
Mitochondrial and Endoplasmic Reticulum Alterations in a Case of Amyotrophic Lateral Sclerosis Caused by TDP-43 A382T Mutation
 ,
, 
 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Clinical Findings
2.2. Biological Assessments
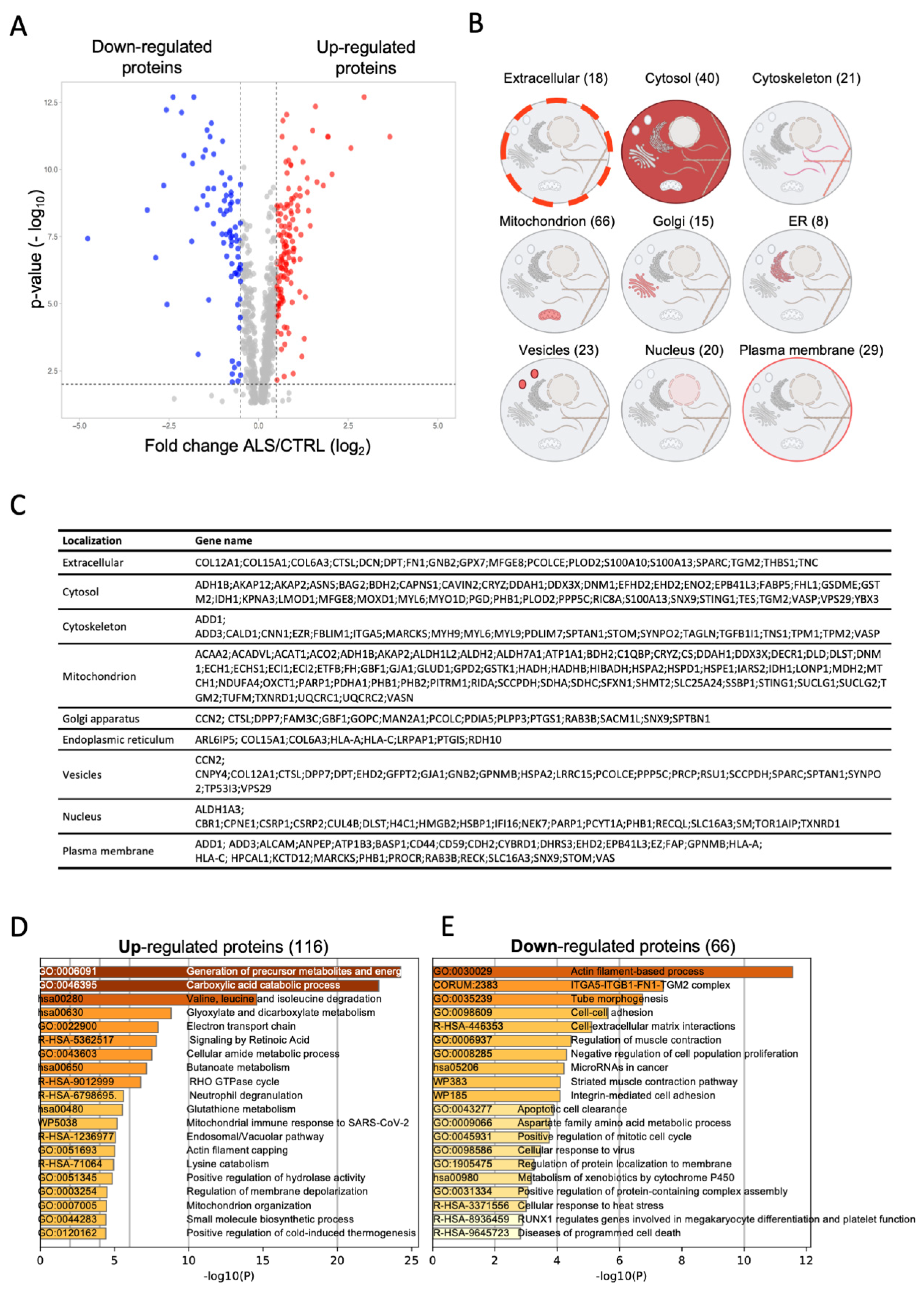
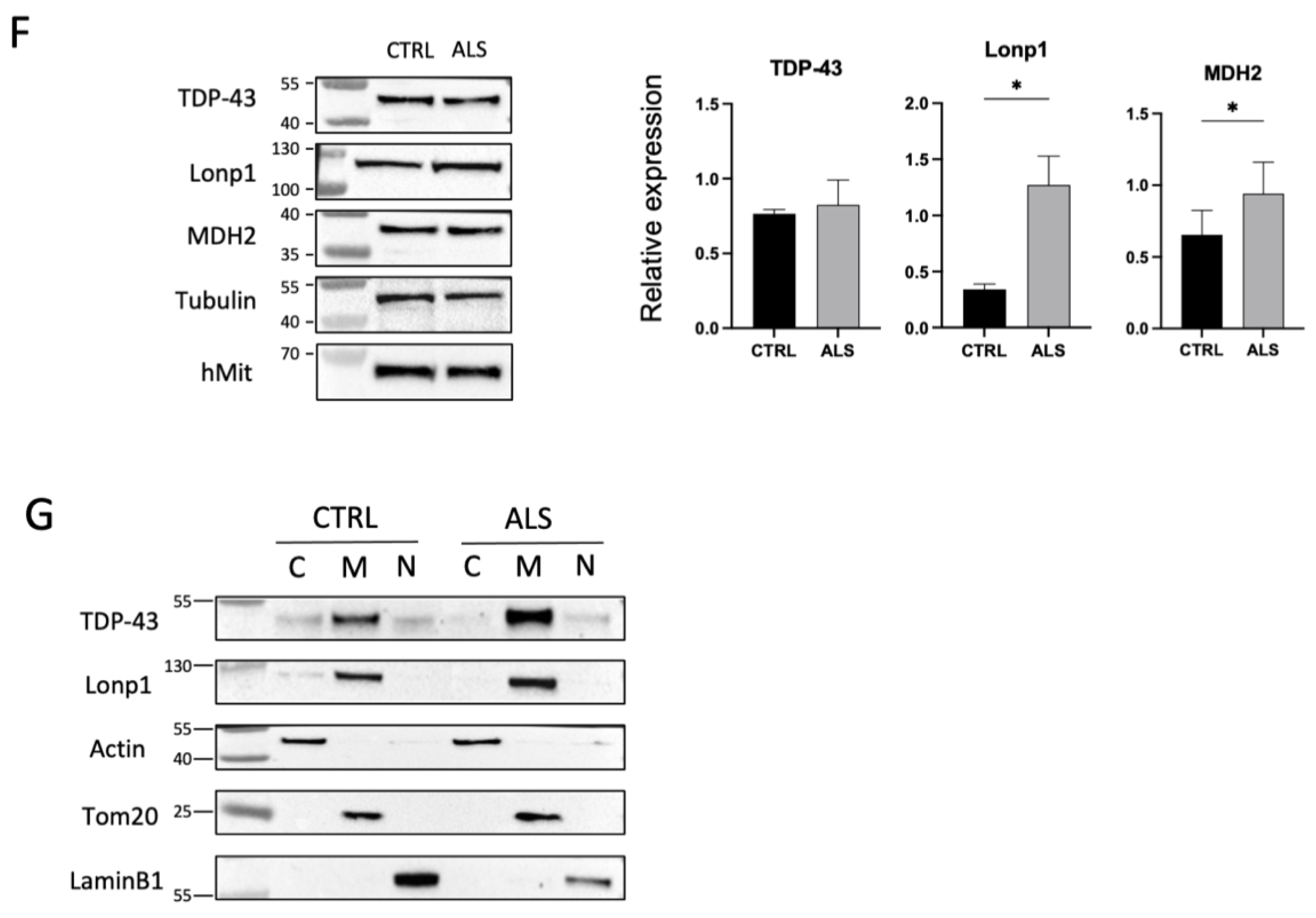
2.2.1. Alterations of the Proteome of Fibroblasts from the Proband
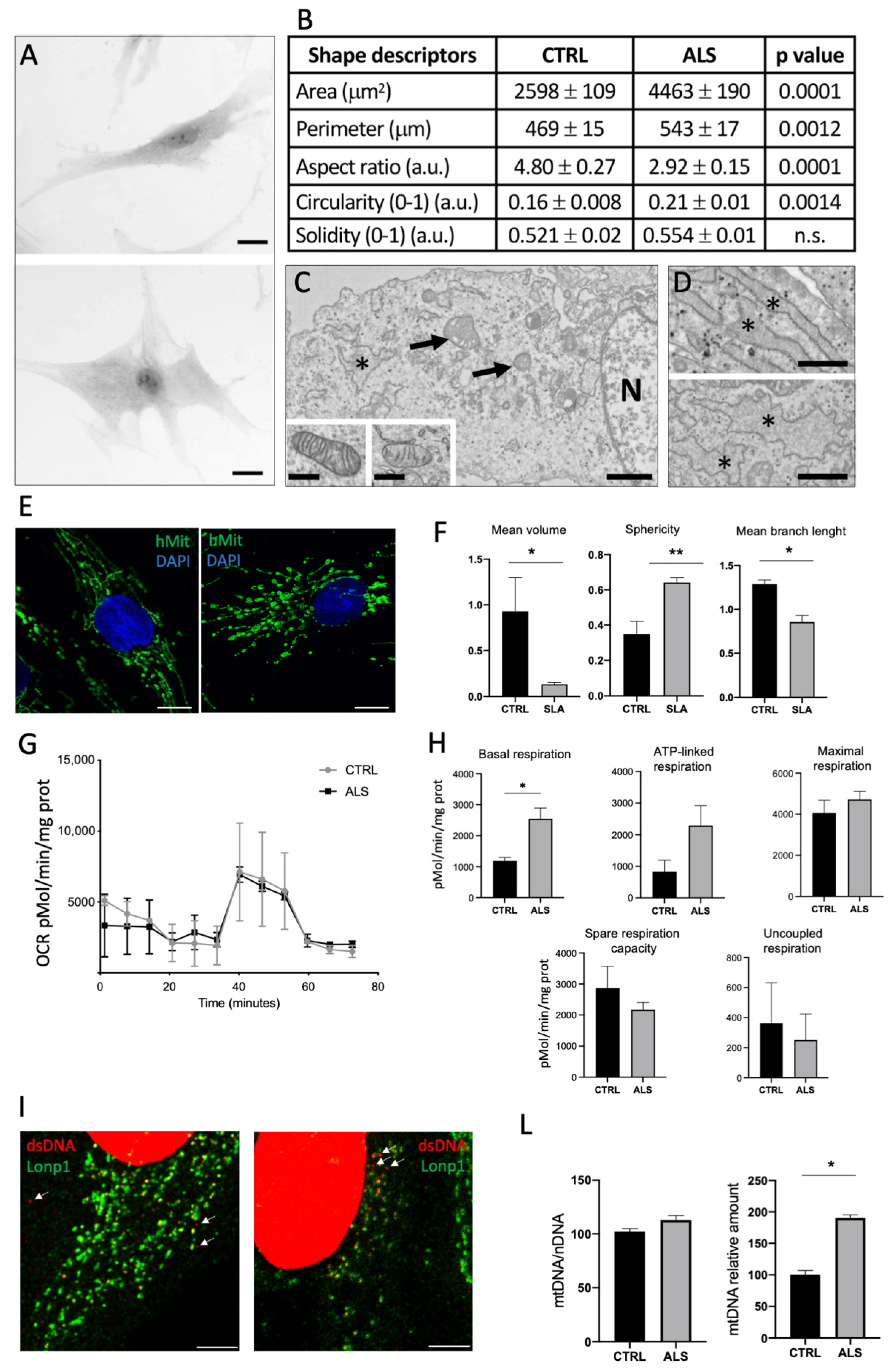
2.2.2. Morphological, Ultrastructural and Functional Alteration of Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Isolation and Culture of Fibroblasts
4.3. Microscopy and Analysis
4.3.1. Light Microscopy
4.3.2. Transmission Electron Microscopy
4.4. Proteomic Analysis
4.4.1. Sample Preparation
4.4.2. Protein Identification and Quantification
4.5. Sub Cellular and Extracellular Localization and Pathway Enrichment Analysis of DEPs
4.6. Immunoblot
4.7. Confocal Microscopy
4.8. Mitochondrial Network Analysis
4.9. Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR)
4.10. Quantification of Mitochondrial DNA
4.11. Relative Quantification of mRNA Expression
4.12. Reactive Oxygen Species Measurement
4.13. Quantification of Cytokines in Cell Supernatants
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Ratti, A.; Gellera, C.; Buratti, E.; Castellotti, B.; Carlomagno, Y.; Ticozzi, N.; Mazzini, L.; Testa, L.; Taroni, F.; et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 2009, 30, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Pesiridis, G.S.; Lee, V.M.; Trojanowski, J.Q. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, R156–R162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef]
- Corcia, P.; Valdmanis, P.; Millecamps, S.; Lionnet, C.; Blasco, H.; Mouzat, K.; Daoud, H.; Belzil, V.; Morales, R.; Pageot, N.; et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology 2012, 78, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Gianferrari, G.; Martinelli, I.; Zucchi, E.; Simonini, C.; Fini, N.; Vinceti, M.; Ferro, S.; Gessani, A.; Canali, E.; Valzania, F.; et al. Epidemiological, Clinical and Genetic Features of ALS in the Last Decade: A Prospective Population-Based Study in the Emilia Romagna Region of Italy. Biomedicines 2022, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Benajiba, L.; Le Ber, I.; Camuzat, A.; Lacoste, M.; Thomas-Anterion, C.; Couratier, P.; Legallic, S.; Salachas, F.; Hannequin, D.; Decousus, M.; et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 2009, 65, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Parish, L.D.; Occhineri, P.; Cau, T.B.; et al. Genetic architecture of ALS in Sardinia. Neurobiol. Aging 2014, 35, 2882.e7–2882.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, A.; Borghero, G.; Pugliatti, M.; Ticca, A.; Calvo, A.; Moglia, C.; Mutani, R.; Brunetti, M.; Ossola, I.; Marrosu, M.G.; et al. Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch. Neurol. 2011, 68, 594–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannas, A.; Borghero, G.; Floris, G.L.; Solla, P.; Chio, A.; Traynor, B.J.; Calvo, A.; Restagno, G.; Majounie, E.; Costantino, E.; et al. The p.A382T TARDBP gene mutation in Sardinian patients affected by Parkinson’s disease and other degenerative parkinsonisms. Neurogenetics 2013, 14, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kaneko, K.; Yamanaka, K. Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J. Biol. Chem. 2013, 288, 3641–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, T.H.; Choi, M.; Banfai, B.; Liu, Y.; MacLean, B.X.; Dunkley, T.; Vitek, O. Selection of Features with Consistent Profiles Improves Relative Protein Quantification in Mass Spectrometry Experiments. Mol. Cell Proteom. 2020, 19, 944–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.; Wang, S.; Zhang, Z.; Morgens, D.W.; Hayes, L.R.; Lee, S.; Portz, B.; Xie, Y.; Nguyen, B.V.; Haney, M.S.; et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron 2019, 104, 885–898.e8. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, F.; West, P.K.; Brennan, S.; Petrovic, B.; Hooshmand, K.; Akkari, P.A.; Keon, M.; Guennewig, B. New perspectives on cytoskeletal dysregulation and mitochondrial mislocalization in amyotrophic lateral sclerosis. Transl. Neurodegener. 2021, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Bel Hadj, S.; Durham, H.D.; Vande Velde, C.; et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2010, 19, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Mandrioli, J.; Mediani, L.; Alberti, S.; Carra, S. ALS and FTD: Where RNA metabolism meets protein quality control. Semin. Cell Dev. Biol. 2020, 99, 183–192. [Google Scholar] [CrossRef]
- Giannini, M.; Bayona-Feliu, A.; Sproviero, D.; Barroso, S.I.; Cereda, C.; Aguilera, A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLoS Genet. 2020, 16, e1009260. [Google Scholar] [CrossRef]
- Orru, S.; Coni, P.; Floris, A.; Littera, R.; Carcassi, C.; Sogos, V.; Brancia, C. Reduced stress granule formation and cell death in fibroblasts with the A382T mutation of TARDBP gene: Evidence for loss of TDP-43 nuclear function. Hum. Mol. Genet. 2016, 25, 4473–4483. [Google Scholar] [CrossRef]
- Mutihac, R.; Alegre-Abarrategui, J.; Gordon, D.; Farrimond, L.; Yamasaki-Mann, M.; Talbot, K.; Wade-Martins, R. TARDBP pathogenic mutations increase cytoplasmic translocation of TDP-43 and cause reduction of endoplasmic reticulum Ca(2)(+) signaling in motor neurons. Neurobiol. Dis. 2015, 75, 64–77. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marco, G.; Lomartire, A.; Calvo, A.; Risso, A.; De Luca, E.; Mostert, M.; Mandrioli, J.; Caponnetto, C.; Borghero, G.; Manera, U.; et al. Monocytes of patients with amyotrophic lateral sclerosis linked to gene mutations display altered TDP-43 subcellular distribution. Neuropathol. Appl. Neurobiol. 2017, 43, 133–153. [Google Scholar] [CrossRef] [PubMed]
- Borghero, G.; Floris, G.; Cannas, A.; Marrosu, M.G.; Murru, M.R.; Costantino, E.; Parish, L.D.; Pugliatti, M.; Ticca, A.; Traynor, B.J.; et al. A patient carrying a homozygous p.A382T TARDBP missense mutation shows a syndrome including ALS, extrapyramidal symptoms, and FTD. Neurobiol. Aging 2011, 32, 2327.e1–2327.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goris, A.; van Setten, J.; Diekstra, F.; Ripke, S.; Patsopoulos, N.A.; Sawcer, S.J.; International Multiple Sclerosis Genetics Consortium; van Es, M.; Australia and New Zealand MS Genetics Consortium; Andersen, P.M.; et al. No evidence for shared genetic basis of common variants in multiple sclerosis and amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 1916–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, C.; Longinetti, E.; Larsson, H.; Andersson, J.; Pawitan, Y.; Piehl, F.; Fang, F. Associations between autoimmune diseases and amyotrophic lateral sclerosis: A register-based study. Amyotroph. Lateral Scler. Frontotemporal Degener. 2021, 22, 211–219. [Google Scholar] [CrossRef]
- Turner, M.R.; Goldacre, R.; Ramagopalan, S.; Talbot, K.; Goldacre, M.J. Autoimmune disease preceding amyotrophic lateral sclerosis: An epidemiologic study. Neurology 2013, 81, 1222–1225. [Google Scholar] [CrossRef] [Green Version]
- Etemadifar, M.; Abtahi, S.H.; Akbari, M.; Maghzi, A.H. Multiple sclerosis and amyotrophic lateral sclerosis: Is there a link? Mult. Scler. 2012, 18, 902–904. [Google Scholar] [CrossRef]
- Hemminki, K.; Li, X.; Sundquist, J.; Sundquist, K. Familial risks for amyotrophic lateral sclerosis and autoimmune diseases. Neurogenetics 2009, 10, 111–116. [Google Scholar] [CrossRef]
- Longinetti, E.; Sveinsson, O.; Press, R.; Ye, W.; Ingre, C.; Piehl, F.; Fang, F. ALS patients with concurrent neuroinflammatory disorders; a nationwide clinical records study. Amyotroph. Lateral Scler. Frontotemporal Degener. 2022, 23, 209–219. [Google Scholar] [CrossRef]
- Francois-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-translational Modifications. Front. Mol. Neurosci. 2019, 12, 301. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Wu, H.C.; Burchell, V.S.; Preza, E.; Wray, S.; Mahoney, C.J.; Fox, N.C.; Calvo, A.; Canosa, A.; Moglia, C.; et al. Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 2013, 78, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzo, F.; Mirra, A.; Carri, M.T. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef]
- Gibellini, L.; Pinti, M.; Boraldi, F.; Giorgio, V.; Bernardi, P.; Bartolomeo, R.; Nasi, M.; De Biasi, S.; Missiroli, S.; Carnevale, G.; et al. Silencing of mitochondrial Lon protease deeply impairs mitochondrial proteome and function in colon cancer cells. FASEB J. 2014, 28, 5122–5135. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Lu, B.; Lee, I.; Ondrovicova, G.; Kutejova, E.; Suzuki, C.K. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J. Biol. Chem. 2004, 279, 13902–13910. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Yadav, S.; Shah, P.G.; Liu, T.; Tian, B.; Pukszta, S.; Villaluna, N.; Kutejova, E.; Newlon, C.S.; Santos, J.H.; et al. Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J. Biol. Chem. 2007, 282, 17363–17374. [Google Scholar] [CrossRef] [Green Version]
- Lofaro, F.D.; Boraldi, F.; Garcia-Fernandez, M.; Estrella, L.; Valdivielso, P.; Quaglino, D. Relationship Between Mitochondrial Structure and Bioenergetics in Pseudoxanthoma elasticum Dermal Fibroblasts. Front. Cell Dev. Biol. 2020, 8, 610266. [Google Scholar] [CrossRef]
- Lofaro, F.D.; Cisterna, B.; Lacavalla, M.A.; Boschi, F.; Malatesta, M.; Quaglino, D.; Zancanaro, C.; Boraldi, F. Age-Related Changes in the Matrisome of the Mouse Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 564. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Gibellini, L.; Losi, L.; De Biasi, S.; Nasi, M.; Lo Tartaro, D.; Pecorini, S.; Patergnani, S.; Pinton, P.; De Gaetano, A.; Carnevale, G.; et al. LonP1 Differently Modulates Mitochondrial Function and Bioenergetics of Primary Versus Metastatic Colon Cancer Cells. Front. Oncol. 2018, 8, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; De Biasi, S.; Nasi, M.; Torelli, F.; et al. Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Shi, R.; Luciani, D.S. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E87–E101. [Google Scholar] [CrossRef]
- Nasi, M.; Bianchini, E.; De Biasi, S.; Gibellini, L.; Neroni, A.; Mattioli, M.; Pinti, M.; Iannone, A.; Mattioli, A.V.; Simone, A.M.; et al. Increased plasma levels of mitochondrial DNA and pro-inflammatory cytokines in patients with progressive multiple sclerosis. J. Neuroimmunol. 2020, 338, 577107. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Timeline | Patient’s Symptoms | Neurological Examination |
|---|---|---|
| Eleven months after symptoms onset (at diagnosis) | Right hand muscles weakness Right hand muscles hypotrophy Widespread fasciculations and cramps Walking impairment | Severe weakness of intrinsic hand muscles (MRC * score 2 on the right and 3 on the left) Moderate weakness of foreharm muscles (MRC score 3–4). Intrinsic hand muscles atrophy Distal lower limbs weakness (MRC score 4–5) Widespread fasciculations Brisk tendon reflexes both at upper and lower limbs |
| Thirty-six months from symptoms onset | Dysarthria and dysphagia Pseudobulbar affect Upper and lower limbs weakness and atrophy Widespread fasciculations Orthopnoea requiring non-invasive ventilation a few months before | Dysarthria and tongue fasciculations. Mild weakness of soft palate Severe weakness of intrinsic hand muscles (MRC score 0 on the right and 1 on the left) Severe weakness of foreharm (MRC score 1–2) and arm (MRC score 2–3) muscles Bilateral foot drop and moderate proximal lower limbs weakness (MRC score 3) Distal limbs muscles atrophy Widespread fasciculation Brisk tendon reflexes at lower limbs |
| Forty months from symptoms onset | Anarthria, requiring eye-tracking communication system Dysphagia with nutrition possible only by percutaneous endoscopic gastrostomy Pseudobulbar affect Sialorrhea Severe tetraparesis with limbs atrophy Dyspnea and orthopnea requiring continuous invasive ventilation through tracheostomy | Anarthria; tongue weakness and fasciculations Palatal palsy Severe tetraparesis (MRC score 0–1) Widespread muscle atrophy Widespread fasciculations Brisk tendon reflexes at lower limbs with mild spasticity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanini, G.; Selleri, V.; Nasi, M.; De Gaetano, A.; Martinelli, I.; Gianferrari, G.; Lofaro, F.D.; Boraldi, F.; Mandrioli, J.; Pinti, M. Mitochondrial and Endoplasmic Reticulum Alterations in a Case of Amyotrophic Lateral Sclerosis Caused by TDP-43 A382T Mutation. Int. J. Mol. Sci. 2022, 23, 11881. https://doi.org/10.3390/ijms231911881
Zanini G, Selleri V, Nasi M, De Gaetano A, Martinelli I, Gianferrari G, Lofaro FD, Boraldi F, Mandrioli J, Pinti M. Mitochondrial and Endoplasmic Reticulum Alterations in a Case of Amyotrophic Lateral Sclerosis Caused by TDP-43 A382T Mutation. International Journal of Molecular Sciences. 2022; 23(19):11881. https://doi.org/10.3390/ijms231911881
Chicago/Turabian StyleZanini, Giada, Valentina Selleri, Milena Nasi, Anna De Gaetano, Ilaria Martinelli, Giulia Gianferrari, Francesco Demetrio Lofaro, Federica Boraldi, Jessica Mandrioli, and Marcello Pinti. 2022. "Mitochondrial and Endoplasmic Reticulum Alterations in a Case of Amyotrophic Lateral Sclerosis Caused by TDP-43 A382T Mutation" International Journal of Molecular Sciences 23, no. 19: 11881. https://doi.org/10.3390/ijms231911881