
Alternation of Organ-Specific Exposure in LPS-Induced Pneumonia Mice after the Inhalation of Tetrandrine Is Governed by Metabolizing Enzyme Suppression and Lysosomal Trapping
 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Features of the ALI Mouse Model Induced by LPS
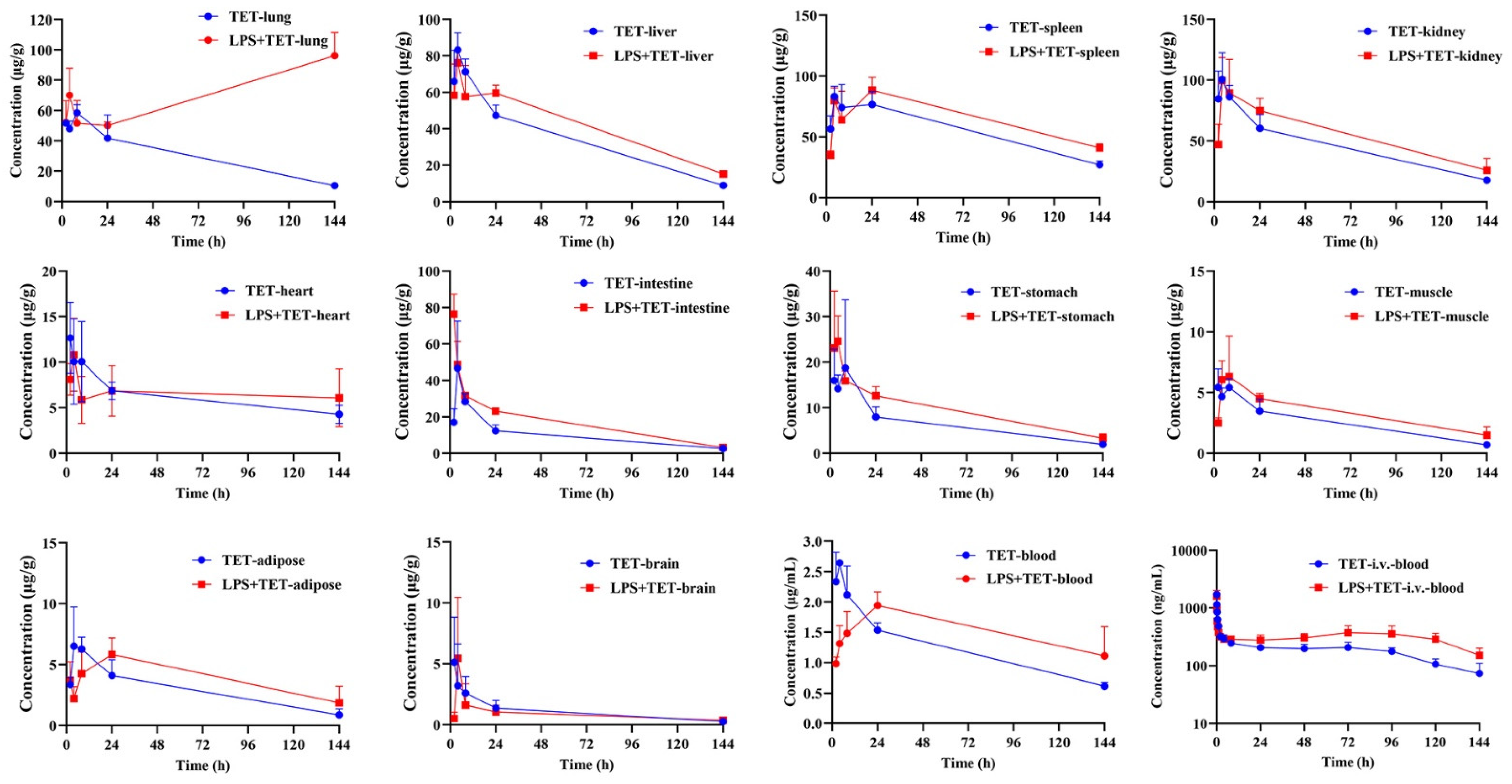
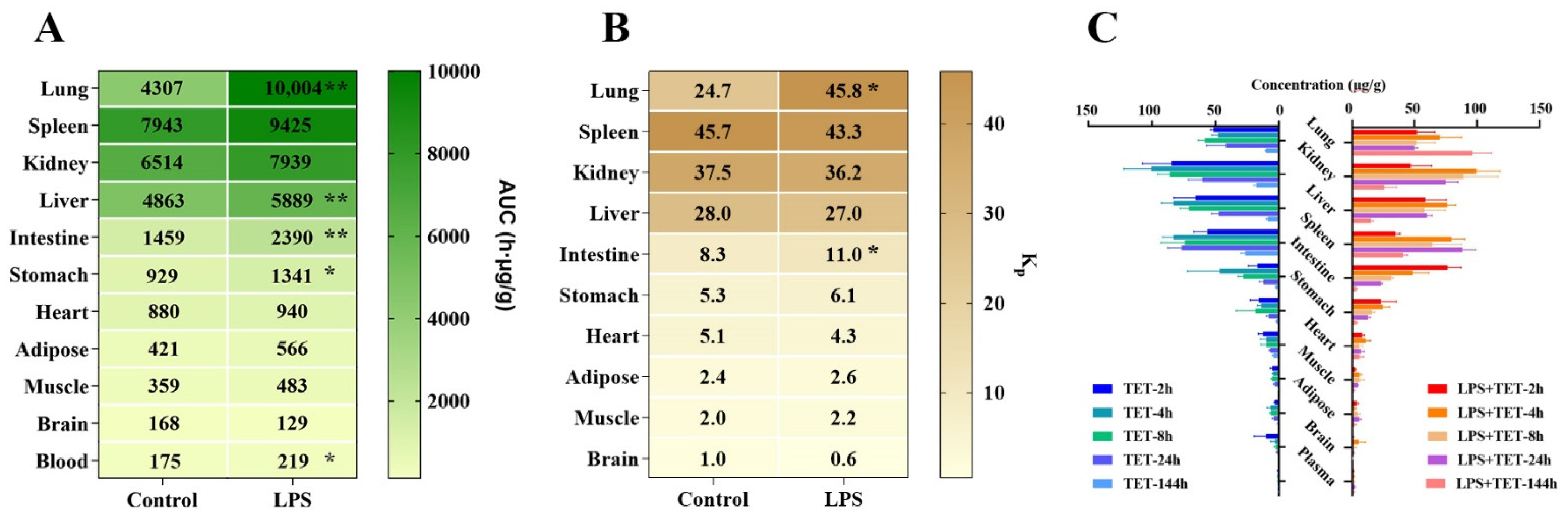
2.2. Effect of ALI on TET PK Behavior and Biodistribution
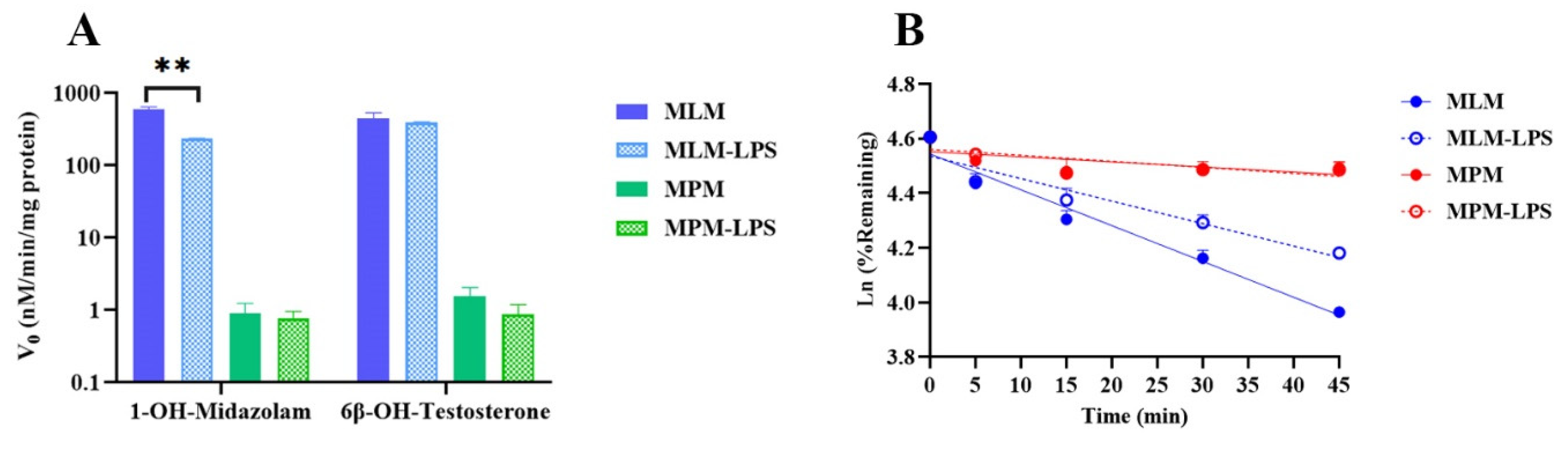
2.3. Impact of Inflammation on CYP-Associated TET Metabolism
2.4. The Role of Lysosomes in the TET Cellular Distribution and the Effects of Inflammation
2.5. Roles of Drug Transporters Involved in TET Cellular Accumulation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals
4.3. Construction of the LPS-Induced Mouse Model of ALI
4.4. Cell Culturse
4.5. In Vivo Experiments
4.5.1. Pharmacokinetics after i.v. Administration
4.5.2. Tissue Distribution after i.t. Delivery
4.6. In Vitro Experiments
4.6.1. Preparation of Mouse Liver Microsomes and Pulmonary Microsomes
4.6.2. Metabolic Stability of TET in MLM and MPM from Normal and Acute Pneumonia Mice
4.6.3. Quantification of Lysosomal TET Content in Normal and LPS-Induced Cell Lines
4.6.4. Uptake Study of TET by OATP1B1 and OATP1B3 Expressing the HEK293 Cell Line
4.6.5. Bidirectional Transport Study of TET by MDR1 Expressing the LLC-PK1 Cell Line
4.7. Bioanalytical Methods
4.8. Data Processing and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xie, Q.M.; Tang, H.F.; Chen, J.Q.; Bian, R.L. Pharmacological actions of tetrandrine in inflammatory pulmonary diseases. Acta Pharmacol. Sin. 2002, 23, 1107–1113. [Google Scholar]
- Banks, D.E.; Cheng, Y.H.; Weber, S.L.; Ma, J.K. Strategies for the treatment of pneumoconiosis. Occup. Med. 1993, 8, 205–232. [Google Scholar] [PubMed]
- Jiang, Y.; Liu, M.; Liu, H.; Liu, S. A critical review: Traditional uses, phytochemistry, pharmacology and toxicology of Stephania tetrandra S. Moore (Fen Fang Ji). Phytochem. Rev. 2020, 19, 449–489. [Google Scholar] [CrossRef] [PubMed]
- Luan, F.; He, X.; Zeng, N. Tetrandrine: A review of its anticancer potentials, clinical settings, pharmacokinetics and drug delivery systems. J. Pharm. Pharmacol. 2020, 72, 1491–1512. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Liu, Y.S.; Guo, Y.R.; Zhong, W.X.; Guo, Y.P.; Guo, L. Nano-Liposomes Double Loaded with Curcumin and Tetrandrine: Preparation, Characterization, Hepatotoxicity and Anti-Tumor Effects. Int. J. Mol. Sci. 2022, 23, 6858. [Google Scholar] [CrossRef]
- Heister, P.M.; Poston, R.N. Pharmacological hypothesis: TPC2 antagonist tetrandrine as a potential therapeutic agent for COVID-19. Pharmacol. Res. Perspect. 2020, 8, e00653. [Google Scholar] [CrossRef]
- Grimm, C.; Tang, R. Could an endo-lysosomal ion channel be the Achilles heel of SARS-CoV2? Cell Calcium. 2020, 88, 102212. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Liang, Y.; Meng, Z.; Chen, X.; Lu, M.; Han, X.; Deng, X.; Zhang, Q.; Zhu, H.; Fu, T. Inhalation of Tetrandrine-hydroxypropyl-beta-cyclodextrin Inclusion Complexes for Pulmonary Fibrosis Treatment. Mol. Pharm. 2020, 17, 1596–1607. [Google Scholar] [CrossRef]
- Plaunt, A.J.; Islam, S.; Macaluso, T.; Gauani, H.; Baker, T.; Chun, D.; Viramontes, V.; Chang, C.; Corboz, M.R.; Chapman, R.W.; et al. Development and Characterization of Treprostinil Palmitil Inhalation Aerosol for the Investigational Treatment of Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 548. [Google Scholar] [CrossRef]
- Wang, L.; Rao, Y.; Liu, X.; Sun, L.; Gong, J.; Zhang, H.; Shen, L.; Bao, A.; Yang, H. Administration route governs the therapeutic efficacy, biodistribution and macrophage targeting of anti-inflammatory nanoparticles in the lung. J. Nanobiotechnol. 2021, 19, 56. [Google Scholar] [CrossRef]
- Aitken, A.E.; Richardson, T.A.; Morgan, E.T. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 123–149. [Google Scholar] [CrossRef]
- Morgan, E.T. Regulation of Drug-Metabolizing Enzymes and Drug Metabolism by Inflammatory Responses. In Drug Metabolism in Diseases; Academic Press: Cambridge, MA, USA, 2017; pp. 21–58. [Google Scholar]
- Morgan, E.T.; Goralski, K.B.; Piquette-Miller, M.; Renton, K.W.; Robertson, G.R.; Chaluvadi, M.R.; Charles, K.A.; Clarke, S.J.; Kacevska, M.; Liddle, C.; et al. Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab. Dispos. 2008, 36, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saib, S.; Delavenne, X. Inflammation Induces Changes in the Functional Expression of P-gp, BCRP, and MRP2: An Overview of Different Models and Consequences for Drug Disposition. Pharmaceutics 2021, 13, 1544. [Google Scholar] [CrossRef] [PubMed]
- Seifert, S.M.; Castillo-Mancilla, J.R.; Erlandson, K.M.; Anderson, P.L. Inflammation and pharmacokinetics: Potential implications for HIV-infection. Expert Opin. Drug Metab. Toxicol. 2017, 13, 641–650. [Google Scholar] [CrossRef]
- Norgard, N.B.; Monte, S.V. Obesity and Inflammation and Altered Clopidogrel Pharmacokinetics and Pharmacodynamics. Drug Metab. Lett. 2017, 11, 3–13. [Google Scholar] [CrossRef] [PubMed]
- De-Oliveira, A.; Paumgartten, F.J.R. Malaria-induced Alterations of Drug Kinetics and Metabolism in Rodents and Humans. Curr. Drug Metab. 2021, 22, 127–138. [Google Scholar] [CrossRef]
- Cavaillon, J.M.; Adib-Conquy, M. The Pro-Inflammatory Cytokine Cascade. Update Intensive Care Emerg. Med. 2002, 31, 37–66. [Google Scholar]
- De-Oliveira, A.C.; Poça, K.S.; Totino, P.R.; Paumgartten, F.J. Modulation of cytochrome P450 2A5 activity by lipopolysaccharide: Low-dose effects and non-monotonic dose-response relationship. PLoS ONE 2015, 10, e0117842. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.N.; Greene, J.; Kenna, L.; Kissell, L.; Kuhn, M. The Impact of Infection and Inflammation on Drug Metabolism, Active Transport, and Systemic Drug Concentrations in Veterinary Species. Drug Metab. Dispos. 2020, 48, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.S.; Byron, P.R. Inhaling medicines: Delivering drugs to the body through the lungs. Nat. Rev. Drug Discov. 2007, 6, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.; Sjögren, E.; Thörn, H.; Rubin, K.; Bäckman, P.; Lennernäs, H. Pulmonary absorption-estimation of effective pulmonary permeability and tissue retention of ten drugs using an ex vivo rat model and computational analysis. Eur. J. Pharm. Biopharm. 2018, 124, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M. An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, W.A.; Bickel, M.H.; Honegger, U.E. The contribution of lysosomal trapping in the uptake of desipramine and chloroquine by different tissues. Pharmacol. Toxicol. 1995, 77, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.V.; Lienau, P.; Fricker, G.; Reichel, A. Quantitation of Lysosomal Trapping of Basic Lipophilic Compounds Using In Vitro Assays and In Silico Predictions Based on the Determination of the Full pH Profile of the Endo-/Lysosomal System in Rat Hepatocytes. Drug Metab. Dispos. 2019, 47, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, A.; Lindberg, L.; Sawalha, S.; Nilsson, U.; Stridsman, C.; Lundbäck, B.; Backman, H. Large underreporting of COPD as cause of death-results from a population-based cohort study. Respir. Med. 2021, 186, 106518. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F. Global, regional, and national under-5 mortality, adult mortality, age-specific mortality, and life expectancy, 1970–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1084–1150. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Q.; Liu, X.; Yang, T.; Cui, K.; Kong, L.; Yang, C.; Zhang, Z. Nanomedicine for acute respiratory distress syndrome: The latest application, targeting strategy, and rational design. Acta Pharm. Sin. B 2021, 11, 3060–3091. [Google Scholar] [CrossRef]
- Zoulikha, M.; Xiao, Q.; Boafo, G.F.; Sallam, M.A.; Chen, Z.; He, W. Pulmonary delivery of siRNA against acute lung injury/acute respiratory distress syndrome. Acta Pharm. Sin. B 2022, 12, 600–620. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, H.S.; Min, K.R.; Kim, Y.; Lim, H.K.; Chang, Y.K.; Chung, M.W. Anti-inflammatory effects of fangchinoline and tetrandrine. J. Ethnopharmacol. 2000, 69, 173–179. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heumann, D.; Roger, T. Initial responses to endotoxins and Gram-negative bacteria. Clin. Chim. Acta 2002, 323, 59–72. [Google Scholar] [CrossRef]
- Ndolo, R.A.; Luan, Y.; Duan, S.; Forrest, M.L.; Krise, J.P. Lysosomotropic properties of weakly basic anticancer agents promote cancer cell selectivity in vitro. PLoS ONE 2012, 7, e49366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.C.; Lin, C.J. The regulation of drug-metabolizing enzymes and membrane transporters by inflammation: Evidences in inflammatory diseases and age-related disorders. J. Food Drug Anal. 2019, 27, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Aowicki, D.; Huczyński, A. Structure and antimicrobial properties of monensin A and its derivatives: Summary of the achievements. Biomed. Res. Int. 2013, 2013, 742149. [Google Scholar]
- Hartz, A.M.; Bauer, B.; Fricker, G.; Miller, D.S. Rapid modulation of P-glycoprotein-mediated transport at the blood-brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol. Pharmacol. 2006, 69, 462–470. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Miller, D.S. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol. Pharmacol. 2007, 71, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Amar-Costesec, A.; Beaufay, H.; Wibo, M.; Thinès-Sempoux, D.; Feytmans, E.; Robbi, M.; Berthet, J. Analytical study of microsomes and isolated subcellular membranes from rat liver. II. Preparation and composition of the microsomal fraction. J. Cell Biol. 1974, 61, 201–212. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Unit | Normal Mice | LPS-Treated Mice |
|---|---|---|---|
| t1/2 | h | 30.7 ± 5.21 | 93.2 ± 26.4 ** |
| C0 | ng/mL | 2273 ± 270 | 2300 ± 935 |
| AUC(0-t) | h·ng/mL | 26253 ± 3417 | 44098 ± 8457 ** |
| AUC(0-∞) | h·ng/mL | 29451 ± 3794 | 65735 ± 17527 ** |
| MRT(0-t) | h | 72.7 ± 6.40 | 137 ± 40.4 ** |
| Vss | L/kg | 7.71 ± 2.01 | 10.5 ± 1.77 |
| Vz | L/kg | 12.5 ± 1.63 | 10.3 ± 1.75 * |
| CL | mL/h/kg | 172 ± 22.1 | 80.9 ± 22.3 ** |
| Parameters | Unit | Spleen | Kidney | Liver | Lung | Intestine | Heart | Stomach | Adipose | Muscle | Brain | Blood |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal mice | ||||||||||||
| t1/2 | h | 83.8 ± 22.7 | 63.4 ± 8.09 | 47.5 ± 5.47 | 65.1 ± 18.3 | 44.7 ± 1.94 | 96.8 ± 6.46 | 64.0 ± 16.5 | 54.0 ± 21.5 | 49.2 ± 4.06 | 51.6 ± 17.2 | 83.2 ± 15.4 |
| Tmax | h | 12.00 ± 10.6 | 3.33 ± 1.15 | 4.00 ± 0 | 13.3 ± 9.24 | 4.00 ± 0 | 2.67 ± 1.15 | 4.67 ± 3.06 | 5.33 ± 2.31 | 4.00 ± 3.16 | 2.00 ± 0 | 3.33 ± 1.15 |
| Cmax | μg/mL | 89.7 ± 7.09 | 107 ± 16.6 | 83.3 ± 9.29 | 60.3 ± 3.21 | 46.7 ± 25.9 | 14.2 ± 3.16 | 22.3 ± 12.4 | 7.38 ± 1.79 | 6.12 ± 0.44 | 10.4 ± 9.53 | 2.67 ± 0.05 |
| AUC(0-t) | h·μg/mL | 7943 ± 665 | 6514 ± 691 | 4863 ± 334 | 4307 ± 1003 | 1459 ± 134 | 880 ± 22.2 | 929 ± 163 | 421 ± 94.4 | 359 ± 79.8 | 168 ± 60.6 | 175 ± 12.3 |
| AUC(0-∞) | h·μg/mL | 11,282 ± 629 | 8159 ± 422 | 5485 ± 237 | 5295 ± 721 | 1632 ± 130 | 1407 ± 21.5 | 1121 ± 207 | 501 ± 88.3 | 410 ± 80.1 | 190 ± 51.4 | 249 ± 16.4 |
| MRT | h | 112 ± 34.7 | 80.1 ± 16.6 | 53.9 ± 8.29 | 78.3 ± 24.5 | 50.5 ± 2.31 | 137 ± 0.09 | 70.0 ± 14.0 | 67.7 ± 37.1 | 57.7 ± 7.81 | 56.8 ± 26.5 | 111 ± 23.1 |
| Kp | / | 45.7 ± 6.62 | 37.5 ± 5.70 | 28.0 ± 3.49 | 24.7 ± 6.06 | 8.35 ± 0.67 | 5.05 ± 0.38 | 5.29 ± 0.66 | 2.39 ± 0.40 | 2.04 ± 0.38 | 0.97 ± 0.39 | 1.00 |
| LPS-treated mice | ||||||||||||
| t1/2 | h | 111 ± 18.3 | 77.8 ± 18.1 | 69.3 ± 14.8 | / | 41.7 ± 4.93 | 449 ± 259 | 61.4 ± 13.0 | 75.2 ± 35.2 | 78.8 ± 31.3 | 92.6 ± 27.2 | 95.5 ± 17.6 |
| Tmax | h | 17.3 ± 11.5 | 5.33 ± 2.31 | 3.33 ± 1.15 | 144 ± 0.00 ** | 2.00 ± 0.00 ** | 4.00 ± 0.00 | 3.33 ± 1.15 | 18.7 ± 9.24 | 6.67 ± 2.31 | 5.33 ± 2.31 | 24.0 ± 0.00 ** |
| Cmax | μg/mL | 89.7 ± 10.7 | 112 ± 11.5 | 76.3 ± 7.02 | 96.2 ± 15.3 * | 76.5 ± 10.9 | 10.8 ± 4.01 | 27.2 ± 9.32 | 5.92 ± 1.41 | 7.51 ± 2.11 | 6.43 ± 3.79 | 1.94 ± 0.225 ** |
| AUC(0-t) | h·μg/mL | 9425 ± 942 | 7939 ± 1171 | 5889 ± 178 ** | 10,004 ± 1109 ** | 2390 ± 196 ** | 940 ± 381 | 1340 ± 192 * | 566 ± 163 | 483 ± 89.5 | 129 ± 46.2 | 219 ± 17.7 |
| AUC(0-∞) | h·μg/mL | 15,987 ± 1318 ** | 11,010 ± 3084 | 7427 ± 632 ** | / | 2591 ± 271 ** | 4089 ± 3400 | 1645 ± 323 | 816 ± 429 | 674 ± 240 | 179 ± 38.4 | 326 ± 12.9 * |
| MRT | h | 157 ± 29.5 | 101 ± 30.0 | 82.1 ± 15.6 | / | 42.4 ± 6.38 | 643 ± 376 | 73.3 ± 17.7 | 102 ± 57.9 | 101 ± 44.4 | 112 ± 35.4 | 133 ± 26.1 |
| Kp | / | 43.3 ± 6.88 | 36.2 ± 3.83 | 27.0 ± 1.72 | 45.8 ± 6.49 * | 11.0 ± 1.35 * | 4.27 ± 1.59 | 6.13 ± 0.87 | 2.57 ± 0.62 | 2.20 ± 0.31 | 0.59 ± 0.17 | 1.00 |
| Groups | In Vitro | In Vivo | ||
|---|---|---|---|---|
| t1/2 (min) | CLint,scaled (mL/min/mg Protein) | CLhep,blood (mL/h/kg) | CLblood (mL/h/kg) | |
| Normal mice | 52.8 ± 1.75 | 103 ± 3.40 | 140 ± 4.50 | 172 ± 22.1 |
| LPS-treated mice | 84.0 ± 0.780 ** | 65.0 ± 0.603 ** | 89.0 ± 0.814 ** | 80.9 ± 22.3 ** |
| OATP | TET | Positive Control | ||||
|---|---|---|---|---|---|---|
| Uptake Clearance (nM/min/mg Protein) | Uptake Ratio | Inhibition Ratio | Uptake Clearance (nM/min/mg Protein) | Uptake Ratio | Inhibition Ratio | |
| MOCK | 131 ± 11.3 | 1.21 | 11.1 | 2.4 ± 0.03 # | 18.4 | 97.7 |
| OATP1B1 | 158 ± 35.1 | 44.2 ± 1.65 **, ## | ||||
| MOCK+RIF | 117 ± 12.8 | 1.2 | 2.63 ± 0.08 | 1.36 | ||
| OATP1B1+RIF | 141 ± 13.7 | 3.57± 0.03 ** | ||||
| MOCK | 126 ± 10.9 | 1.2 | 20.6 | 2.7 ± 0.07 | 11.1 | 96.6 |
| OATP1B3 | 151 ± 25.5 | 30.0 ± 1.46 **, ## | ||||
| MOCK+RIF | 119 ± 8.03 | 1.01 | 2.73 ± 0.08 | 1.34 | ||
| OATP1B3+RIF | 120 ± 8.61 | 3.66 ± 0.03 ** | ||||
| Mock Cell | Papp | ER | MDR1-Transfected | Papp | ER | ||
|---|---|---|---|---|---|---|---|
| A-B | B-A | A-B | B-A | ||||
| Atenolol | 0.00 | 0.12 ± 0.01 | \ | Atenolol | 1.23 ± 0.08 ** | 0.12 ± 0.01 ** | 0.84 |
| Metoprolol | 32.22 ± 2.80 | 24.99 ± 0.30 | 0.7 | Metoprolol | 28.23 ± 0.65 | 28.32 ± 2.07 | 1.00 |
| Digoxin | 2.25 ± 0.26 | 1.74 ± 0.20 | 0.77 | Digoxin | 0.98 ± 0.11 **,## | 11.27 ± 0.77 **,## | 11.54 |
| Digoxin + TAR | 2.08 ± 0.09 | 1.66 ± 0.21 | 0.80 | Digoxin + TAR | 5.14 ± 1.02 ** | 5.31 ± 0.85 ** | 1.03 |
| TET | 1.22 ± 0.07 ## | 3.83 ± 0.12 # | 3.13 | TET | 0.52 ± 0.19 ** | 14.02 ± 2.95 **,# | 32.46 |
| TET + TAR | 1.88 ± 0.14 | 5.21 ± 0.73 | 2.17 | TET + TAR | 0.37 ± 0.05 * | 6.03 ± 1.50 | 16.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Jiang, X.; Yang, Z.; Fu, S.; Yao, S.; Wang, L.; Lv, Y.; Zhang, W.; Ding, R.; Zhuang, X. Alternation of Organ-Specific Exposure in LPS-Induced Pneumonia Mice after the Inhalation of Tetrandrine Is Governed by Metabolizing Enzyme Suppression and Lysosomal Trapping. Int. J. Mol. Sci. 2022, 23, 12948. https://doi.org/10.3390/ijms232112948
Wang F, Jiang X, Yang Z, Fu S, Yao S, Wang L, Lv Y, Zhang W, Ding R, Zhuang X. Alternation of Organ-Specific Exposure in LPS-Induced Pneumonia Mice after the Inhalation of Tetrandrine Is Governed by Metabolizing Enzyme Suppression and Lysosomal Trapping. International Journal of Molecular Sciences. 2022; 23(21):12948. https://doi.org/10.3390/ijms232112948
Chicago/Turabian StyleWang, Furun, Xue Jiang, Zengxu Yang, Shuang Fu, Shi Yao, Lingchao Wang, Yue Lv, Wenpeng Zhang, Rigao Ding, and Xiaomei Zhuang. 2022. "Alternation of Organ-Specific Exposure in LPS-Induced Pneumonia Mice after the Inhalation of Tetrandrine Is Governed by Metabolizing Enzyme Suppression and Lysosomal Trapping" International Journal of Molecular Sciences 23, no. 21: 12948. https://doi.org/10.3390/ijms232112948