssel, P.R.; Snoeckx, L.H.; de Jong, W.W. alpha B-crystallin and hsp25 in neonatal cardiac cells--differences in cellular localization under stress conditions. Eur. J. Cell Biol. 1998, 75, 38–45. [Google Scholar] [CrossRef]
- van de Klundert, F.A.; Smulders, R.H.; Gijsen, M.L.; Lindner, R.A.; Jaenicke, R.; Carver, J.A.; de Jong, W.W. The mammalian small heat-shock protein Hsp20 forms dimers and is a poor chaperone. Eur. J. Biochem. 1998, 258, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- Bukach, O.V.; Seit-Nebi, A.S.; Marston, S.B.; Gusev, N.B. Some properties of human small heat shock protein Hsp20 (HspB6). Eur. J. Biochem. 2004, 271, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Delbecq, S.P.; Rosenbaum, J.C.; Klevit, R.E. A Mechanism of Subunit Recruitment in Human Small Heat Shock Protein Oligomers. Biochemistry 2015, 54, 4276–4284. [Google Scholar] [CrossRef] [Green Version]
- Bukach, O.V.; Glukhova, A.E.; Seit-Nebi, A.S.; Gusev, N.B. Heterooligomeric complexes formed by human small heat shock proteins HspB1 (Hsp27) and HspB6 (Hsp20). Biochim. Biophys. Acta 2009, 1794, 486–495. [Google Scholar] [CrossRef]
- Heirbaut, M.; Lermyte, F.; Martin, E.M.; Beelen, S.; Verschueren, T.; Sobott, F.; Strelkov, S.V.; Weeks, S.D. The preferential heterodimerization of human small heat shock proteins HSPB1 and HSPB6 is dictated by the N-terminal domain. Arch. Biochem. Biophys. 2016, 610, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Mymrikov, E.V.; Riedl, M.; Peters, C.; Weinkauf, S.; Haslbeck, M.; Buchner, J. Regulation of small heat-shock proteins by hetero-oligomer formation. J. Biol. Chem. 2020, 295, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Shatov, V.M.; Gusev, N.B. Physico-chemical properties of two point mutants of small heat shock protein HspB6 (Hsp20) with abrogated cardioprotection. Biochimie 2020, 174, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Shatov, V.M.; Strelkov, S.V.; Gusev, N.B. The Heterooligomerization of Human Small Heat Shock Proteins Is Controlled by Conserved Motif Located in the N-Terminal Domain. Int. J. Mol. Sci. 2020, 21, 4248. [Google Scholar] [CrossRef] [PubMed]
- Muranova, L.K.; Shatov, V.M.; Bukach, O.V.; Gusev, N.B. Cardio-Vascular Heat Shock Protein (cvHsp, HspB7), an Unusual Representative of Small Heat Shock Protein Family. Biochemistry 2021, 86 (Suppl. 1), S1–S11. [Google Scholar] [CrossRef]
- Muranova, L.K.; Shatov, V.M.; Slushchev, A.V.; Gusev, N.B. Quaternary Structure and Hetero-Oligomerization of Recombinant Human Small Heat Shock Protein HspB7 (cvHsp). Int. J. Mol. Sci. 2021, 22, 7777. [Google Scholar] [CrossRef]
- Fuchs, M.; Luthold, C.; Guilbert, S.M.; Varlet, A.A.; Lambert, H.; Jette, A.; Elowe, S.; Landry, J.; Lavoie, J.N. A Role for the Chaperone Complex BAG3-HSPB8 in Actin Dynamics, Spindle Orientation and Proper Chromosome Segregation during Mitosis. PLoS Genet. 2015, 11, e1005582. [Google Scholar] [CrossRef]
- Fuchs, M.; Poirier, D.J.; Seguin, S.J.; Lambert, H.; Carra, S.; Charette, S.J.; Landry, J. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem J. 2009, 425, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Shemetov, A.A.; Gusev, N.B. Biochemical characterization of small heat shock protein HspB8 (Hsp22)-Bag3 interaction. Arch Biochem. Biophys. 2011, 513, 1–9. [Google Scholar] [CrossRef]
- Rauch, J.N.; Tse, E.; Freilich, R.; Mok, S.A.; Makley, L.N.; Southworth, D.R.; Gestwicki, J.E. BAG3 Is a Modular, Scaffolding Protein that physically Links Heat Shock Protein 70 (Hsp70) to the Small Heat Shock Proteins. J. Mol. Biol. 2017, 429, 128–141. [Google Scholar] [CrossRef] [Green Version]
- Fan, G.H.; Qi, C.; Chen, S.D. Heat shock proteins reduce toxicity of 1-methyl-4-phenylpyridinium ion in SK-N-SH cells. J. Neurosci. Res. 2005, 82, 551–562. [Google Scholar] [CrossRef]
- Komalavilas, P.; Penn, R.B.; Flynn, C.R.; Thresher, J.; Lopes, L.B.; Furnish, E.J.; Guo, M.; Pallero, M.A.; Murphy-Ullrich, J.E.; Brophy, C.M. The small heat shock-related protein, HSP20, is a cAMP-dependent protein kinase substrate that is involved in airway smooth muscle relaxation. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L69–L78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, E.K.; Macintyre, D.A.; Smith, R.; Chan, E.C.; Read, M. Evidence that a protein kinase A substrate, small heat-shock protein 20, modulates myometrial relaxation in human pregnancy. Endocrinology 2008, 149, 6157–6165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karolczak-Bayatti, M.; Sweeney, M.; Cheng, J.; Edey, L.; Robson, S.C.; Ulrich, S.M.; Treumann, A.; Taggart, M.J.; Europe-Finner, G.N. Acetylation of heat shock protein 20 (Hsp20) regulates human myometrial activity. J. Biol. Chem. 2011, 286, 34346–34355. [Google Scholar] [CrossRef] [Green Version]
- Rembold, C.M.; Foster, D.B.; Strauss, J.D.; Wingard, C.J.; Eyk, J.E. cGMP-mediated phosphorylation of heat shock protein 20 may cause smooth muscle relaxation without myosin light chain dephosphorylation in swine carotid artery. J. Physiol. 2000, 524 Pt 3, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Rembold, C.M.; O’Connor, M. Caldesmon and heat shock protein 20 phosphorylation in nitroglycerin- and magnesium-induced relaxation of swine carotid artery. Biochim. Biophys. Acta 2000, 1500, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Islamovic, E.; Duncan, A.; Bers, D.M.; Gerthoffer, W.T.; Mestril, R. Importance of small heat shock protein 20 (hsp20) C-terminal extension in cardioprotection. J. Mol. Cell Cardiol. 2007, 42, 862–869. [Google Scholar] [CrossRef]
- Nicolaou, P.; Knoll, R.; Haghighi, K.; Fan, G.C.; Dorn, G.W., 2nd; Hasenfub, G.; Kranias, E.G. Human mutation in the anti-apoptotic heat shock protein 20 abrogates its cardioprotective effects. J. Biol. Chem. 2008, 283, 33465–33471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.S.; Zhu, H.; Cai, W.F.; Wang, X.; Jiang, M.; Essandoh, K.; Vafiadaki, E.; Haghighi, K.; Lam, C.K.; Gardner, G.; et al. Regulation of BECN1-mediated autophagy by HSPB6: Insights from a human HSPB6(S10F) mutant. Autophagy 2018, 14, 80–97. [Google Scholar] [CrossRef] [Green Version]
- Verschuure, P.; Tatard, C.; Boelens, W.C.; Grongnet, J.F.; David, J.C. Expression of small heat shock proteins HspB2, HspB8, Hsp20 and cvHsp in different tissues of the perinatal developing pig. Eur. J. Cell Biol. 2003, 82, 523–530. [Google Scholar] [CrossRef]
- Bartelt-Kirbach, B.; Golenhofen, N. Reaction of small heat-shock proteins to different kinds of cellular stress in cultured rat hippocampal neurons. Cell Stress Chaperones 2014, 19, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Gortz, A.L.; Peferoen, L.A.N.; Gerritsen, W.H.; van Noort, J.M.; Bugiani, M.; Amor, S. Heat shock protein expression in cerebral X-linked adrenoleukodystrophy reveals astrocyte stress prior to myelin loss. Neuropathol. Appl. Neurobiol. 2018, 44, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Peferoen, L.A.; Gerritsen, W.H.; Breur, M.; Ummenthum, K.M.; Peferoen-Baert, R.M.; van der Valk, P.; van Noort, J.M.; Amor, S. Small heat shock proteins are induced during multiple sclerosis lesion development in white but not grey matter. Acta Neuropathol. Comm. 2015, 3, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, A.Q.; Zhang, Y.H.; Qi, Q.D.; Liu, Y.H.; Zhu, J.L. Overexpressed HspB6 Underlines a Novel Inhibitory Role in Kainic Acid-Induced Epileptic Seizure in Rats by Activating the cAMP-PKA Pathway. Cell Mol. Neurobiol. 2019, 39, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Carson, K.; Rice-Ficht, A.; Good, T. Hsp20, a novel alpha-crystallin, prevents Abeta fibril formation and toxicity. Protein Sci. 2005, 14, 593–601. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Boelens, W.C.; Otte-Holler, I.; Kamps, B.; Kusters, B.; Maat-Schieman, M.L.; de Waal, R.M.; Verbeek, M.M. Small heat shock protein HspB8: Its distribution in Alzheimer’s disease brains and its inhibition of amyloid-beta protein aggregation and cerebrovascular amyloid-beta toxicity. Acta Neuropathol. 2006, 111, 139–149. [Google Scholar] [CrossRef]
- Bruinsma, I.B.; Bruggink, K.A.; Kinast, K.; Versleijen, A.A.; Segers-Nolten, I.M.; Subramaniam, V.; Kuiperij, H.B.; Boelens, W.; de Waal, R.M.; Verbeek, M.M. Inhibition of alpha-synuclein aggregation by small heat shock proteins. Proteins 2011, 79, 2956–2967. [Google Scholar] [CrossRef]
- Krief, S.; Faivre, J.F.; Robert, P.; Le Douarin, B.; Brument-Larignon, N.; Lefrere, I.; Bouzyk, M.M.; Anderson, K.M.; Greller, L.D.; Tobin, F.L.; et al. Identification and characterization of cvHsp. A novel human small stress protein selectively expressed in cardiovascular and insulin-sensitive tissues. J. Biol. Chem. 1999, 274, 36592–36600. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Fontaine, J.M.; Rest, J.S.; Shelden, E.A.; Welsh, M.J.; Benndorf, R. Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J. Biol. Chem. 2004, 279, 2394–2402. [Google Scholar] [CrossRef] [Green Version]
- Vos, M.J.; Zijlstra, M.P.; Kanon, B.; van Waarde-Verhagen, M.A.; Brunt, E.R.; Oosterveld-Hut, H.M.; Carra, S.; Sibon, O.C.; Kampinga, H.H. HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet. 2010, 19, 4677–4693. [Google Scholar] [CrossRef] [Green Version]
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: Implications for a proteasome-to-autophagy switch. Autophagy 2014, 10, 1603–1621. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Vonk, J.J.; Salles, F.; Vonk, D.; Haslbeck, M.; Melki, R.; Bergink, S.; Kampinga, H.H. The N terminus of the small heat shock protein HSPB7 drives its polyQ aggregation-suppressing activity. J. Biol. Chem. 2019, 294, 9985–9994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juo, L.Y.; Liao, W.C.; Shih, Y.L.; Yang, B.Y.; Liu, A.B.; Yan, Y.T. HSPB7 interacts with dimerized FLNC and its absence results in progressive myopathy in skeletal muscles. J. Cell Sci. 2016, 129, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.C.; Juo, L.Y.; Shih, Y.L.; Chen, Y.H.; Yan, Y.T. HSPB7 prevents cardiac conduction system defect through maintaining intercalated disc integrity. PLoS Genet. 2017, 13, e1006984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.S.; Chang, Y.H.; Pan, C.C. High expression of heat shock proteins and heat shock factor-1 distinguishes an aggressive subset of clear cell renal cell carcinoma. Histopathology 2017, 71, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Mercer, E.J.; Lin, Y.F.; Cohen-Gould, L.; Evans, T. Hspb7 is a cardioprotective chaperone facilitating sarcomeric proteostasis. Dev. Biol. 2018, 435, 41–55. [Google Scholar] [CrossRef]
- Carra, S.; Seguin, S.J.; Landry, J. HspB8 and Bag3: A new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 2008, 4, 237–239. [Google Scholar] [CrossRef] [Green Version]
- Crippa, V.; Carra, S.; Rusmini, P.; Sau, D.; Bolzoni, E.; Bendotti, C.; De Biasi, S.; Poletti, A. A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy 2010, 6, 958–960. [Google Scholar] [CrossRef] [Green Version]
- Crippa, V.; Sau, D.; Rusmini, P.; Boncoraglio, A.; Onesto, E.; Bolzoni, E.; Galbiati, M.; Fontana, E.; Marino, M.; Carra, S.; et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 2010, 19, 3440–3456. [Google Scholar] [CrossRef] [Green Version]
- Cristofani, R.; Crippa, V.; Rusmini, P.; Cicardi, M.E.; Meroni, M.; Licata, N.V.; Sala, G.; Giorgetti, E.; Grunseich, C.; Galbiati, M.; et al. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 2017, 13, 1280–1303. [Google Scholar] [CrossRef]
- Cristofani, R.; Crippa, V.; Cicardi, M.E.; Tedesco, B.; Ferrari, V.; Chierichetti, M.; Casarotto, E.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. A Crucial Role for the Protein Quality Control System in Motor Neuron Diseases. Front. Aging Neurosci. 2020, 12, 191. [Google Scholar] [CrossRef]
- Cristofani, R.; Piccolella, M.; Crippa, V.; Tedesco, B.; Montagnani Marelli, M.; Poletti, A.; Moretti, R.M. The Role of HSPB8, a Component of the Chaperone-Assisted Selective Autophagy Machinery, in Cancer. Cells 2021, 10, 335. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Xiao, M.; Li, W.; Sun, X.; Bai, Y.; Meng, F.; Zhu, Z.; Yuan, W.; Sun, K. SHP2 Inhibitors Show Anti-Myeloma Activity and Synergize With Bortezomib in the Treatment of Multiple Myeloma. Front. Pharmacol. 2022, 13, 841308. [Google Scholar] [CrossRef] [PubMed]
- Yew, E.H.; Cheung, N.S.; Choy, M.S.; Qi, R.Z.; Lee, A.Y.; Peng, Z.F.; Melendez, A.J.; Manikandan, J.; Koay, E.S.; Chiu, L.L.; et al. Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: A microarray analysis. J. Neurochem. 2005, 94, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to Autophagy and Proteasome Ameliorates its Aggregation in Amyotrophic Lateral Sclerosis Target Cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef] [PubMed]
- Crippa, V.; Cicardi, M.E.; Ramesh, N.; Seguin, S.J.; Ganassi, M.; Bigi, I.; Diacci, C.; Zelotti, E.; Baratashvili, M.; Gregory, J.M.; et al. The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity. Hum. Mol. Genet. 2016, 25, 3908–3924. [Google Scholar] [CrossRef]
- Crippa, V.; D’Agostino, V.G.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Loffredo, R.; Pancher, M.; Piccolella, M.; Galbiati, M.; et al. Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 2016, 6, 22827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofani, R.; Crippa, V.; Vezzoli, G.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Tedesco, B.; Piccolella, M.; et al. The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones 2018, 23, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, G.; Akbar, M.T.; Paul, P.; Angelinetta, C.; Steiner, T.J.; de Belleroche, J. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 2010, 31, 969–985. [Google Scholar] [CrossRef]
- Marino, M.; Papa, S.; Crippa, V.; Nardo, G.; Peviani, M.; Cheroni, C.; Trolese, M.C.; Lauranzano, E.; Bonetto, V.; Poletti, A.; et al. Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 492–504. [Google Scholar] [CrossRef]
- Crippa, V.; Boncoraglio, A.; Galbiati, M.; Aggarwal, T.; Rusmini, P.; Giorgetti, E.; Cristofani, R.; Carra, S.; Pennuto, M.; Poletti, A. Differential autophagy power in the spinal cord and muscle of transgenic ALS mice. Front. Cell Neurosci. 2013, 7, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez Zobel, A.T.; Loranger, A.; Marceau, N.; Theriault, J.R.; Lambert, H.; Landry, J. Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G alphaB-crystallin mutant. Hum. Mol. Genet. 2003, 12, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Seguin, S.J.; Lambert, H.; Landry, J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem. 2008, 283, 1437–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carra, S.; Brunsting, J.F.; Lambert, H.; Landry, J.; Kampinga, H.H. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J. Biol. Chem. 2009, 284, 5523–5532. [Google Scholar] [CrossRef]
- Seidel, K.; Vinet, J.; Dunnen, W.F.; Brunt, E.R.; Meister, M.; Boncoraglio, A.; Zijlstra, M.P.; Boddeke, H.W.; Rub, U.; Kampinga, H.H.; et al. The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol. Appl. Neurobiol. 2012, 38, 39–53. [Google Scholar] [CrossRef]
- Rusmini, P.; Crippa, V.; Giorgetti, E.; Boncoraglio, A.; Cristofani, R.; Carra, S.; Poletti, A. Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol. Aging 2013, 34, 2585–2603. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.V.; Seit-Nebi, A.S.; Marston, S.B.; Gusev, N.B. Some properties of human small heat shock protein Hsp22 (H11 or HspB8). Biochem Biophys Res Comm 2004, 315, 796–801. [Google Scholar] [CrossRef]
- Fontaine, J.M.; Sun, X.; Benndorf, R.; Welsh, M.J. Interactions of HSP22 (HSPB8) with HSP20, alphaB-crystallin, and HSPB3. Biochem. Biophys. Res. Comm. 2005, 337, 1006–1011. [Google Scholar] [CrossRef]
- Shatov, V.M.; Sluchanko, N.N.; Gusev, N.B. Replacement of Arg in the conserved N-terminal RLFDQxFG motif affects physico-chemical properties and chaperone-like activity of human small heat shock protein HspB8 (Hsp22). PLoS ONE 2021, 16, e0253432. [Google Scholar] [CrossRef]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Furst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef]
- Carra, S. The stress-inducible HspB8-Bag3 complex induces the eIF2alpha kinase pathway: Implications for protein quality control and viral factory degradation? Autophagy 2009, 5, 428–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriaenssens, E.; Tedesco, B.; Mediani, L.; Asselbergh, B.; Crippa, V.; Antoniani, F.; Carra, S.; Poletti, A.; Timmerman, V. BAG3 Pro209 mutants associated with myopathy and neuropathy relocate chaperones of the CASA-complex to aggresomes. Sci. Rep. 2020, 10, 8755. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.E.; Cristofani, R.; Crippa, V.; Ferrari, V.; Tedesco, B.; Casarotto, E.; Chierichetti, M.; Galbiati, M.; Piccolella, M.; Messi, E.; et al. Autophagic and Proteasomal Mediated Removal of Mutant Androgen Receptor in Muscle Models of Spinal and Bulbar Muscular Atrophy. Front. Endocrinol. 2019, 10, 569. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, A.; Arndt, V.; Hohfeld, J. Chaperone-assisted proteostasis is essential for mechanotransduction in mammalian cells. Commun. Integr. Biol. 2013, 6, e24925. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, A.; Gehlert, S.; Leciejewski, B.; Schiffer, T.; Bloch, W.; Hohfeld, J. Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy 2015, 11, 538–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, T.; Ramaglia, V.; Abdel-Nour, M.; Bianchi, A.A.; Tsalikis, J.; Chau, H.N.; Kalia, S.K.; Kalia, L.V.; Chen, J.J.; Arnoult, D.; et al. The eIF2alpha kinase HRI triggers the autophagic clearance of cytosolic protein aggregates. J. Biol. Chem. 2021, 296, 100050. [Google Scholar] [CrossRef]
- Abdel-Nour, M.; Carneiro, L.A.M.; Downey, J.; Tsalikis, J.; Outlioua, A.; Prescott, D.; Da Costa, L.S.; Hovingh, E.S.; Farahvash, A.; Gaudet, R.G.; et al. The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 2019, 365, eaaw4144. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Gamerdinger, M.; Carra, S.; Behl, C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: Focus on BAG proteins. J. Mol. Med. 2011, 89, 1175–1182. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Kaya, A.M.; Wolfrum, U.; Clement, A.M.; Behl, C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011, 12, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Rusmini, P.; Cristofani, R.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Messi, E.; Piccolella, M.; et al. The Role of the Heat Shock Protein B8 (HSPB8) in Motoneuron Diseases. Front. Mol. Neurosci. 2017, 10, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Graham, K.; Foote, M.; Liang, F.; Rizkallah, R.; Hurt, M.; Wang, Y.; Wu, Y.; Zhou, Y. 14-3-3 protein targets misfolded chaperone-associated proteins to aggresomes. J. Cell Sci. 2013, 126 Pt 18, 4173–4186. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Wu, Y.; Zhou, Y. 14-3-3 and aggresome formation: Implications in neurodegenerative diseases. Prion 2014, 8, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. BAG3 and friends: Co-chaperones in selective autophagy during aging and disease. Autophagy 2011, 7, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, A.; Christianson, J.C.; Bucci, M.; Ellerby, L.M.; Nukina, N.; Forno, L.S.; Kopito, R.R. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 13135–13140. [Google Scholar] [CrossRef] [Green Version]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mata, R.; Bebok, Z.; Sorscher, E.J.; Sztul, E.S. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 1999, 146, 1239–1254. [Google Scholar] [CrossRef] [Green Version]
- Rusmini, P.; Sau, D.; Crippa, V.; Palazzolo, I.; Simonini, F.; Onesto, E.; Martini, L.; Poletti, A. Aggregation and proteasome: The case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol aging 2007, 28, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Bolzoni, E.; Crippa, V.; Onesto, E.; Sau, D.; Galbiati, M.; Piccolella, M.; Poletti, A. Proteasomal and autophagic degradative activities in spinal and bulbar muscular atrophy. Neurobiol. Dis. 2010, 40, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol. Sci. 2016, 37, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Ritson, G.P.; Custer, S.K.; Freibaum, B.D.; Guinto, J.B.; Geffel, D.; Moore, J.; Tang, W.; Winton, M.J.; Neumann, M.; Trojanowski, J.Q.; et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 2010, 30, 7729–7739. [Google Scholar] [CrossRef] [PubMed]
- Bouhy, D.; Juneja, M.; Katona, I.; Holmgren, A.; Asselbergh, B.; De Winter, V.; Hochepied, T.; Goossens, S.; Haigh, J.J.; Libert, C.; et al. A knock-in/knock-out mouse model of HSPB8-associated distal hereditary motor neuropathy and myopathy reveals toxic gain-of-function of mutant Hspb8. Acta Neuropathol. 2018, 135, 131–148. [Google Scholar] [CrossRef]
- Fontaine, J.M.; Sun, X.; Hoppe, A.D.; Simon, S.; Vicart, P.; Welsh, M.J.; Benndorf, R. Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J. 2006, 20, 2168–2170. [Google Scholar] [CrossRef]
- Irobi, J.; Almeida-Souza, L.; Asselbergh, B.; De Winter, V.; Goethals, S.; Dierick, I.; Krishnan, J.; Timmermans, J.P.; Robberecht, W.; De Jonghe, P.; et al. Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum. Mol. Genet. 2010, 19, 3254–3265. [Google Scholar] [CrossRef]
- Ghaoui, R.; Palmio, J.; Brewer, J.; Lek, M.; Needham, M.; Evila, A.; Hackman, P.; Jonson, P.H.; Penttila, S.; Vihola, A.; et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 2016, 86, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Adriaenssens, E.; Geuens, T.; Baets, J.; Echaniz-Laguna, A.; Timmerman, V. Novel insights in the disease biology of mutant small heat shock proteins in neuromuscular diseases. Brain 2017, 140, 2541–2549. [Google Scholar] [CrossRef] [Green Version]
- Guilbert, S.M.; Lambert, H.; Rodrigue, M.A.; Fuchs, M.; Landry, J.; Lavoie, J.N. HSPB8 and BAG3 cooperate to promote spatial sequestration of ubiquitinated proteins and coordinate the cellular adaptive response to proteasome insufficiency. FASEB J. 2018, 32, 3518–3535. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Konersman, C.G.; Bordini, B.J.; Scharer, G.; Lawlor, M.W.; Zangwill, S.; Southern, J.F.; Amos, L.; Geddes, G.C.; Kliegman, R.; Collins, M.P. BAG3 myofibrillar myopathy presenting with cardiomyopathy. Neuromusc. Dis. 2015, 25, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Pegoraro, E.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irobi, J.; Van Impe, K.; Seeman, P.; Jordanova, A.; Dierick, I.; Verpoorten, N.; Michalik, A.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004, 36, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.S.; Zhao, G.H.; Luo, W.; Xia, K.; Cai, F.; Pan, Q.; Zhang, R.X.; Zhang, F.F.; Liu, X.M.; Chen, B.; et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum. Genet. 2005, 116, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Nakhro, K.; Park, J.M.; Kim, Y.J.; Yoon, B.R.; Yoo, J.H.; Koo, H.; Choi, B.O.; Chung, K.W. A novel Lys141Thr mutation in small heat shock protein 22 (HSPB8) gene in Charcot-Marie-Tooth disease type 2L. Neuromusc. Dis. 2013, 23, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Geuens, T.; Petiot, P.; Pereon, Y.; Adriaenssens, E.; Haidar, M.; Capponi, S.; Maisonobe, T.; Fournier, E.; Dubourg, O.; et al. Axonal Neuropathies due to Mutations in Small Heat Shock Proteins: Clinical, Genetic, and Functional Insights into Novel Mutations. Hum. Mutat. 2017, 38, 556–568. [Google Scholar] [CrossRef]
- Al-Tahan, S.; Weiss, L.; Yu, H.; Tang, S.; Saporta, M.; Vihola, A.; Mozaffar, T.; Udd, B.; Kimonis, V. New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol. Genet. 2019, 5, e349. [Google Scholar] [CrossRef] [Green Version]
- Nicolau, S.; Liewluck, T.; Elliott, J.L.; Engel, A.G.; Milone, M. A novel heterozygous mutation in the C-terminal region of HSPB8 leads to limb-girdle rimmed vacuolar myopathy. Neuromusc. Dis. 2020, 30, 236–240. [Google Scholar] [CrossRef]
- Inoue-Shibui, A.; Niihori, T.; Kobayashi, M.; Suzuki, N.; Izumi, R.; Warita, H.; Hara, K.; Shirota, M.; Funayama, R.; Nakayama, K.; et al. A novel deletion in the C-terminal region of HSPB8 in a family with rimmed vacuolar myopathy. J. Hum. Genet. 2021, 66, 965–972. [Google Scholar] [CrossRef]
- Irobi, J.; Holmgren, A.; De Winter, V.; Asselbergh, B.; Gettemans, J.; Adriaensen, D.; Ceuterick-de Groote, C.; Van Coster, R.; De Jonghe, P.; Timmerman, V. Mutant HSPB8 causes protein aggregates and a reduced mitochondrial membrane potential in dermal fibroblasts from distal hereditary motor neuropathy patients. Neuromusc. Dis. 2012, 22, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Sanbe, A.; Daicho, T.; Mizutani, R.; Endo, T.; Miyauchi, N.; Yamauchi, J.; Tanonaka, K.; Glabe, C.; Tanoue, A. Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS ONE 2009, 4, e5351. [Google Scholar] [CrossRef] [PubMed]
- Sanbe, A.; Yamauchi, J.; Miyamoto, Y.; Fujiwara, Y.; Murabe, M.; Tanoue, A. Interruption of CryAB-amyloid oligomer formation by HSP22. J. Biol. Chem. 2007, 282, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.D.; Cen, Z.D.; Cheng, H.P.; Shi, K.; Bai, J.; Xie, F.; Wu, H.W.; Li, B.B.; Luo, W. L-3-n-Butylphthalide Protects HSPB8 K141N Mutation-Induced Oxidative Stress by Modulating the Mitochondrial Apoptotic and Nrf2 Pathways. Front. Neurosci. 2017, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Piccolella, M.; Crippa, V.; Cristofani, R.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferri, N.; Morelli, F.F.; Carra, S.; et al. The small heat shock protein B8 (HSPB8) modulates proliferation and migration of breast cancer cells. Oncotarget 2017, 8, 10400–10415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Fontaine, J.M.; Bartl, I.; Behnam, B.; Welsh, M.J.; Benndorf, R. Induction of Hsp22 (HspB8) by estrogen and the metalloestrogen cadmium in estrogen receptor-positive breast cancer cells. Cell Stress Chaperones 2007, 12, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldink, J.H.; Bar, P.R.; Joosten, E.A.; Otten, M.; Wokke, J.H.; van den Berg, L.H. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromusc. Dis. 2003, 13, 737–743. [Google Scholar] [CrossRef]
- Mandrioli, J.; Crippa, V.; Cereda, C.; Bonetto, V.; Zucchi, E.; Gessani, A.; Ceroni, M.; Chio, A.; D’Amico, R.; Monsurro, M.R.; et al. Proteostasis and ALS: Protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open 2019, 9, e028486. [Google Scholar] [CrossRef]
- Giorgetti, E.; Rusmini, P.; Crippa, V.; Cristofani, R.; Boncoraglio, A.; Cicardi, M.E.; Galbiati, M.; Poletti, A. Synergic prodegradative activity of Bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2015, 24, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Comm. 2017, 8, 14338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.C.; Lin, C.H.; Tao, Y.C.; Yang, J.M.; Hsu, K.C.; Huang, Y.J.; Huang, S.H.; Kung, P.J.; Chen, W.L.; Wang, C.M.; et al. The potential of lactulose and melibiose, two novel trehalase-indigestible and autophagy-inducing disaccharides, for polyQ-mediated neurodegenerative disease treatment. Neurotoxicology 2015, 48, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Wu, Y.R.; Yang, J.M.; Chen, W.L.; Chao, C.Y.; Chen, I.C.; Lin, T.H.; Wu, Y.C.; Hsu, K.C.; Chen, C.M.; et al. Novel Lactulose and Melibiose Targeting Autophagy to Reduce PolyQ Aggregation in Cell Models of Spinocerebellar Ataxia 3. CNS Neurol. Dis. Drug Targets 2016, 15, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Liu, R.; Barkhordarian, H.; Emadi, S.; Park, C.B.; Sierks, M.R. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol. Dis. 2005, 20, 74–81. [Google Scholar] [CrossRef]
- Davies, J.E.; Sarkar, S.; Rubinsztein, D.C. Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2006, 15, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Navarro, J.A.; Rodriguez, L.; Casarejos, M.J.; Solano, R.M.; Gomez, A.; Perucho, J.; Cuervo, A.M.; Garcia de Yebenes, J.; Mena, M.A. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 2010, 39, 423–438. [Google Scholar] [CrossRef]
- Perucho, J.; Casarejos, M.J.; Gomez, A.; Solano, R.M.; de Yebenes, J.G.; Mena, M.A. Trehalose protects from aggravation of amyloid pathology induced by isoflurane anesthesia in APP(swe) mutant mice. Curr. Alzheimer. Res. 2012, 9, 334–343. [Google Scholar] [CrossRef]
- Schaeffer, V.; Goedert, M. Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy 2012, 8, 1686–1687. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135 Pt 7, 2169–2177. [Google Scholar] [CrossRef] [Green Version]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; van Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Chigurupati, S.; Raymick, J.; Mann, D.; Bowyer, J.F.; Schmitt, T.; Beger, R.D.; Hanig, J.P.; Schmued, L.C.; Paule, M.G. Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP-induced Parkinson’s disease mouse model. Neurotoxicology 2014, 44, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Y.; Wang, X.; Yu, X.; Duan, W.; Hong, K.; Wang, J.; Han, H.; Li, C. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 2015, 298, 12–25. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Koprich, J.B.; Wang, Y.; Yu, W.B.; Xiao, B.G.; Brotchie, J.M.; Wang, J. Treatment with Trehalose Prevents Behavioral and Neurochemical Deficits Produced in an AAV alpha-Synuclein Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2016, 53, 2258–2268. [Google Scholar] [CrossRef]
- Debnath, K.; Pradhan, N.; Singh, B.K.; Jana, N.R.; Jana, N.R. Poly(trehalose) Nanoparticles Prevent Amyloid Aggregation and Suppress Polyglutamine Aggregation in a Huntington’s Disease Model Mouse. ACS Appl. Mater. Interfaces 2017, 9, 24126–24139. [Google Scholar] [CrossRef] [Green Version]
- Mobini, M.; Radbakhsh, S.; Kubaski, F.; Eshraghi, P.; Vakili, S.; Vakili, R.; Khalili, M.; Varesvazirian, M.; Jamialahmadi, T.; Alamdaran, S.A.; et al. Impact of Intravenous Trehalose Administration in Patients with Niemann-Pick Disease Types A and B. J. Clin. Med. 2022, 11, 247. [Google Scholar] [CrossRef]
- van Marion, D.M.S.; Dorsch, L.; Hoogstra-Berends, F.; Kakuchaya, T.; Bockeria, L.; de Groot, N.M.S.; Brundel, B. Oral geranylgeranylacetone treatment increases heat shock protein expression in human atrial tissue. Heart Rhythm. 2020, 17, 115–122. [Google Scholar] [CrossRef]
- Marunouchi, T.; Inomata, S.; Sanbe, A.; Takagi, N.; Tanonaka, K. Protective effect of geranylgeranylacetone via enhanced induction of HSPB1 and HSPB8 in mitochondria of the failing heart following myocardial infarction in rats. Eur. J. Pharmacol. 2014, 730, 140–147. [Google Scholar] [CrossRef]
- Ramos, E.; Romero, A.; Marco-Contelles, J.; Lopez-Munoz, F.; Del Pino, J. Modulation of Heat Shock Response Proteins by ASS234, Targeted for Neurodegenerative Diseases Therapy. Chem. Res. Toxicol. 2018, 31, 839–842. [Google Scholar] [CrossRef]
- Bolea, I.; Juarez-Jimenez, J.; de Los Rios, C.; Chioua, M.; Pouplana, R.; Luque, F.J.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2011, 54, 8251–8270. [Google Scholar] [PubMed]
- Serrano, M.P.; Herrero-Labrador, R.; Futch, H.S.; Serrano, J.; Romero, A.; Fernandez, A.P.; Samadi, A.; Unzeta, M.; Marco-Contelles, J.; Martinez-Murillo, R. The proof-of-concept of ASS234: Peripherally administered ASS234 enters the central nervous system and reduces pathology in a male mouse model of Alzheimer disease. J. Psychiatry Neurosci. 2017, 42, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, A.; Marco-Contelles, J.; Ramos, E. Highlights of ASS234: A novel and promising therapeutic agent for Alzheimer’s disease therapy. Neural. Reg. Res. 2020, 15, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Suzuki, K.; Matsushima, T.; Yamakawa, N.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s disease-related phenotypes by geranylgeranylacetone in mice. PLoS ONE 2013, 8, e76306. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.R.; Chen, S. Suppression of Alzheimer’s disease-related phenotypes by the heat shock protein 70 inducer, geranylgeranylacetone, in APP/PS1 transgenic mice via the ERK/p38 MAPK signaling pathway. Exp. Ther. Med. 2017, 14, 5267–5274. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, S.; Yoshinaga, T.; Matsuzaki, J.; Suzuki, H. A Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy of Teprenone in Patients with Alzheimer’s Disease. J. Alzheimer’s. Dis. 2019, 71, 1187–1199. [Google Scholar] [CrossRef]
- Luthold, C.; Lambert, H.; Guilbert, S.M.; Rodrigue, M.A.; Fuchs, M.; Varlet, A.A.; Fradet-Turcotte, A.; Lavoie, J.N. CDK1-Mediated Phosphorylation of BAG3 Promotes Mitotic Cell Shape Remodeling and the Molecular Assembly of Mitotic p62 Bodies. Cells 2021, 10, 2638. [Google Scholar] [CrossRef]
- Varlet, A.A.; Fuchs, M.; Luthold, C.; Lambert, H.; Landry, J.; Lavoie, J.N. Fine-tuning of actin dynamics by the HSPB8-BAG3 chaperone complex facilitates cytokinesis and contributes to its impact on cell division. Cell Stress Chaperones 2017, 22, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Fontaine, J.M.; Hoppe, A.D.; Carra, S.; DeGuzman, C.; Martin, J.L.; Simon, S.; Vicart, P.; Welsh, M.J.; Landry, J.; et al. Abnormal interaction of motor neuropathy-associated mutant HspB8 (Hsp22) forms with the RNA helicase Ddx20 (gemin3). Cell Stress Chaperones 2010, 15, 567–582. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, J.M.; Rest, J.S.; Welsh, M.J.; Benndorf, R. The sperm outer dense fiber protein is the 10th member of the superfamily of mammalian small stress proteins. Cell Stress Chaperones 2003, 8, 62–69. [Google Scholar] [CrossRef]
- Kappe, G.; Verschuure, P.; Philipsen, R.L.; Staalduinen, A.A.; Van de Boogaart, P.; Boelens, W.C.; De Jong, W.W. Characterization of two novel human small heat shock proteins: Protein kinase-related HspB8 and testis-specific HspB9. Biochim. Biophys. Acta 2001, 1520, 1–6. [Google Scholar] [CrossRef]
- Okano, H. A combined stem-cell-gene therapy strategy for ALS. Nat. Med. 2022, 28, 1751–1752. [Google Scholar] [CrossRef] [PubMed]
- Meijboom, K.E.; Brown, R.H. Approaches to Gene Modulation Therapy for ALS. Neurotherapeutics 2022, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Amado, D.A.; Davidson, B.L. Gene therapy for ALS: A review. Mol. Ther. 2021, 29, 3345–3358. [Google Scholar] [CrossRef]
- Poletti, A.; Fischbeck, K.H. Combinatorial treatment for spinal muscular atrophy: An Editorial for ‘Combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in Spinal Muscular Atrophy cells’ on page 264. J. Neurochem. 2020, 153, 146–149. [Google Scholar]
- Demarest, T.G.; Biferi, M.G. Translation of gene therapy strategies for amyotrophic lateral sclerosis. Trends Mol. Med. 2022, 28, 795–796. [Google Scholar] [CrossRef]
- Boros, B.D.; Schoch, K.M.; Kreple, C.J.; Miller, T.M. Antisense Oligonucleotides for the Study and Treatment of ALS. Neurotherapeutics 2022, 1–14. [Google Scholar] [CrossRef]
- Holm, A.; Hansen, S.N.; Klitgaard, H.; Kauppinen, S. Clinical advances of RNA therapeutics for treatment of neurological and neuromuscular diseases. RNA Biol. 2022, 19, 594–608. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef]
- Tran, H.; Moazami, M.P.; Yang, H.; McKenna-Yasek, D.; Douthwright, C.L.; Pinto, C.; Metterville, J.; Shin, M.; Sanil, N.; Dooley, C.; et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 2022, 28, 117–124. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tedesco, B.; Ferrari, V.; Cozzi, M.; Chierichetti, M.; Casarotto, E.; Pramaggiore, P.; Mina, F.; Galbiati, M.; Rusmini, P.; Crippa, V.; et al. The Role of Small Heat Shock Proteins in Protein Misfolding Associated Motoneuron Diseases. Int. J. Mol. Sci. 2022, 23, 11759. https://doi.org/10.3390/ijms231911759
Tedesco B, Ferrari V, Cozzi M, Chierichetti M, Casarotto E, Pramaggiore P, Mina F, Galbiati M, Rusmini P, Crippa V, et al. The Role of Small Heat Shock Proteins in Protein Misfolding Associated Motoneuron Diseases. International Journal of Molecular Sciences. 2022; 23(19):11759. https://doi.org/10.3390/ijms231911759
Chicago/Turabian StyleTedesco, Barbara, Veronica Ferrari, Marta Cozzi, Marta Chierichetti, Elena Casarotto, Paola Pramaggiore, Francesco Mina, Mariarita Galbiati, Paola Rusmini, Valeria Crippa, and et al. 2022. "The Role of Small Heat Shock Proteins in Protein Misfolding Associated Motoneuron Diseases" International Journal of Molecular Sciences 23, no. 19: 11759. https://doi.org/10.3390/ijms231911759