
Reactive Centre Loop Mutagenesis of SerpinB3 to Target TMPRSS2 and Furin: Inhibition of SARS-CoV-2 Cell Entry and Replication
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Designer SerpinB3—RCL Mutagenesis
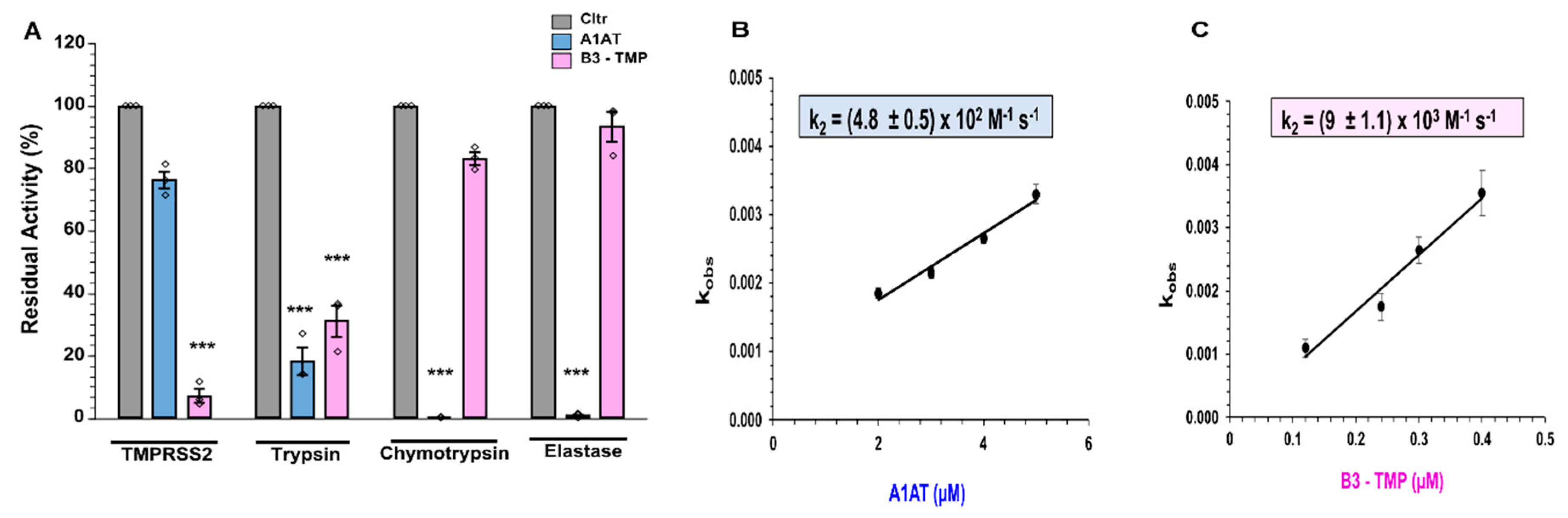
2.2. Inhibition of Cathepsin L, TMPRSS2 and Furin by SerpinB3 Variants
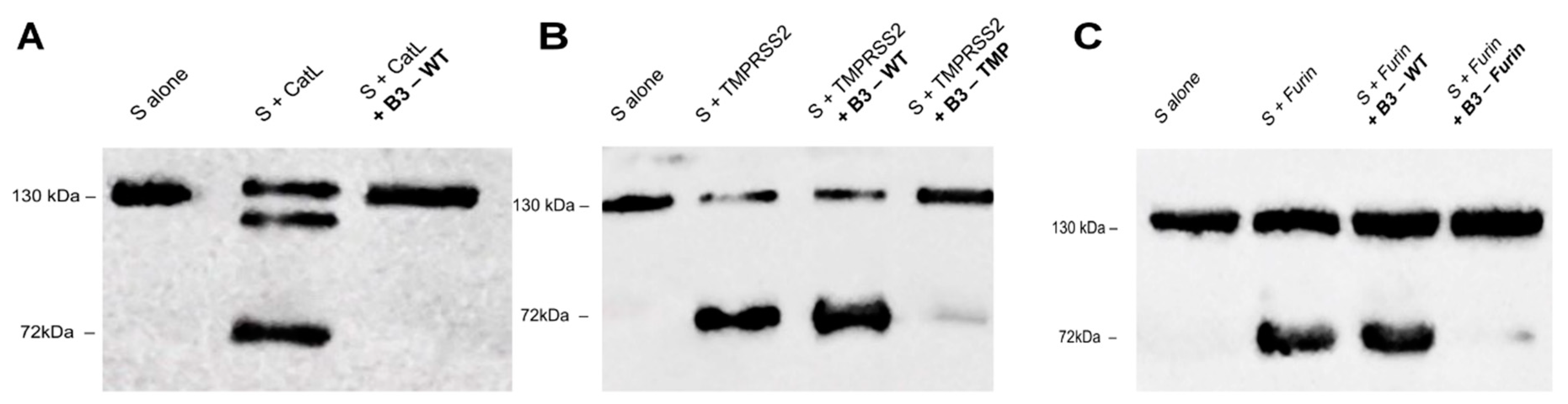
2.3. SerpinB3 Variants Can Prevent SARS-CoV-2 Spike Degradation In Vitro
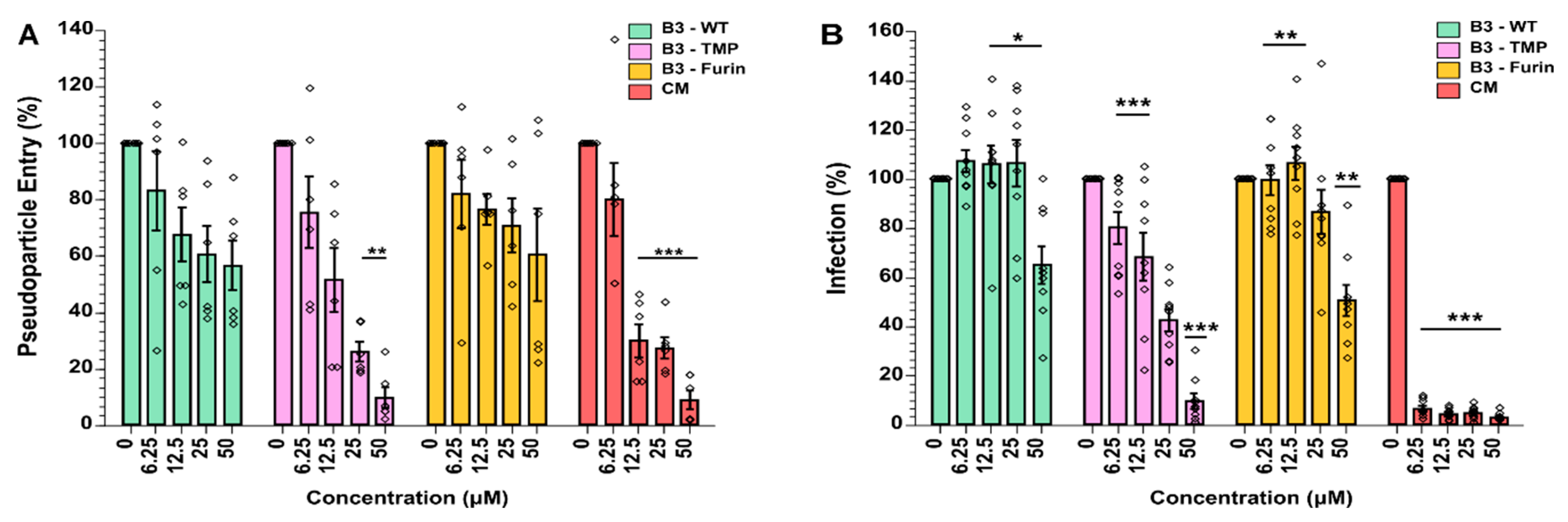
2.4. SerpinB3 and Modified Variants Inhibit SARS-CoV-2 Spike Mediated Pseudovirus Cell Entry
2.5. SerpinB3 Anti-TMPRSS2 Inhibited SARS-CoV-2 Infection in VeroE6-TMPRSS2 Cells
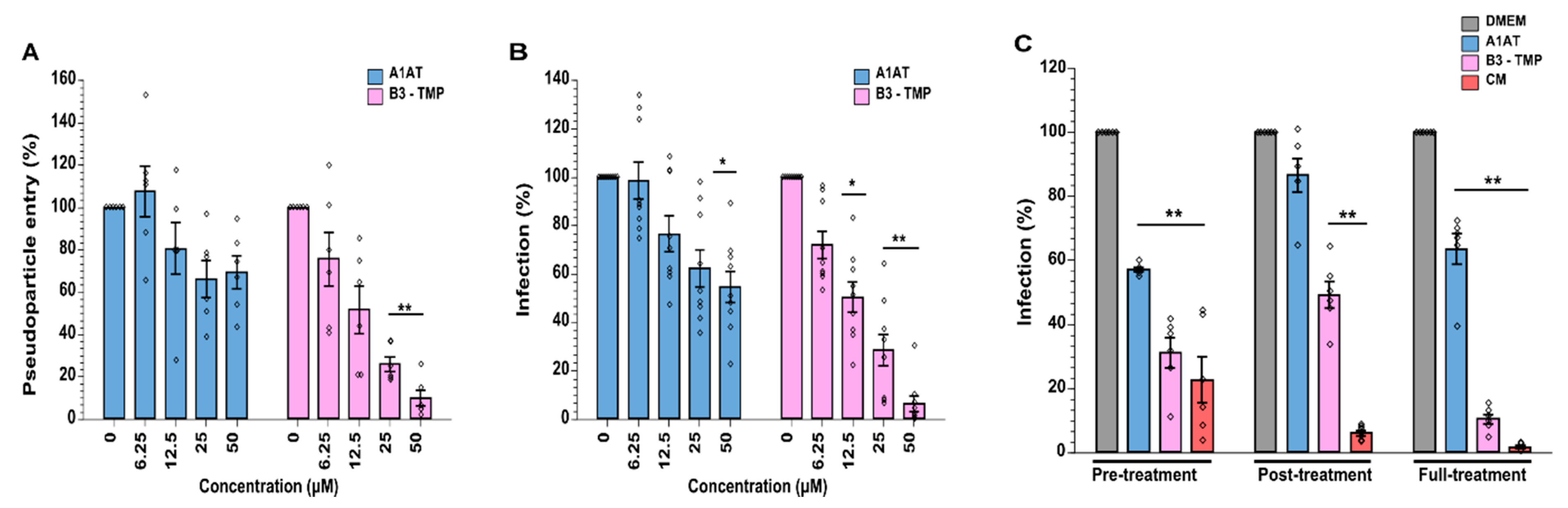
2.6. SerpinB3 Anti-TMPRSS2 (B3-TMP) Is More Effective Than A1AT at Inhibiting TMPRSS2 and Supressing SARS-CoV-2 Infection
3. Discussion
4. Materials and Methods
4.1. Mutagenesis of SerpinB3
4.2. Expression and Purification of Recombinant Proteins
4.3. Protease Inhibition Assays
4.4. Spike Protein Cleavage
4.5. Cell Culture
4.6. Generation of Lentiviral Pseudoparticles
4.7. Assay for Pseudoparticle Cell Entry Inhibition
4.8. SARS-CoV-2 Clinical Isolates
4.9. VeroE6-TMPRSS2 Viral Infection Assay
4.10. Flow Cytometry Analysis of SARS-CoV-2 Infected Cells
4.11. Cell Viability Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A1AT | Alpha-1 antitrypsin |
| ACE2 | Angiotensin-converting enzyme 2 |
| CM | Camostat mesylate |
| CatL | Cathepsin L |
| COVID-19 | Coronavirus disease 2019 |
| IMAC | Immobilized metal affinity chromatography |
| k2 | Second order inhibition rate constant |
| NE | No-glycoprotein, no-enveloped pseudoparticle control |
| RCL | Reactive centre loop |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SDM | Site-directed mutagenesis |
| SERPIN | Serine protease inhibitor |
| S | Spike protein |
| Spp | Spike pseudoparticles |
| TMPRSS2 | Transmembrane protease serine 2 |
| WT | Wild type |
References
- Lee, S.J.; Kim, Y.J.; Ahn, D.G. Distinct Molecular Mechanisms Characterizing Pathogenesis of SARS-CoV-2. J. Microbiol. Biotechnol. 2022, 32, 1073–1085. [Google Scholar] [CrossRef]
- O’Reilly, S.; Angeliadis, M.; Murtagh, R.; Gautier, V.W. Drug repurposing and other strategies for rapid coronavirus antiviral development: Lessons from the early stage of the COVID-19 pandemic. Eur. Respir. Monogr. 2021, 4, 39–68. [Google Scholar]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pohlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e775. [Google Scholar] [CrossRef]
- Jaimes, J.; Millet, J.; Whittaker, G. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. IScience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Laporte, M.; Naesens, L. Airway proteases: An emerging drug target for influenza and other respiratory virus infections. Curr. Opin. Virol. 2017, 24, 16–24. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [Green Version]
- Cioccarelli, C.; Sánchez-Rodríguez, R.; Angioni, R.; Venegas, F.C.; Bertoldi, N.; Munari, F.; Cattelan, A.; Molon, B.; Viola, A. IL1beta Promotes TMPRSS2 Expression and SARS-CoV-2 Cell Entry Through the p38 MAPK-GATA2 Axis. Front. Immunol. 2021, 12, 781352. [Google Scholar] [CrossRef]
- Koch, J.; Uckeley, Z.M.; Doldan, P.; Stanifer, M.; Boulant, S.; Lozach, P. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821. [Google Scholar] [CrossRef]
- Saadat, S.; Tehrani, Z.R.; Logue, J.; Newman, M.; Frieman, M.B.; Harris, A.D.; Sajadi, M.M. Binding and Neutralization Antibody Titers After a Single Vaccine Dose in Health Care Workers Previously Infected With SARS-CoV-2. JAMA 2021, 325, 1467–1469. [Google Scholar] [CrossRef]
- Mahoney, M.; Damalanka, V.C.; Tartell, M.A.; Chung, D.H.; Lourenço, A.L.; Pwee, D.; Bridwell, A.E.M.; Hoffmann, M.; Voss, J.; Karmakar, P.; et al. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS-CoV viral entry and protect human epithelial lung cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2108728118. [Google Scholar] [CrossRef]
- Shapira, T.; Monreal, I.A.; Dion, S.P.; Buchholz, D.W.; Imbiakha, B.; Olmstead, A.D.; Jager, M.; Désilets, A.; Gao, G.; Martins, M.; et al. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature 2022, 605, 340–348. [Google Scholar] [CrossRef]
- Azouz, N.P.; Klingler, A.; Callahan, V.; Akhrymuk, I.; Elez, K.; Raich, L.; Henry, B.; Benoit, J.; Benoit, S.; Noé, F.; et al. Alpha 1 Antitrypsin is an Inhibitor of the SARS-CoV-2-Priming Protease TMPRSS2. Pathog. Immun. 2021, 6, 55–74. [Google Scholar] [CrossRef]
- Wettstein, L.; Weil, T.; Conzelmann, C.; Müller, J.A.; Groß, R.; Hirschenberger, M.; Seidel, A.; Klute, S.; Zech, F.; Bozzo, C.P.; et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat. Commun. 2021, 12, 1726. [Google Scholar] [CrossRef]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef]
- Schick, C.; Brömme, D.; Bartuski, A.J.; Uemura, Y.; Schechter, N.M.; Silverman, G.A. The reactive site loop of the serpin SCCA1 is essential for cysteine proteinase inhibition. Proc. Natl. Acad. Sci. USA 1998, 95, 13465–13470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlen, J.R.; Jean, F.; Thomas, G.; Foster, D.C.; Kisiel, W. Inhibition of soluble recombinant furin by human proteinase inhibitor 8. J. Biol. Chem. 1998, 273, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, G.; Qi, L.; Lima, M.; Olson, S.T. Identification of serpin determinants of specificity and selectivity for furin inhibition through studies of alpha1PDX (alpha1-protease inhibitor Portland)-serpin B8 and furin active-site loop chimeras. J. Biol. Chem. 2013, 288, 21802–21814. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef]
- Zhou, A.; Stein, P.E.; Huntington, J.A.; Carrell, R.W. Serpin polymerization is prevented by a hydrogen bond network that is centered on his-334 and stabilized by glycerol. J. Biol. Chem. 2003, 278, 15116–15122. [Google Scholar] [CrossRef] [Green Version]
- Scott, B.M.; Sheffield, W.P. Engineering the serpin alpha1-antitrypsin: A diversity of goals and techniques. Protein. Sci. 2020, 29, 856–871. [Google Scholar] [CrossRef]
- Takeda, M. Proteolytic activation of SARS-CoV-2 spike protein. Microbiol. Immunol. 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Zhao, M.-M.; Zhu, Y.; Zhang, L.; Zhong, G.; Tai, L.; Liu, S.; Yin, G.; Lu, J.; He, Q.; Li, M.-J.; et al. Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies. Cell Discov. 2022, 8, 53. [Google Scholar] [CrossRef]
- Cowden, D.I.; Fisher, G.E.; Weeks, R.L. A pilot study comparing the purity, functionality and isoform composition of alpha-1-proteinase inhibitor (human) products. Curr. Med. Res. Opin. 2005, 21, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Huntington, J.A.; Carrell, R.W. The serpins: Nature’s molecular mousetraps. Sci. Prog. 2001, 84, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.C.; Brennan, S.O.; Lewis, J.H.; Carrell, R.W. Mutation of antitrypsin to antithrombin. alpha 1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N. Engl. J. Med. 1983, 309, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Sulikowski, T.; Bauer, B.A.; Patston, P.A. alpha(1)-Proteinase inhibitor mutants with specificity for plasma kallikrein and C1s but not C1. Protein. Sci. 2002, 11, 2230–2236. [Google Scholar] [CrossRef] [PubMed]
- Marcet-Palacios, M.; Ewen, C.; Pittman, E.; Duggan, B.; Carmine-Simmen, K.; Fahlman, R.P.; Bleackley, R.C. Design and characterization of a novel human Granzyme B inhibitor. Protein. Eng. Des. Sel. 2015, 28, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Polderdijk, S.G.I.; Baglin, T.P.; Huntington, J.A. Targeting activated protein C to treat hemophilia. Curr. Opin. Hematol. 2017, 24, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K.; et al. Structure and activity of human TMPRSS2 protease implicated in SARS-CoV-2 activation. Nat. Chem. Biol. 2022, 18, 963–971. [Google Scholar] [CrossRef]
- Sanrattana, W.; Maas, C.; de Maat, S. SERPINs-From Trap to Treatment. Front. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Marijanovic, E.M.; Fodor, J.; Riley, B.T.; Porebski, B.T.; Costa, M.G.S.; Kass, I.; Hoke, D.E.; McGowan, S.; Buckle, A.M. Reactive centre loop dynamics and serpin specificity. Sci. Rep. 2019, 9, 3870. [Google Scholar] [CrossRef] [Green Version]
- Shimeld, S.M.; Donoghue, P.C. Evolutionary crossroads in developmental biology: Cyclostomes (lamprey and hagfish). Development 2012, 139, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Scott, B.M.; Matochko, W.L.; Gierczak, R.F.; Bhakta, V.; Derda, R.; Sheffield, W.P. Phage display of the serpin alpha-1 proteinase inhibitor randomized at consecutive residues in the reactive centre loop and biopanned with or without thrombin. PLoS ONE 2014, 9, e84491. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Feng, Q.; Wang, X. Computational analysis of TMPRSS2 expression in normal and SARS-CoV-2-infected human tissues. Chem. Biol. Interact. 2021, 346, 109583. [Google Scholar] [CrossRef]
- Thomas, G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.P. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef]
- Kim, T.S.; Heinlein, C.; Hackman, R.C.; Nelson, P.S. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell. Biol. 2006, 26, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, L.M.; List, K. The role of type II transmembrane serine protease-mediated signaling in cancer. FEBS J. 2017, 284, 1421–1436. [Google Scholar] [CrossRef] [Green Version]
- Mollica, V.; Rizzo, A.; Massari, F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. 2020, 16, 2029–2033. [Google Scholar] [CrossRef]
- Maas, C.; de Maat, S. Therapeutic SERPINs: Improving on Nature. Front. Cardiovasc. Med. 2021, 8, 648349. [Google Scholar] [CrossRef]
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef]
- Higgins, W.J.; Fox, D.M.; Kowalski, P.S.; Nielsen, J.E.; Worrall, D.M. Heparin enhances serpin inhibition of the cysteine protease cathepsin L. J. Biol. Chem. 2010, 285, 3722–3729. [Google Scholar] [CrossRef] [Green Version]
- Rihn, S.J.; Merits, A.; Bakshi, S.; Turnbull, M.L.; Wickenhagen, A.; Alexander, A.J.T.; Baillie, C.; Brennan, B.; Brown, F.; Brunker, K.; et al. A plasmid DNA-launched SARS-CoV-2 reverse genetics system and coronavirus toolkit for COVID-19 research. PLoS Biol. 2021, 19, e3001091. [Google Scholar] [CrossRef] [PubMed]
- Conceicao, C.; Thakur, N.; Human, S.; Kelly, J.T.; Logan, L.; Bialy, D.; Bhat, S.; Stevenson-Leggett, P.; Zagrajek, A.K.; Hollinghurst, P.; et al. The SARS-CoV-2 Spike protein has a broad tropism for mammalian ACE2 proteins. PLoS Biol. 2020, 18, e3001016. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; O’Reilly, S.; Gewaid, H.; Bowie, A.G.; Gautier, V.; Worrall, D.M. Reactive Centre Loop Mutagenesis of SerpinB3 to Target TMPRSS2 and Furin: Inhibition of SARS-CoV-2 Cell Entry and Replication. Int. J. Mol. Sci. 2022, 23, 12522. https://doi.org/10.3390/ijms232012522
Singh S, O’Reilly S, Gewaid H, Bowie AG, Gautier V, Worrall DM. Reactive Centre Loop Mutagenesis of SerpinB3 to Target TMPRSS2 and Furin: Inhibition of SARS-CoV-2 Cell Entry and Replication. International Journal of Molecular Sciences. 2022; 23(20):12522. https://doi.org/10.3390/ijms232012522
Chicago/Turabian StyleSingh, Saravjeet, Sophie O’Reilly, Hossam Gewaid, Andrew G. Bowie, Virginie Gautier, and D. Margaret Worrall. 2022. "Reactive Centre Loop Mutagenesis of SerpinB3 to Target TMPRSS2 and Furin: Inhibition of SARS-CoV-2 Cell Entry and Replication" International Journal of Molecular Sciences 23, no. 20: 12522. https://doi.org/10.3390/ijms232012522