

A Cross-Sectional and Longitudinal Analysis of Pre-Diagnostic Blood Plasma Biomarkers for Early Detection of Pancreatic Cancer

 , , , and
, , , and
Abstract
1. Introduction
2. Results
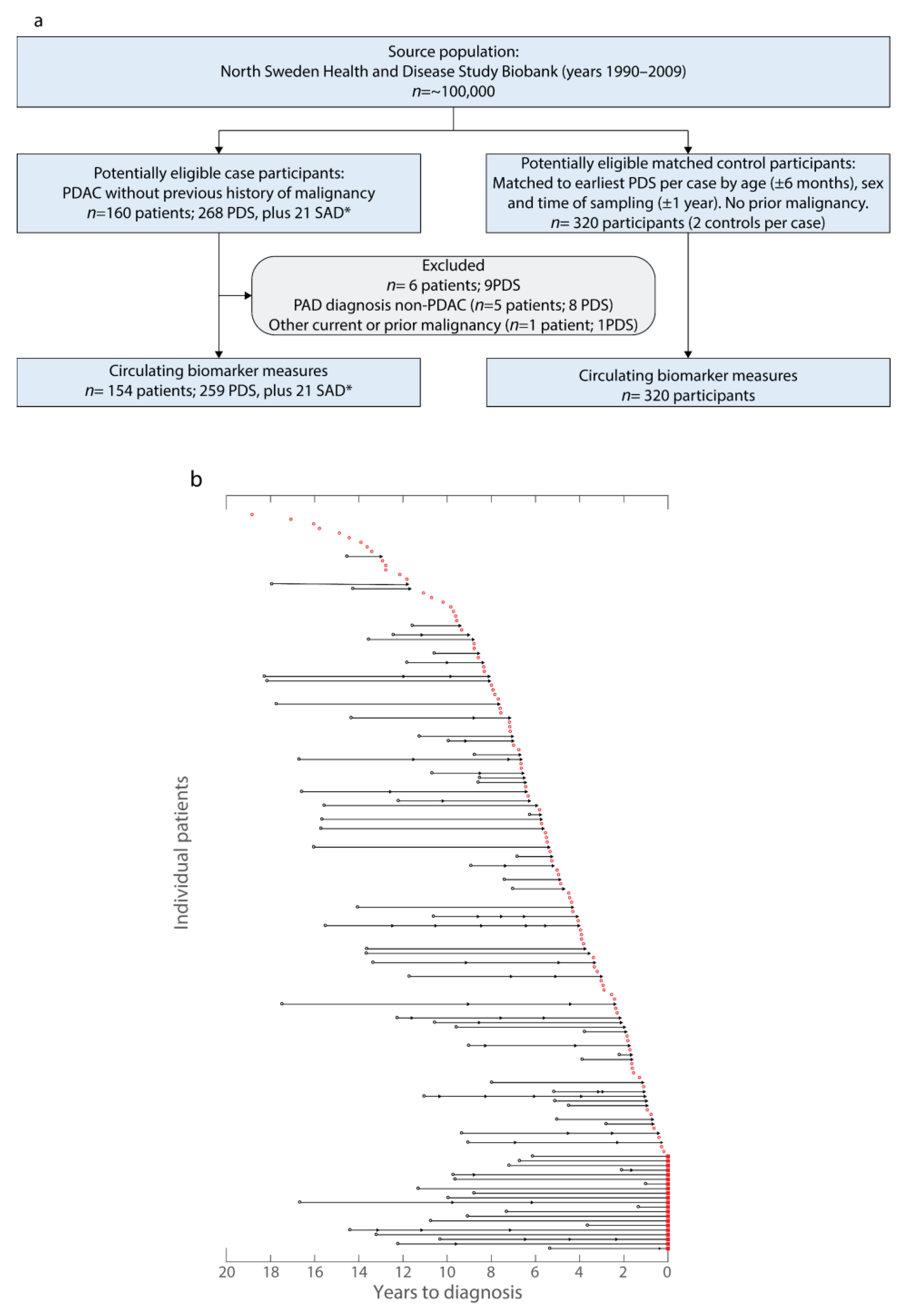
2.1. NSHDS Cohort Description
2.2. Cross-Sectional Analysis
2.3. Longitudinal Analysis
3. Discussion
4. Materials and Methods
4.1. Ethical Considerations
4.2. Pre-Diagnostic Sample Cohort
4.3. Samples at PDAC Diagnosis Cohort
4.4. ELISA and Multiplex Biomarker Assays
4.5. Cross-Sectional Analysis and Model Generation
4.6. Samples at Diagnosis—Analysis and Model Generation
4.7. Longitudinal Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Kalser, M.H.; Barkin, J.; MacIntyre, J.M. Pancreatic cancer. Assessment of prognosis by clinical presentation. Cancer 1985, 56, 397–402. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Yu, J.; Blackford, A.L.; dal Molin, M.; Wolfgang, C.L.; Goggins, M. Time to progression of pancreatic ductal adenocarcinoma from low-to-high tumour stages. Gut 2015, 64, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Le, N.; Sund, M.; Vinci, A.; Beyer, G.; Javed, M.A.; Krug, S.; Neessee, A.; Schober, M. Prognostic and predictive markers in pancreatic adenocarcinoma. Dig. Liver Dis. 2016, 48, 223–230. [Google Scholar] [CrossRef]
- van Manen, L.; Groen, J.V.; Putter, H.; Vahrmeijer, A.L.; Swijnenburg, R.J.; Bonsing, B.A.; Mieog, J.S.D. Elevated CEA and CA19-9 serum levels independently predict advanced pancreatic cancer at diagnosis. Biomarkers 2020, 25, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Firpo, M.A.; Adler, D.G.; Mulvihill, S.J. Screening for pancreatic cancer: Why, how, and who? Ann. Surg. 2013, 257, 17–26. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Franklin, O.; Ohlund, D.; Lundin, C.; Oman, M.; Naredi, P.; Wang, W.; Sund, M. Combining conventional and stroma-derived tumour markers in pancreatic ductal adenocarcinoma. Cancer Biomark. 2015, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Ardnor, B.; Oman, M.; Naredi, P.; Sund, M. Expression pattern and circulating levels of endostatin in patients with pancreas cancer. Int. J. Cancer 2008, 122, 2805–2810. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Lundin, C.; Ardnor, B.; Oman, M.; Naredi, P.; Sund, M. Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br. J. Cancer 2009, 101, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Pukkala, E.; Andersen, A.; Berglund, G.; Gislefoss, R.; Gudnason, V.; Hallmans, G.; Jellum, E.; Jousilahti, P.; Knekt, P.; Koskela, P.; et al. Nordic biological specimen banks as basis for studies of cancer causes and control—More than 2 million sample donors, 25 million person years and 100,000 prospective cancers. Acta Oncol. 2007, 46, 286–307. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Ali, S.M.; Leitzel, K.; Drabick, J.J.; Yee, N.; Polimera, H.V.; Nagabhairu, V.; Krecko, L.; Ali, A.; Maddukuri, A.; et al. Collagen fragments quantified in serum as measures of desmoplasia associate with survival outcome in patients with advanced pancreatic cancer. Sci. Rep. 2019, 9, 19761. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Gay, D.Z.; Brown, K.; Mulvihill, J.D.; Boucher, K.M.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. The clinical utility of CA 19-9 in pancreatic adenocarcinoma: Diagnostic and prognostic updates. Curr. Mol. Med. 2013, 13, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Brailski, K.; Mendizova, A.; Milkov, V. The carcinoembryonic antigen in chronic pancreatitis. Vutreshni Boles. 1990, 29, 26–30. [Google Scholar]
- Shao, Y.; Sun, X.; He, Y.; Liu, C.; Liu, H. Elevated Levels of Serum Tumor Markers CEA and CA15-3 Are Prognostic Parameters for Different Molecular Subtypes of Breast Cancer. PLoS ONE 2015, 10, e0133830. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J. Carcinoembryonic antigen as a marker for colorectal cancer: Is it clinically useful? Clin. Chem. 2001, 47, 624–630. [Google Scholar] [CrossRef]
- O’Brien, D.P.; Sandanayake, N.S.; Jenkinson, C.; Gentry-Maharaj, A.; Apostolidou, S.; Fourkala, E.O.; Camuzeaux, S.; Blyuss, O.; Gunu, R.; Dawnay, A.; et al. Serum CA19-9 is significantly upregulated up to 2 years before diagnosis with pancreatic cancer: Implications for early disease detection. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Katzke, V.A.; Hüsing, A.; Okaya, S.; Shoji, H.; Onidani, K.; Olsen, A.; Tjønneland, A.; Overvad, K.; Weiderpass, E.; et al. CA19-9 and apolipoprotein-A2 isoforms as detection markers for pancreatic cancer: A prospective evaluation. Int. J. Cancer 2019, 144, 1877–1887. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, K.T.; Lee, J.K.; Paik, S.W.; Rhee, J.C.; Choi, K.W. Clinical usefulness of carbohydrate antigen 19-9 as a screening test for pancreatic cancer in an asymptomatic population. J. Gastroenterol. Hepatol. 2004, 19, 182–186. [Google Scholar] [CrossRef]
- Lee, D.H.; Yoon, W.; Lee, A.; Han, Y.; Byun, Y.; Kang, J.S.; Kim, H.; Kwon, W.; Suh, Y.A.; Choi, Y.; et al. Multi-biomarker panel prediction model for diagnosis of pancreatic cancer. J. Hepato-Biliary-Pancreat. Sci. 2021, 00, 1–11. [Google Scholar] [CrossRef]
- Franklin, O.; Jonsson, P.; Billing, O.; Lundberg, E.; Ohlund, D.; Nystrom, H.; Lundin, C.; Antti, H.; Sund, M. Plasma Micro-RNA Alterations Appear Late in Pancreatic Cancer. Ann. Surg. 2017, 267, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, D.K.; Gress, T.M.; Langer, P. Familial pancreatic cancer—current knowledge. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 445–453. [Google Scholar] [CrossRef]
- Latenstein, A.E.J.; van der Geest, L.G.M.; Bonsing, B.A.; Groot Koerkamp, B.; Haj Mohammad, N.; de Hingh, I.H.J.T.; de Meijer, V.E.; Molenaar, I.Q.; van Santvoort, H.C.; van Tienhoven, G.; et al. Nationwide trends in incidence, treatment and survival of pancreatic ductal adenocarcinoma. Eur. J. Cancer 2020, 125, 83–93. [Google Scholar] [CrossRef]
- Mellby, L.D.; Nyberg, A.P.; Johansen, J.S.; Wingren, C.; Nordestgaard, B.G.; Bojesen, S.E.; Mitchell, B.L.; Sheppard, B.C.; Sears, R.C.; Borrebaeck, C.A.K. Serum Biomarker Signature-Based Liquid Biopsy for Diagnosis of Early-Stage Pancreatic Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Unger, K.; Mehta, K.Y.; Kaur, P.; Wang, Y.; Menon, S.S.; Jain, S.K.; Moonjelly, R.A.; Suman, S.; Datta, K.; Singh, R.; et al. Metabolomics based predictive classifier for early detection of pancreatic ductal adenocarcinoma. Oncotarget 2018, 9, 23078–23090. [Google Scholar] [CrossRef]
- Kerachian, M.A.; Poudineh, A.; Thiery, J.P. Cell free circulating tumor nucleic acids, a revolution in personalized cancer medicine. Crit. Rev. Oncol./Hematol. 2019, 144, 102827. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Controls | Cases | p Value | Average Time from Sample Collection to Diagnosis, Years (Range) | Average Survival of Cases from Diagnosis, Days |
|---|---|---|---|---|---|
| Number | 320 | 160 a | |||
| Overall Survival | 8 (0–18) | 267 | |||
| Age at initial blood sample collection | >0.999 | ||||
| <45 | 32 | 16 | 8 (2–18) | 252 | |
| 45–54 | 106 | 53 | 9 (0–18) | 255 | |
| 55–64 | 162 | 81 | 7 (0–18) | 295 | |
| ≥65 | 20 | 10 | 8 (1–12) | 132 | |
| Sex | >0.999 | ||||
| Male | 116 | 58 | 9 (0–18) | 247 | |
| Female | 204 | 102 | 7 (0–18) | 279 | |
| Smoking | 0.158 | ||||
| Current or previous smokers | 123 | 72 | 8 (0–18) | 266 | |
| Non-smokers | 184 | 78 | 7 (0–17) | 279 | |
| Information missing | 13 | 10 | 10 (2–18) | 178 | |
| Disease stage at diagnosis | |||||
| Confined to pancreas (I) | N/A | 12 | 7 (2–14) | 864 | |
| Locally advanced and/or regional lymph node metastasis (II) | N/A | 12 | 7 (1–15) | 413 | |
| Large vessels involved (III) | N/A | 37 | 7 (0–17) | 348 | |
| Distant metastasis (IV) | N/A | 99 | 8 (0–18) | 147 | |
| Tumor Grade | |||||
| Low | N/A | 33 | 8 (0–17) | 137 | |
| Intermediate | N/A | 43 | 6 (0–15) | 438 | |
| High | N/A | 4 | 7 (2–14) | 614/1314 b | |
| Information missing | N/A | 80 | 9 (0–18) | 177 | |
| Surgical treatment | |||||
| Curative intent | N/A | 23 | 8 (1–15) | 681 | |
| Palliative | N/A | 25 | 7 (0–17) | 159 | |
| No Surgery | N/A | 111 | 8 (0–18) | 207 | |
| Information missing | N/A | 1 | 14 (N/A) | 123 | |
| Systemic treatment | |||||
| Neoadjuvant and adjuvant chemotherapy | N/A | 2 | 6 (5–7) | 400 | |
| Adjuvant chemotherapy | N/A | 11 | 11 (5–15) | 312 | |
| Palliative chemotherapy | N/A | 68 | 9 (0–18) | 287 | |
| Palliative intraperitoneal 5-FU | N/A | 18 | 8 (0–18) | 339 | |
| Palliative radiotherapy | N/A | 2 | 6 (1–10) | 79 | |
| No systemic treatment | N/A | 59 | 6 (0–17) | 216 | |
| Information missing | N/A | 0 | N/A | N/A |
| Descriptor | PDAC (n) | Breast Cancer (n) | Colorectal Cancer (n) | Chronic Pancreatitis (n) | Controls without Malignant Disease (n) | p Value |
|---|---|---|---|---|---|---|
| Patients | 16 | 9 | 10 | 5 | 16 | N/A |
| Age at blood sample collection | 0.117 | |||||
| <45 | 0 | 0 | 0 | 0 | 0 | |
| 45–54 | 3 | 1 | 0 | 1 | 3 | |
| 55–64 | 7 | 4 | 0 | 1 | 7 | |
| ≥65 | 6 | 4 | 10 | 3 | 6 | |
| Sex | 0.105 (0.981 a) | |||||
| Male | 8 | 0 | 5 | 2 | 8 | |
| Female | 8 | 9 | 5 | 3 | 8 | |
| Disease stage at diagnosis | 0.002 | |||||
| i | 4 | 6 | 3 | N/A | N/A | |
| ii | 0 | 3 | 3 | N/A | N/A | |
| iii | 4 | 0 | 4 | N/A | N/A | |
| iv | 8 | 0 | 0 | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mason, J.; Lundberg, E.; Jonsson, P.; Nyström, H.; Franklin, O.; Lundin, C.; Naredi, P.; Antti, H.; Sund, M.; Öhlund, D. A Cross-Sectional and Longitudinal Analysis of Pre-Diagnostic Blood Plasma Biomarkers for Early Detection of Pancreatic Cancer. Int. J. Mol. Sci. 2022, 23, 12969. https://doi.org/10.3390/ijms232112969
Mason J, Lundberg E, Jonsson P, Nyström H, Franklin O, Lundin C, Naredi P, Antti H, Sund M, Öhlund D. A Cross-Sectional and Longitudinal Analysis of Pre-Diagnostic Blood Plasma Biomarkers for Early Detection of Pancreatic Cancer. International Journal of Molecular Sciences. 2022; 23(21):12969. https://doi.org/10.3390/ijms232112969
Chicago/Turabian StyleMason, James, Erik Lundberg, Pär Jonsson, Hanna Nyström, Oskar Franklin, Christina Lundin, Peter Naredi, Henrik Antti, Malin Sund, and Daniel Öhlund. 2022. "A Cross-Sectional and Longitudinal Analysis of Pre-Diagnostic Blood Plasma Biomarkers for Early Detection of Pancreatic Cancer" International Journal of Molecular Sciences 23, no. 21: 12969. https://doi.org/10.3390/ijms232112969
APA StyleMason, J., Lundberg, E., Jonsson, P., Nyström, H., Franklin, O., Lundin, C., Naredi, P., Antti, H., Sund, M., & Öhlund, D. (2022). A Cross-Sectional and Longitudinal Analysis of Pre-Diagnostic Blood Plasma Biomarkers for Early Detection of Pancreatic Cancer. International Journal of Molecular Sciences, 23(21), 12969. https://doi.org/10.3390/ijms232112969