
Epigenetic Regulation by microRNAs in Hyperhomocysteinemia-Accelerated Atherosclerosis
 ,
,  , and
, and
Abstract
:1. Introduction
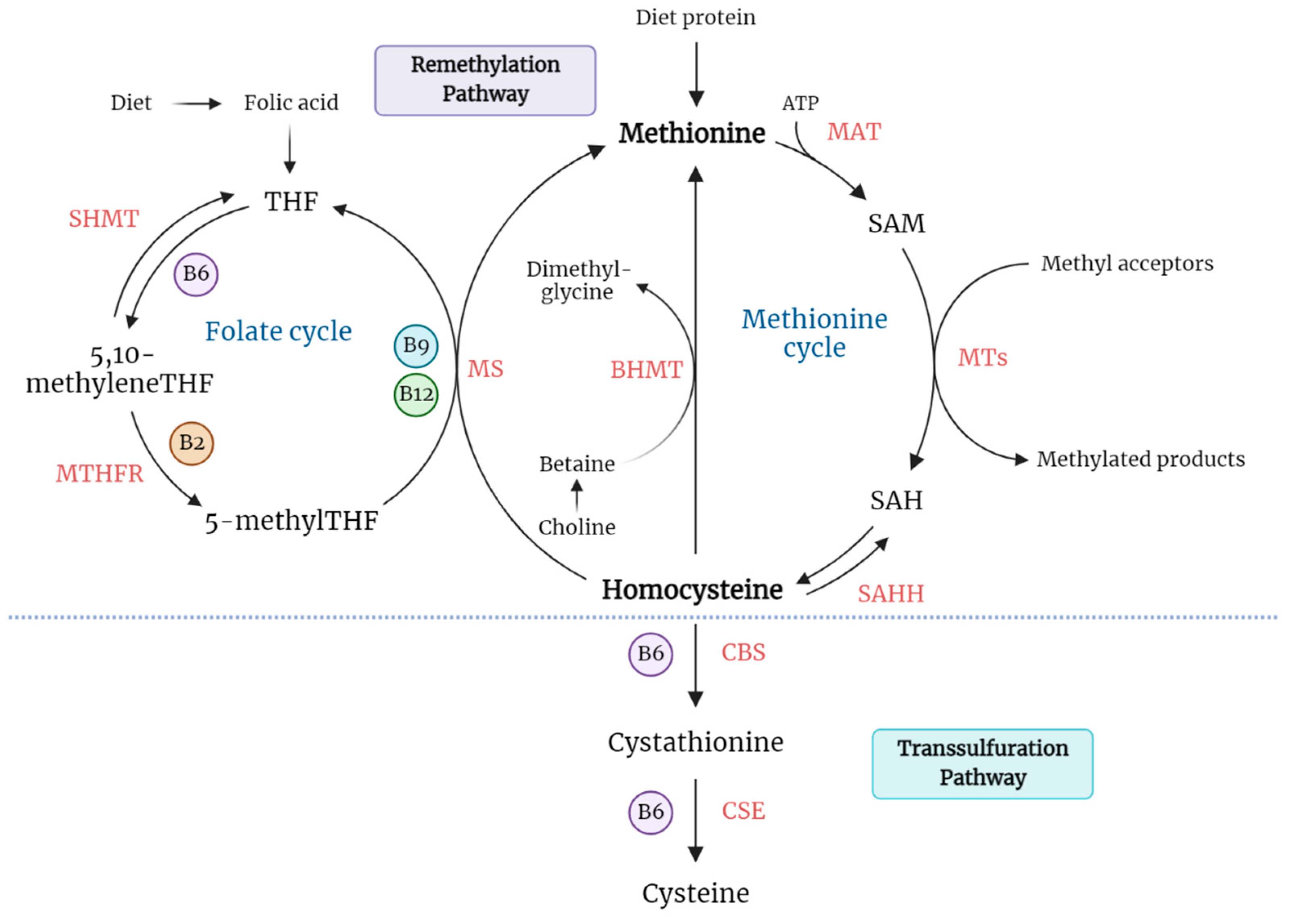
1.1. Homocysteine Biosynthesis and Catabolism
1.2. Hyperhomocysteinemia as a Risk Factor in CVD
1.3. Atherosclerosis-Related Hyperhomocysteinemia
2. Epigenetic Regulation of Hyperhomocysteinemia-Related Atherosclerosis
miRNA Regulation of Hyperhomocysteinemia-Related Atherosclerosis
3. Target Therapies in HHcy-Related Atherosclerosis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Wald, D.S. Homocysteine and cardiovascular disease: Evidence on causality from a meta-analysis. BMJ 2002, 325, 1202–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma Homocysteine as a Risk Factor for Dementia and Alzheimer’s Disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.A.; Hewedi, D.H.; Eissa, A.M.; Frydecka, D.; Misiak, B. Homocysteine levels in schizophrenia and affective disorders—focus on cognition. Front. Behav. Neurosci. 2014, 8, 343. [Google Scholar] [CrossRef] [Green Version]
- Daly, S.; Cotter, A.; Molloy, A.E.; Scott, J. Homocysteine and Folic Acid: Implications for Pregnancy. Semin. Vasc. Med. 2005, 5, 190–200. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, R.; Qu, Y.Y.; Mei, X.Y.; Zhang, X.; Zhou, Q.; Li, Y.; Yang, S.B.; Zuo, Z.G.; Chen, Y.M.; et al. Colonic Lysine Homocysteinylation Induced by High-Fat Diet Suppresses DNA Damage Repair. Cell Rep. 2018, 25, 398–412.e6. [Google Scholar] [CrossRef] [Green Version]
- Jakubowski, H. Protein N-Homocysteinylation and Colorectal Cancer. Trends Cancer 2019, 5, 7–10. [Google Scholar] [CrossRef]
- García-Tevijano, E.R.; Berasain, C.; Rodríguez, J.A.; Corrales, F.J.; Arias, R.; Martín-Duce, A.; Caballería, J.; Mato, J.M.; Avila, M.A. Hyperhomocysteinemia in Liver Cirrhosis. Hypertension 2001, 38, 1217–1221. [Google Scholar] [CrossRef] [Green Version]
- Gjesdal, C.G. Plasma Total Homocysteine Level and Bone Mineral Density. Arch. Intern. Med. 2006, 166, 88. [Google Scholar] [CrossRef] [Green Version]
- Homocysteine Studies Collaboration. Homocysteine and Risk of Ischemic Heart Disease and Stroke. JAMA 2002, 288, 2015–2022. [Google Scholar] [CrossRef]
- Feinberg, M.W.; Moore, K.J. MicroRNA Regulation of Atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Bønaa, K.H.; Njølstad, I.; Ueland, P.M.; Schirmer, H.; Tverdal, A.; Steigen, T.; Wang, H.; Nordrehaug, J.E.; Arnesen, E.; Rasmussen, K. Homocysteine Lowering and Cardiovascular Events after Acute Myocardial Infarction. N. Engl. J. Med. 2006, 354, 1578–1588. [Google Scholar] [CrossRef] [Green Version]
- Albert, C.M.; Cook, N.R.; Gaziano, J.M.; Zaharris, E.; MacFadyen, J.; Danielson, E.; Buring, J.E.; Manson, J.E. Effect of Folic Acid and B Vitamins on Risk of Cardiovascular Events and Total Mortality Among Women at High Risk for Cardiovascular Disease. JAMA 2008, 299, 2027. [Google Scholar] [CrossRef] [Green Version]
- Ebbing, M.; Bleie, Ø.; Ueland, P.M.; Nordrehaug, J.E.; Nilsen, D.W.; Vollset, S.E.; Refsum, H.; Ringdal Pedersen, E.K.; Nygård, O. Mortality and Cardiovascular Events in Patients Treated With Homocysteine-Lowering B Vitamins After Coronary Angiography. JAMA 2008, 300, 795. [Google Scholar] [CrossRef] [Green Version]
- Kaye, A.D.; Jeha, G.M.; Pham, A.D.; Fuller, M.C.; Lerner, Z.I.; Sibley, G.T.; Cornett, E.M.; Urits, I.; Viswanath, O.; Kevil, C.G. Folic Acid Supplementation in Patients with Elevated Homocysteine Levels. Adv. Ther. 2020, 37, 4149. [Google Scholar] [CrossRef]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halašová, E.; Lehotský, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2012, 17, 1733. [Google Scholar] [CrossRef] [Green Version]
- Schalinske, K.L.; Smazal, A.L. Homocysteine Imbalance: A Pathological Metabolic Marker. Adv. Nutr. 2012, 3, 755. [Google Scholar] [CrossRef] [Green Version]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef] [Green Version]
- Durand, P.; Prost, M.; Loreau, N.; Lussier-Cacan, S.; Blache, D. Impaired homocysteine metabolism and atherothrombotic disease. Lab. Investig. 2001, 81, 645–672. [Google Scholar] [CrossRef]
- Castro, R.; Rivera, I.; Blom, H.J.; Jakobs, C.; de Almeida, I.T. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J. Inherit. Metab. Dis. 2006, 29, 3–20. [Google Scholar] [CrossRef]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.; Caruso, P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 231. [Google Scholar] [CrossRef] [Green Version]
- Tinelli, C.; di Pino, A.; Ficulle, E.; Marcelli, S.; Feligioni, M. Hyperhomocysteinemia as a Risk Factor and Potential Nutraceutical Target for Certain Pathologies. Front. Nutr. 2019, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Stipanuk, M.H.; Ueki, I. Dealing with methionine/homocysteine sulfur: Cysteine metabolism to taurine and inorganic sulfur. J. Inherit. Metab. Dis. 2011, 34, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Capelli, I.; Cianciolo, G.; Gasperoni, L.; Zappulo, F.; Tondolo, F.; Cappuccilli, M.; la Manna, G. Folic Acid and Vitamin B12 Administration in CKD, Why Not? Nutrients 2019, 11, 383. [Google Scholar] [CrossRef] [Green Version]
- Bajic, Z.; Sobot, T.; Skrbic, R.; Stojiljkovic, M.P.; Ponorac, N.; Matavulj, A.; Djuric, D.M. Homocysteine, Vitamins B6 and Folic Acid in Experimental Models of Myocardial Infarction and Heart Failure-How Strong Is That Link? Biomolecules 2022, 12, 536. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, X.; Kong, W. Hyperhomocysteinaemia and vascular injury: Advances in mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1173–1189. [Google Scholar] [CrossRef] [Green Version]
- Lip, G.Y.H.; Edmunds, E.; Martin, S.C.; Jones, A.F.; Blann, A.D.; Beevers, D.G. A pilot study of homocyst(e)ine levels in essential hypertension: Relationship to von Willebrand factor, an index of endothelial damage. Am. J. Hypertens. 2001, 14, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Fang, K.; Chen, Z.; Liu, M.; Peng, J.; Wu, P. Apoptosis and calcification of vascular endothelial cell under hyperhomocysteinemia. Med. Oncol. 2015, 32, 403. [Google Scholar] [CrossRef]
- Liu, Z.; Luo, H.; Zhang, L.; Huang, Y.; Liu, B.; Ma, K.; Feng, J.; Xie, J.; Zheng, J.; Hu, J.; et al. Hyperhomocysteinemia exaggerates adventitial inflammation and angiotensin II-induced abdominal aortic aneurysm in mice. Circ. Res. 2012, 111, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2013581/ (accessed on 9 September 2022).
- Kim, J.; Kim, H.; Roh, H.; Kwon, Y. Causes of hyperhomocysteinemia and its pathological significance. Arch. Pharm. Res. 2018, 41, 372–383. [Google Scholar] [CrossRef]
- Levin, B.L.; Varga, E. MTHFR: Addressing Genetic Counseling Dilemmas Using Evidence-Based Literature. J. Genet. Couns. 2016, 25, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Makris, M. Hyperhomocysteinemia and thrombosis. Clin. Lab. Haematol. 2000, 22, 133–143. [Google Scholar] [CrossRef]
- Cattaneo, M. Hyperhomocysteinemia, atherosclerosis and thrombosis. Thromb. Haemost. 1999, 81, 165–176. [Google Scholar] [CrossRef]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, W.; Herrmann, M. The Controversial Role of HCY and Vitamin B Deficiency in Cardiovascular Diseases. Nutrients 2022, 14, 1412. [Google Scholar] [CrossRef]
- Hankey, G.J.; Eikelboom, J.W. Homocysteine and vascular disease. Lancet 1999, 354, 407–413. [Google Scholar] [CrossRef]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Daly, L.; Robinson, K.; Naughten, E.; Cahalane, S.; Fowler, B.; Graham, I. Hyperhomocysteinemia: An independent risk factor for vascular disease. N. Engl. J. Med. 1991, 324, 1149–1155. [Google Scholar] [CrossRef]
- Mousavi, S.A.; Ghasemi, M.; Hoseini, T. Association between plasma homocysteine concentrations and extracranial carotid stenosis. Ann. Saudi Med. 2006, 26, 120–122. [Google Scholar] [CrossRef]
- Ueland, P.M.; Refsum, H.; Beresford, S.A.A.; Vollset, S.E. The controversy over homocysteine and cardiovascular risk. Am. J. Clin. Nutr. 2000, 72, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Falk, E.; Zhou, J.; Møller, J. Homocysteine and atherothrombosis. Lipids 2001, 36, S3–S11. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, H.; Liu, N.; Chen, J.; Gu, Y.; Chen, J.; Yang, K. Role of Hyperhomocysteinemia and Hyperuricemia in Pathogenesis of Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2017, 26, 2695–2699. [Google Scholar] [CrossRef]
- Balint, B.; Jepchumba, V.K.; Guéant, J.L.; Guéant-Rodriguez, R.M. Mechanisms of homocysteine-induced damage to the endothelial, medial and adventitial layers of the arterial wall. Biochimie 2020, 173, 100–106. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef]
- Dayal, S.; Lentz, S.R. ADMA and hyperhomocysteinemia. Vasc. Med. 2005, 10, S27–S33. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, F.; Tan, H.; Liao, D.; Bryan, R.M.; Randhawa, J.K.; Rumbaut, R.E.; Durante, W.; Schafer, A.I.; Yang, X.; et al. Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arter. Thromb. Vasc. Biol. 2005, 25, 2515–2521. [Google Scholar] [CrossRef] [Green Version]
- Yan, T.T.; Li, Q.; Zhang, X.H.; Wu, W.K.; Sun, J.; Li, L.; Zhang, Q.; Tan, H.M. Homocysteine impaired endothelial function through compromised vascular endothelial growth factor/Akt/endothelial nitric oxide synthase signalling. Clin. Exp. Pharmacol. Physiol. 2010, 37, 1071–1077. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Ali, F.; Bailey, L.; Moreno, L.; Harrington, L.S. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp. Physiol. 2008, 93, 141–147. [Google Scholar] [CrossRef]
- Nording, H.M.; Seizer, P.; Langer, H.F. Platelets in inflammation and atherogenesis. Front. Immunol. 2015, 6, 98. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Ye, Z.X.; Wang, X.F.; Chang, J.; Yang, M.W.; Zhong, H.H.; Hong, F.F.; Yang, S.L. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed. Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Gorica, E.; Brogi, S.; Testai, L.; Calderone, V. Role of hydrogen sulfide in endothelial dysfunction: Pathophysiology and therapeutic approaches. J. Adv. Res. 2020, 27, 99–113. [Google Scholar] [CrossRef]
- Paganelli, F.; Mottola, G.; Fromonot, J.; Marlinge, M.; Deharo, P.; Guieu, R.; Ruf, J. Hyperhomocysteinemia and Cardiovascular Disease: Is the Adenosinergic System the Missing Link? Int. J. Mol. Sci. 2021, 22, 1690. [Google Scholar] [CrossRef]
- Esse, R.; Barroso, M.; de Almeida, I.T.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Jacobsen, D.W. Hyperhomocysteinemia and Oxidative Stress. Arter. Thromb. Vasc. Biol. 2000, 20, 1182–1184. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.K.C.; Kan, M.Y. Homocysteine-Induced Endothelial Dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef]
- Chan, S.-H.; Hung, C.-H.; Shih, J.-Y.; Chu, P.-M.; Cheng, Y.-H.; Lin, H.-C.; Hsieh, P.-L.; Tsai, K.-L. Exercise intervention attenuates hyperhomocysteinemia-induced aortic endothelial oxidative injury by regulating SIRT1 through mitigating NADPH oxidase/LOX-1 signaling. Redox Biol. 2018, 14, 116–125. [Google Scholar] [CrossRef]
- Sipkens, J.A.; Hahn, N.; den Brand, C.S.; Meischl, C.; Cillessen, S.A.G.M.; Smith, D.E.C.; Juffermans, L.J.M.; Musters, R.J.P.; Roos, D.; Jakobs, C.; et al. Homocysteine-Induced Apoptosis in Endothelial Cells Coincides With Nuclear NOX2 and Peri-nuclear NOX4 Activity. Cell Bio. Biophys 2013, 67, 341–352. [Google Scholar] [CrossRef]
- Richard, E.; Gallego-Villar, L.; Rivera-Barahona, A.; Oyarzábal, A.; Pérez, B.; Rodríguez-Pombo, P.; Desviat, L.R. Altered Redox Homeostasis in Branched-Chain Amino Acid Disorders, Organic Acidurias, and Homocystinuria. Oxid. Med. Cell. Longev. 2018, 2018, 1246069. [Google Scholar] [CrossRef] [Green Version]
- Austin, R.C.; Sood, S.K.; Dorward, A.M.; Singh, G.; Shaughnessy, S.G.; Pamidi, S.; Outinen, P.A.; Weitz, J.I. Homocysteine-dependent Alterations in Mitochondrial Gene Expression, Function and Structure. J. Biol. Chem. 1998, 273, 30808–30817. [Google Scholar] [CrossRef] [Green Version]
- Topal, G.; Brunet, A.; Millanvoye, E.; Boucher, J.-L.; Rendu, F.; Devynck, M.-A.; David-Dufilho, M. Homocysteine induces oxidative stress by uncoupling of no synthase activity through reduction of tetrahydrobiopterin. Free Radic. Biol. Med. 2004, 36, 1532–1541. [Google Scholar] [CrossRef]
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. -Heart Circ. Physiol. 2005, 289, H2649–H2656. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Zhang, Y.; Loscalzo, J. Homocysteine Down-regulates Cellular Glutathione Peroxidase (GPx1) by Decreasing Translation. J. Biol. Chem. 2005, 280, 15518–15525. [Google Scholar] [CrossRef] [Green Version]
- Faverzani, J.L.; Hammerschmidt, T.G.; Sitta, A.; Deon, M.; Wajner, M.; Vargas, C.R. Oxidative Stress in Homocystinuria Due to Cystathionine ß-Synthase Deficiency: Findings in Patients and in Animal Models. Cell. Mol. Neurobiol. 2017, 37, 1477–1485. [Google Scholar] [CrossRef]
- Sawle, P.; Foresti, R.; Green, C.J.; Motterlini, R. Homocysteine attenuates endothelial haem oxygenase-1 induction by nitric oxide (NO) and hypoxia. FEBS Lett. 2001, 508, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wei, C.; Zhou, Y.; Yan, T.; Wang, Z.; Li, W.; Zhao, L. Homocysteine Induces Apoptosis of Human Umbilical Vein Endothelial Cells via Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Oxid. Med. Cell. Longev. 2017, 2017, 5736506. [Google Scholar] [CrossRef]
- Yang, S.; Wu, M.; Li, X.; Zhao, R.; Zhao, Y.; Liu, L.; Wang, S. Role of Endoplasmic Reticulum Stress in Atherosclerosis and Its Potential as a Therapeutic Target. Oxid. Med. Cell. Longev. 2020, 2020, 9270107. [Google Scholar] [CrossRef]
- Kumar, A.; Palfrey, H.A.; Pathak, R.; Kadowitz, P.J.; Gettys, T.W.; Murthy, S.N. The metabolism and significance of homocysteine in nutrition and health. Nutr. Metab. 2017, 14, 78. [Google Scholar] [CrossRef]
- Becker, A.; Kostense, P.J.; Bos, G.; Heine, R.J.; Dekker, J.M.; Nijpels, G.; Bouter, L.M.; Stehouwer, C.D.A. Hyperhomocysteinaemia is associated with coronary events in type 2 diabetes. J. Intern. Med. 2003, 253, 293–300. [Google Scholar] [CrossRef] [Green Version]
- de Luis, D.A.; Fernandez, N.; Arranz, M.L.; Aller, R.; Izaola, O.; Romero, E. Total homocysteine levels relation with chronic complications of diabetes, body composition, and other cardiovascular risk factors in a population of patients with diabetes mellitus type 2. J. Diabetes Complicat. 2005, 19, 42–46. [Google Scholar] [CrossRef]
- Soinio, M.; Marniemi, J.; Laakso, M.; Lehto, S.; Rönnemaa, T. Elevated Plasma Homocysteine Level Is an Independent Predictor of Coronary Heart Disease Events in Patients with Type 2 Diabetes Mellitus. Ann. Intern. Med. 2004, 140, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Najib, S.; Sanchez-Margalet, V. Homocysteine thiolactone inhibits insulin signaling, and glutathione has a protective effect. J. Mol. Endocrinol. 2001, 27, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, C.; Xu, G.; Wang, N.; Zhu, Y.; Tang, C.; Wang, X. Homocysteine Upregulates Resistin Production From Adipocytes In Vivo and In Vitro. Diabetes 2008, 57, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Huang, Y.; Hu, Q.; Ma, L. Hyperhomocysteinemia stimulates hepatic glucose output and PEPCK expression. Acta Biochim. Biophys. Sin. 2009, 41, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.; Žák, A.; Vecka, M.; Tvrzická, E.; Písaříková, A.; Staňková, B. N-3 fatty acid supplementation decreases plasma homocysteine in diabetic dyslipidemia treated with statin–fibrate combination. J. Nutr. Biochem. 2006, 17, 379–384. [Google Scholar] [CrossRef]
- Huang, T.; Hu, X.; Khan, N.; Yang, J.; Li, D. Effect of Polyunsaturated Fatty Acids on Homocysteine Metabolism through Regulating the Gene Expressions Involved in Methionine Metabolism. Sci. World J. 2013, 2013, 931626. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.C.; Perrella, M.A.; Yoshizumi, M.; Hsieh, C.M.; Haber, E.; Schlegel, R.; Lee, M.E. Promotion of vascular smooth muscle cell growth by homocysteine: A link to atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 6369–6373. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, A.; Verdoia, M.; Cassetti, E.; Marino, P.; Suryapranata, H.; de Luca, G. Relationship between homocysteine and coronary artery disease. Results from a large prospective cohort study. Thromb. Res. 2014, 134, 288–293. [Google Scholar] [CrossRef]
- Tribouilloy, C.M.; Peltier, M.; Peltier, M.C.I.; Trojette, F.; Andrejak, M.; Lesbre, J.-P.M. Plasma Homocysteine and Severity of Thoracic Aortic Atherosclerosis. Chest 2000, 118, 1685–1689. [Google Scholar] [CrossRef]
- Napoli, C.; Crudele, V.; Soricelli, A.; Al-Omran, M.; Vitale, N.; Infante, T.; Mancini, F.P. Primary Prevention of Atherosclerosis. Circulation 2012, 125, 2363–2373. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Ardekani, A.N.M. The Role of MicroRNAs in Human Diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar]
- Zhang, H.-P.; Wang, Y.-H.; Cao, C.-J.; Yang, X.-M.; Ma, S.-C.; Han, X.-B.; Yang, X.-L.; Yang, A.-N.; Tian, J.; Xu, H.; et al. A regulatory circuit involving miR-143 and DNMT3a mediates vascular smooth muscle cell proliferation induced by homocysteine. Mol. Med. Rep. 2016, 13, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Zhang, H.; Zhao, L.; Zhou, L.; Zhang, M.; Xu, H.; Han, X.; Li, G.; Yang, X.; Jiang, Y. miR-125b targets DNMT3b and mediates p53 DNA methylation involving in the vascular smooth muscle cells proliferation induced by homocysteine. Exp. Cell Res. 2016, 347, 95–104. [Google Scholar] [CrossRef]
- Xiaoling, Y.; Li, Z.; ShuQiang, L.; Shengchao, M.; Anning, Y.; Ning, D.; Nan, L.; Yuexia, J.; Xiaoming, Y.; Guizhong, L.; et al. Hyperhomocysteinemia in ApoE-/- Mice Leads to Overexpression of Enhancer of Zeste Homolog 2 via miR-92a Regulation. PLoS ONE 2016, 11, e0167744. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Sun, Y.; Gao, Y.; Yang, S.; Mao, C.; Ding, N.; Deng, M.; Wang, Y.; Yang, X.; Jia, Y.; et al. Reciprocal Regulation Between miR-148a/152 and DNA Methyltransferase 1 Is Associated with Hyperhomocysteinemia-Accelerated Atherosclerosis. DNA Cell Biol. 2017, 36, 462–474. [Google Scholar] [CrossRef]
- Xiong, J.; Ma, F.; Ding, N.; Xu, L.; Ma, S.; Yang, A.; Hao, Y.; Zhang, H.; Jiang, Y. miR-195-3p alleviates homocysteine-mediated atherosclerosis by targeting IL-31 through its epigenetics modifications. Aging Cell 2021, 20, e13485. [Google Scholar] [CrossRef]
- Liu, K.; Xuekelati, S.; Zhang, Y.; Yin, Y.; Li, Y.; Chai, R.; Li, X.; Peng, Y.; Wu, J.; Guo, X. Expression levels of atherosclerosis-associated miR-143 and miR-145 in the plasma of patients with hyperhomocysteinaemia. BMC Cardiovasc. Disord. 2017, 17, 163. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Xuekelati, S.; Zhou, K.; Yan, Z.; Yang, X.; Inayat, A.; Wu, J.; Guo, X. Expression Profiles of Six Atherosclerosis-Associated microRNAs That Cluster in Patients with Hyperhomocysteinemia: A Clinical Study. DNA Cell Biol. 2018, 37, 189–198. [Google Scholar] [CrossRef]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. Efficacy of Folic Acid Therapy in Primary Prevention of Stroke Among Adults With Hypertension in China. JAMA 2015, 313, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, V.A.; Galan, P.; Torrès, M.; Julia, C.; Hercberg, S.; Kesse-Guyot, E. Supplementation with B vitamins or n−3 fatty acids and depressive symptoms in cardiovascular disease survivors: Ancillary findings from the SUpplementation with FOLate, vitamins B-6 and B-12 and/or OMega-3 fatty acids (SU.FOL.OM3) randomized trial. Am. J. Clin. Nutr. 2012, 96, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Lonn, E.; Yusuf, S.; Arnold, M.J.; Sheridan, P.; Pogue, J.; Micks, M.; McQueen, M.J.; Probstfield, J.; Fodor, G.; Held, C.; et al. Homocysteine Lowering with Folic Acid and B Vitamins in Vascular Disease. N. Engl. J. Med. 2006, 354, 1567–1577. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.D. Homocysteine lowering for stroke prevention: Unravelling the complexity of the evidence. Int. J. Stroke 2016, 11, 744–747. [Google Scholar] [CrossRef]
- Spence, J.D.; Hankey, G.J. Problem in the Recent American Heart Association Guideline on Secondary Stroke Prevention: B Vitamins to Lower Homocysteine Do Prevent Stroke. Stroke 2022, 53, 2702–2708. [Google Scholar] [CrossRef]

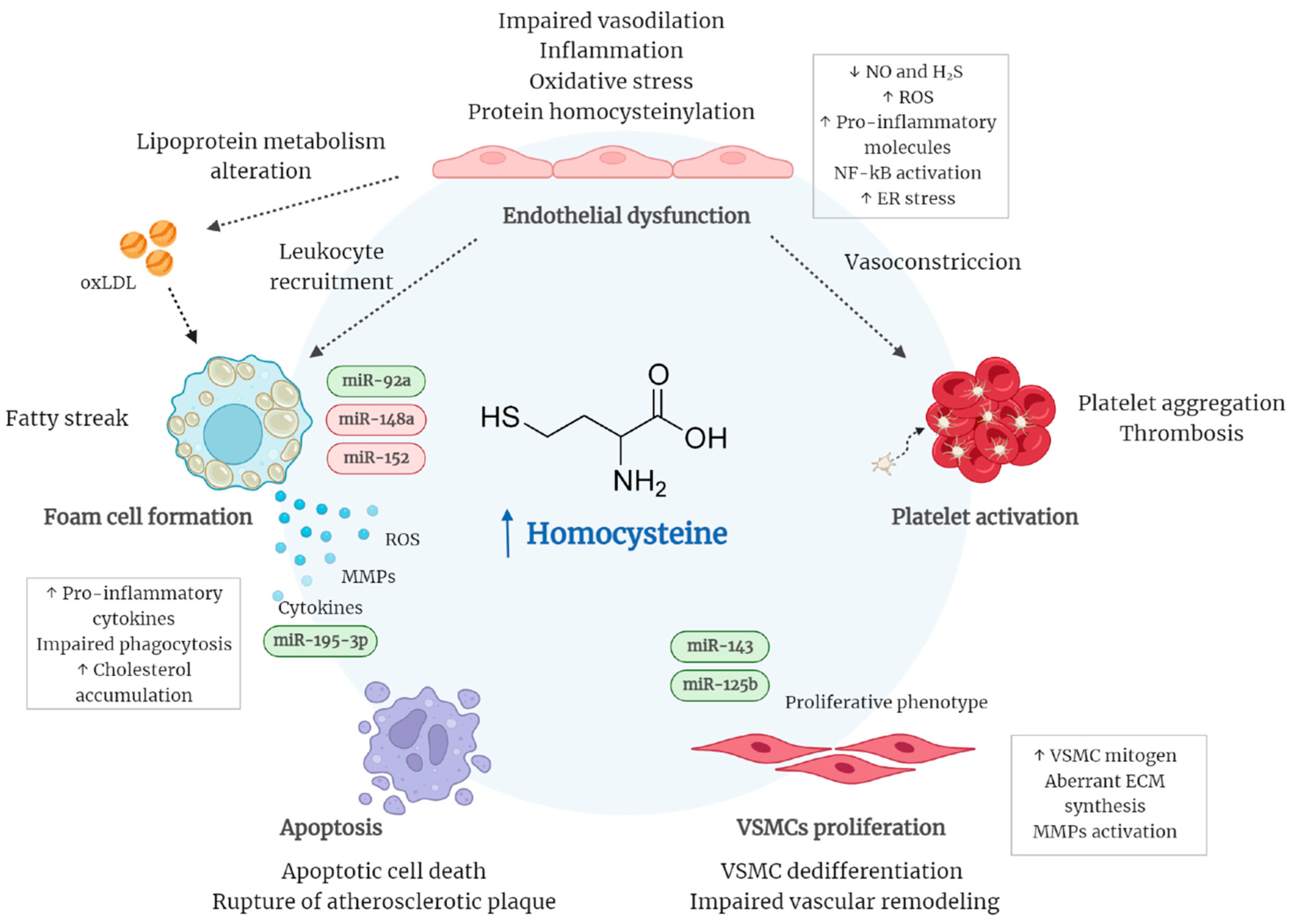

{kind=link}
{kind=link}
| Studies | Treatment | Epigenetic Alterations | Effects | Cell Culture/ Animal Model |
|---|---|---|---|---|
| Ref. [86] | 50, 100, 200, 500 µM of Hcy for 72 h | ↑DNMT3a mRNA ↓miR-143 | ↑Proliferation of VSMCs | Human VSMCs |
| Ref. [87] | 50, 100, 200, 500µM of Hcy for 72 h/ High-Met diet for 15 weeks | ↓miR-125b ↑DNMT3b protein ↑p53 DNA methylation | ↑Proliferation of VSMCs | Human VSMC/ ApoE-/- mice |
| Ref. [88] | High-Met diet for 16 weeks | ↓miR-92a ↑EZH2 mRNA ↑H3K27me3 | ↑Accumulation of total cholesterol and triglycerides in foam cells | ApoE-/- mice |
| Ref. [89] | High-Met diet for 20 weeks | ↓DNMT1 mRNA ↑miR-148a/152 ↑ADRP mRNA | ↑Accumulation of total cholesterol and cholesteryl ester in foam cells | ApoE-/- mice |
| Ref. [90] | High-Met diet for 16 weeks | ↑DNMT3a and HDAC11 mRNA ↓miR-195-3p ↑IL-31 | ↑Macrophage inflammation and atherosclerotic plaque formation | ApoE-/- mice |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griñán, R.; Escolà-Gil, J.C.; Julve, J.; Benítez, S.; Rotllan, N. Epigenetic Regulation by microRNAs in Hyperhomocysteinemia-Accelerated Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 12452. https://doi.org/10.3390/ijms232012452
Griñán R, Escolà-Gil JC, Julve J, Benítez S, Rotllan N. Epigenetic Regulation by microRNAs in Hyperhomocysteinemia-Accelerated Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(20):12452. https://doi.org/10.3390/ijms232012452
Chicago/Turabian StyleGriñán, Raquel, Joan Carles Escolà-Gil, Josep Julve, Sonia Benítez, and Noemí Rotllan. 2022. "Epigenetic Regulation by microRNAs in Hyperhomocysteinemia-Accelerated Atherosclerosis" International Journal of Molecular Sciences 23, no. 20: 12452. https://doi.org/10.3390/ijms232012452