
Activation of Meningeal Afferents Relevant to Trigeminal Headache Pain after Photothrombotic Stroke Lesion: A Pilot Study in Mice
 , ,
, ,
Abstract
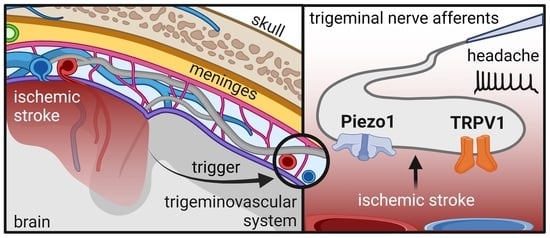
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Cortical Stroke after Photothrombosis
2.2. Motor Function and Mechanical Allodynia Sensitivity after Cortical Photothrombosis
2.3. Electrophysiological Observations in Meningeal Nerves in Relation to Lesion and Edema Formation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cortical Photothrombosis
4.3. Behavioral Testing
4.4. Tissue Preparation
4.5. Electrophysiology
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilkins, E.; Wilson, L.; Wickramasinghe, K.; Bhatnagar, P.; Leal, J.; Luengo-Fernandez, R.; Burns, R.; Rayner, M.; Townsend, N. European Cardiovascular Disease Statistics 2017. Available online: http://www.ehnheart.org/images/CVD-statistics-report-August-2017.pdf (accessed on 12 October 2022).
- Balami, J.S.; Chen, R.-L.; Grunwald, I.Q.; Buchan, A.M. Neurological Complications of Acute Ischaemic Stroke. Lancet Neurol. 2011, 10, 357–371. [Google Scholar] [CrossRef]
- Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd Edition. Cephalalgia 2018, 38, 1–211. [CrossRef] [PubMed]
- Oliveira, F.A.A.; Sampaio Rocha-Filho, P.A. Headaches Attributed to Ischemic Stroke and Transient Ischemic Attack. Headache 2019, 59, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Harriott, A.M.; Karakaya, F.; Ayata, C. Headache after Ischemic Stroke: A Systematic Review and Meta-Analysis. Neurology 2020, 94, e75–e86. [Google Scholar] [CrossRef] [PubMed]
- Leira, R.; Dávalos, A.; Aneiros, A.; Serena, J.; Pumar, J.M.; Castillo, J. Headache as a Surrogate Marker of the Molecular Mechanisms Implicated in Progressing Stroke. Cephalalgia 2002, 22, 303–308. [Google Scholar] [CrossRef]
- Chen, P.-K.; Chiu, P.-Y.; Tsai, I.-J.; Tseng, H.-P.; Chen, J.-R.; Yeh, S.-J.; Yeh, S.-J.; Sheu, J.-J.; Chung, C.-P.; Wu, M.-H.; et al. Onset Headache Predicts Good Outcome in Patients with First-Ever Ischemic Stroke. Stroke 2013, 44, 1852–1858. [Google Scholar] [CrossRef]
- Hansen, A.P.; Marcussen, N.S.; Klit, H.; Kasch, H.; Jensen, T.S.; Finnerup, N.B. Development of Persistent Headache Following Stroke: A 3-Year Follow-Up. Cephalalgia 2015, 35, 399–409. [Google Scholar] [CrossRef]
- Evans, R.W.; Mitsias, P.D. Headache at Onset of Acute Cerebral Ischemia. Headache 2009, 49, 902–908. [Google Scholar] [CrossRef]
- Pietrobon, D.; Moskowitz, M.A. Pathophysiology of Migraine. Annu. Rev. Physiol. 2013, 75, 365–391. [Google Scholar] [CrossRef] [Green Version]
- Noseda, R.; Burstein, R. Migraine Pathophysiology: Anatomy of the Trigeminovascular Pathway and Associated Neurological Symptoms, Cortical Spreading Depression, Sensitization, and Modulation of Pain. Pain 2013, 154 (Suppl 1), S44–S53. [Google Scholar] [CrossRef]
- Mikhailov, N.; Leskinen, J.; Fagerlund, I.; Poguzhelskaya, E.; Giniatullina, R.; Gafurov, O.; Malm, T.; Karjalainen, T.; Gröhn, O.; Giniatullin, R. Mechanosensitive Meningeal Nociception via Piezo Channels: Implications for Pulsatile Pain in Migraine? Neuropharmacology 2019, 149, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Krivoshein, G.; Tolner, E.A.; van den Maagdenberg, A.; Giniatullin, R.A. Migraine-Relevant Sex-Dependent Activation of Mouse Meningeal Afferents by TRPM3 Agonists. J. Headache Pain 2022, 23, 4. [Google Scholar] [CrossRef] [PubMed]
- Giniatullin, R. 5-Hydroxytryptamine in Migraine: The Puzzling Role of Ionotropic 5-HT3 Receptor in the Context of Established Therapeutic Effect of Metabotropic 5-HT1 Subtypes. Br. J. Pharmacol. 2022, 179, 400–415. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Esposito, F.; Conte, F.; Fratello, M.; Caiazzo, G.; Marcuccio, L.; Giordano, A.; Tedeschi, G.; Tessitore, A. Functional Interictal Changes of Pain Processing in Migraine with Ictal Cutaneous Allodynia. Cephalalgia 2017, 37, 305–314. [Google Scholar] [CrossRef]
- Maleki, N.; Szabo, E.; Becerra, L.; Moulton, E.; Scrivani, S.J.; Burstein, R.; Borsook, D. Ictal and Interictal Brain Activation in Episodic Migraine: Neural Basis for Extent of Allodynia. PLoS ONE 2021, 16, e0244320. [Google Scholar] [CrossRef]
- Zakharov, A.; Vitale, C.; Kilinc, E.; Koroleva, K.; Fayuk, D.; Shelukhina, I.; Naumenko, N.; Skorinkin, A.; Khazipov, R.; Giniatullin, R. Hunting for Origins of Migraine Pain: Cluster Analysis of Spontaneous and Capsaicin-Induced Firing in Meningeal Trigeminal Nerve Fibers. Front Cell Neurosci. 2015, 9, 287. [Google Scholar] [CrossRef]
- Dux, M.; Deák, É.; Tassi, N.; Sántha, P.; Jancsó, G. Endovanilloids Are Potential Activators of the Trigeminovascular Nocisensor Complex. J. Headache Pain 2016, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Della Pietra, A.; Savinainen, J.; Giniatullin, R. Inhibiting Endocannabinoid Hydrolysis as Emerging Analgesic Strategy Targeting a Spectrum of Ion Channels Implicated in Migraine Pain. Int. J. Mol. Sci. 2022, 23, 4407. [Google Scholar] [CrossRef]
- Stewart, L.; Turner, N.A. Channelling the Force to Reprogram the Matrix: Mechanosensitive Ion Channels in Cardiac Fibroblasts. Cells 2021, 10, 990. [Google Scholar] [CrossRef]
- Wang, J.; La, J.-H.; Hamill, O.P. PIEZO1 Is Selectively Expressed in Small Diameter Mouse DRG Neurons Distinct from Neurons Strongly Expressing TRPV1. Front Mol. Neurosci. 2019, 12, 178. [Google Scholar] [CrossRef]
- Guo, X.-W.; Lu, Y.; Zhang, H.; Huang, J.-Q.; Li, Y.-W. PIEZO1 Might Be Involved in Cerebral Ischemia-Reperfusion Injury through Ferroptosis Regulation: A Hypothesis. Med. Hypotheses 2021, 146, 110327. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A. Neurogenic Inflammation in the Pathophysiology and Treatment of Migraine. Neurology 1993, 43, S16–S20. [Google Scholar] [PubMed]
- Watson, B.D.; Dietrich, W.D.; Busto, R.; Wachtel, M.S.; Ginsberg, M.D. Induction of Reproducible Brain Infarction by Photochemically Initiated Thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Lipsanen, A.; Flunkert, S.; Kuptsova, K.; Hiltunen, M.; Windisch, M.; Hutter-Paier, B.; Jolkkonen, J. Non-Selective Calcium Channel Blocker Bepridil Decreases Secondary Pathology in Mice after Photothrombotic Cortical Lesion. PLoS ONE 2013, 8, e60235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orset, C.; Macrez, R.; Young, A.R.; Panthou, D.; Angles-Cano, E.; Maubert, E.; Agin, V.; Vivien, D. Mouse Model of in Situ Thromboembolic Stroke and Reperfusion. Stroke 2007, 38, 2771–2778. [Google Scholar] [CrossRef] [Green Version]
- Lipsanen, A.; Jolkkonen, J. Experimental Approaches to Study Functional Recovery Following Cerebral Ischemia. Cell. Mol. Life Sci. 2011, 68, 3007–3017. [Google Scholar] [CrossRef]
- Durukan, A.; Tatlisumak, T. Acute Ischemic Stroke: Overview of Major Experimental Rodent Models, Pathophysiology, and Therapy of Focal Cerebral Ischemia. Pharmacol. Biochem. Behav. 2007, 87, 179–197. [Google Scholar] [CrossRef]
- Kumar, A.; Aakriti; Gupta, V. A Review on Animal Models of Stroke: An Update. Brain Res. Bull. 2016, 122, 35–44. [Google Scholar] [CrossRef]
- Li, F.; Omae, T.; Fisher, M. Spontaneous Hyperthermia and Its Mechanism in the Intraluminal Suture Middle Cerebral Artery Occlusion Model of Rats. Stroke 1999, 30, 2464–2470, discussion 2470–2471. [Google Scholar] [CrossRef] [Green Version]
- Alexis, N.E.; Back, T.; Zhao, W.; Dietrich, W.D.; Watson, B.D.; Ginsberg, M.D. Neurobehavioral Consequences of Induced Spreading Depression Following Photothrombotic Middle Cerebral Artery Occlusion. Brain Res. 1996, 706, 273–282. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Huang, B.S.; Macisaac, S.E.; Mody, I.; Carmichael, S.T. Reducing Excessive GABA-Mediated Tonic Inhibition Promotes Functional Recovery after Stroke. Nature 2010, 468, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Välimäki, N.-N.; Bakreen, A.; Häkli, S.; Dhungana, H.; Keuters, M.H.; Dunlop, Y.; Koskuvi, M.; Keksa-Goldsteine, V.; Oksanen, M.; Jäntti, H.; et al. Astrocyte Progenitors Derived from Patients With Alzheimer Disease Do Not Impair Stroke Recovery in Mice. Stroke 2022, 53, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Corbett, D.; Carmichael, S.T.; Murphy, T.H.; Jones, T.A.; Schwab, M.E.; Jolkkonen, J.; Clarkson, A.N.; Dancause, N.; Wieloch, T.; Johansen-Berg, H.; et al. Enhancing the Alignment of the Preclinical and Clinical Stroke Recovery Research Pipeline: Consensus-Based Core Recommendations from the Stroke Recovery and Rehabilitation Roundtable Translational Working Group. Int. J. Stroke 2017, 12, 462–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Col, R.; Messlinger, K.; Carr, R.W. Repetitive Activity Slows Axonal Conduction Velocity and Concomitantly Increases Mechanical Activation Threshold in Single Axons of the Rat Cranial Dura. J. Physiol. 2012, 590, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Uebner, M.; Carr, R.W.; Messlinger, K.; De Col, R. Activity-Dependent Sensory Signal Processing in Mechanically Responsive Slowly Conducting Meningeal Afferents. J. Neurophysiol. 2014, 112, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.; Devor, M. Chemically Mediated Cross-Excitation in Rat Dorsal Root Ganglia. J. Neurosci. 1996, 16, 4733–4741. [Google Scholar] [CrossRef] [Green Version]
- Thalakoti, S.; Patil, V.V.; Damodaram, S.; Vause, C.V.; Langford, L.E.; Freeman, S.E.; Durham, P.L. Neuron-Glia Signaling in Trigeminal Ganglion: Implications for Migraine Pathology. Headache 2007, 47, 1008–1023, discussion 24–25. [Google Scholar] [CrossRef] [Green Version]
- Goadsby, P.J. Recent Advances in Understanding Migraine Mechanisms, Molecules and Therapeutics. Trends Mol. Med. 2007, 13, 39–44. [Google Scholar] [CrossRef]
- Levy, D. Migraine Pain and Nociceptor Activation--Where Do We Stand? Headache 2010, 50, 909–916. [Google Scholar] [CrossRef]
- Messlinger, K. Migraine: Where and How Does the Pain Originate? Exp. Brain Res. 2009, 196, 179–193. [Google Scholar] [CrossRef]
- Tong, C.-K.; MacDermott, A.B. Synaptic GluN2A and GluN2B Containing NMDA Receptors within the Superficial Dorsal Horn Activated Following Primary Afferent Stimulation. J. Neurosci. 2014, 34, 10808–10820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-F.; Zhu, C.Z.; Thimmapaya, R.; Choi, W.S.; Honore, P.; Scott, V.E.; Kroeger, P.E.; Sullivan, J.P.; Faltynek, C.R.; Gopalakrishnan, M.; et al. Differential Action Potentials and Firing Patterns in Injured and Uninjured Small Dorsal Root Ganglion Neurons after Nerve Injury. Brain Res. 2004, 1009, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Diener, H.C.; Katsarava, Z.; Weimar, C. Headache Associated with Ischemic Cerebrovascular Disease. Rev. Neurol. 2008, 164, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, N.; Plotnikova, L.; Singh, P.; Giniatullin, R.; Hämäläinen, R.H. Functional Characterization of Mechanosensitive Piezo1 Channels in Trigeminal and Somatic Nerves in a Neuron-on-Chip Model. Int. J. Mol. Sci. 2022, 23, 1370. [Google Scholar] [CrossRef] [PubMed]
- Della Pietra, A.; Mikhailov, N.; Giniatullin, R. The Emerging Role of Mechanosensitive Piezo Channels in Migraine Pain. Int. J. Mol. Sci. 2020, 21, E696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syeda, R.; Xu, J.; Dubin, A.E.; Coste, B.; Mathur, J.; Huynh, T.; Matzen, J.; Lao, J.; Tully, D.C.; Engels, I.H.; et al. Chemical Activation of the Mechanotransduction Channel Piezo1. Elife 2015, 4, e07369. [Google Scholar] [CrossRef]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Tomida, M.; Katayama, Y.; Kawakami, Y. Response Durations Encode Nociceptive Stimulus Intensity in the Rat Medial Prefrontal Cortex. Neuroscience 2004, 125, 777–785. [Google Scholar] [CrossRef]
- Simard, J.M.; Sheth, K.N.; Kimberly, W.T.; Stern, B.J.; del Zoppo, G.J.; Jacobson, S.; Gerzanich, V. Glibenclamide in Cerebral Ischemia and Stroke. Neurocrit. Care 2014, 20, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Rungta, R.L.; Choi, H.B.; Tyson, J.R.; Malik, A.; Dissing-Olesen, L.; Lin, P.J.C.; Cain, S.M.; Cullis, P.R.; Snutch, T.P.; MacVicar, B.A. The Cellular Mechanisms of Neuronal Swelling Underlying Cytotoxic Edema. Cell 2015, 161, 610–621. [Google Scholar] [CrossRef]
- Leinonen, V.; Vanninen, R.; Rauramaa, T. Raised Intracranial Pressure and Brain Edema. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 25–37. ISBN 978-0-12-802395-2. [Google Scholar]
- GBD 2015 Mortality and Causes of Death Collaborators Global, Regional, and National Life Expectancy, All-Cause Mortality, and Cause-Specific Mortality for 249 Causes of Death, 1980-2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [CrossRef] [Green Version]
- Qu, S.; Hu, T.; Qiu, O.; Su, Y.; Gu, J.; Xia, Z. Effect of Piezo1 Overexpression on Peritumoral Brain Edema in Glioblastomas. AJNR Am. J. Neuroradiol. 2020, 41, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Zhang, H.; Ma, T.; Lu, Y.; Xie, H.-Y.; Wang, W.; Ma, Y.-H.; Li, G.-H.; Li, Y.-W. Piezo1 Mediates Neuron Oxygen-Glucose Deprivation/Reoxygenation Injury via Ca2+/Calpain Signaling. Biochem. Biophys. Res. Commun. 2019, 513, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wei, Z.; Xin, G.; Li, Y.; Yuan, J.; Ming, Y.; Ji, C.; Sun, Q.; Li, S.; Chen, X.; et al. Piezo1 Initiates Platelet Hyperreactivity and Accelerates Thrombosis in Hypertension. J. Thromb. Haemost. 2021, 19, 3113–3125. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Balasubramanian, A.; Marrelli, S.P. Pharmacologically Induced Hypothermia via TRPV1 Channel Agonism Provides Neuroprotection Following Ischemic Stroke When Initiated 90 Min after Reperfusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R149–R156. [Google Scholar] [CrossRef] [Green Version]
- Muzzi, M.; Felici, R.; Cavone, L.; Gerace, E.; Minassi, A.; Appendino, G.; Moroni, F.; Chiarugi, A. Ischemic Neuroprotection by TRPV1 Receptor-Induced Hypothermia. J. Cereb. Blood Flow. Metab. 2012, 32, 978–982. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Ma, R.; Li, H.; Wang, J.; Guo, X.; Chen, H. Advancement in Research on the Role of the Transient Receptor Potential Vanilloid Channel in Cerebral Ischemic Injury (Review). Exp. Ther. Med. 2021, 22, 881. [Google Scholar] [CrossRef]
- Lee, J.; Saloman, J.L.; Weiland, G.; Auh, Q.-S.; Chung, M.-K.; Ro, J.Y. Functional Interactions between NMDA Receptors and TRPV1 in Trigeminal Sensory Neurons Mediate Mechanical Hyperalgesia in the Rat Masseter Muscle. Pain 2012, 153, 1514–1524. [Google Scholar] [CrossRef] [Green Version]
- Borbiro, I.; Badheka, D.; Rohacs, T. Activation of TRPV1 Channels Inhibits Mechanosensitive Piezo Channel Activity by Depleting Membrane Phosphoinositides. Sci. Signal 2015, 8, ra15. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, J.; Mogil, J.S.; Dirnagl, U. Distinguishing between Exploratory and Confirmatory Preclinical Research Will Improve Translation. PLoS Biol. 2014, 12, e1001863. [Google Scholar] [CrossRef]
- Reglodi, D.; Tamás, A.; Somogyvári-Vigh, A.; Szántó, Z.; Kertes, E.; Lénárd, L.; Arimura, A.; Lengvári, I. Effects of Pretreatment with PACAP on the Infarct Size and Functional Outcome in Rat Permanent Focal Cerebral Ischemia. Peptides 2002, 23, 2227–2234. [Google Scholar] [CrossRef]
- Kuts, R.; Frank, D.; Gruenbaum, B.F.; Grinshpun, J.; Melamed, I.; Knyazer, B.; Tarabrin, O.; Zvenigorodsky, V.; Shelef, I.; Zlotnik, A.; et al. A Novel Method for Assessing Cerebral Edema, Infarcted Zone and Blood-Brain Barrier Breakdown in a Single Post-Stroke Rodent Brain. Front Neurosci. 2019, 13, 1105. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivoshein, G.; Bakreen, A.; van den Maagdenberg, A.M.J.M.; Malm, T.; Giniatullin, R.; Jolkkonen, J. Activation of Meningeal Afferents Relevant to Trigeminal Headache Pain after Photothrombotic Stroke Lesion: A Pilot Study in Mice. Int. J. Mol. Sci. 2022, 23, 12590. https://doi.org/10.3390/ijms232012590
Krivoshein G, Bakreen A, van den Maagdenberg AMJM, Malm T, Giniatullin R, Jolkkonen J. Activation of Meningeal Afferents Relevant to Trigeminal Headache Pain after Photothrombotic Stroke Lesion: A Pilot Study in Mice. International Journal of Molecular Sciences. 2022; 23(20):12590. https://doi.org/10.3390/ijms232012590
Chicago/Turabian StyleKrivoshein, Georgii, Abdulhameed Bakreen, Arn M. J. M. van den Maagdenberg, Tarja Malm, Rashid Giniatullin, and Jukka Jolkkonen. 2022. "Activation of Meningeal Afferents Relevant to Trigeminal Headache Pain after Photothrombotic Stroke Lesion: A Pilot Study in Mice" International Journal of Molecular Sciences 23, no. 20: 12590. https://doi.org/10.3390/ijms232012590