
Design and Characterization of In-One Protease-Esterase PluriZyme

 , ,
, ,  ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
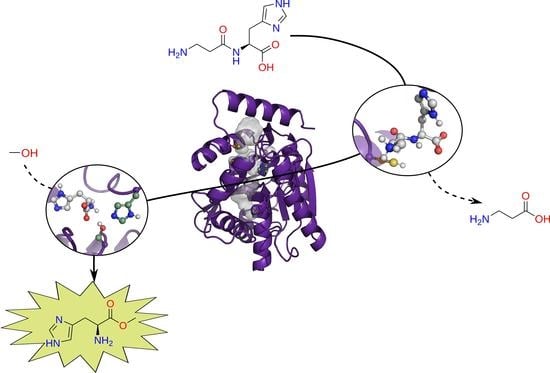
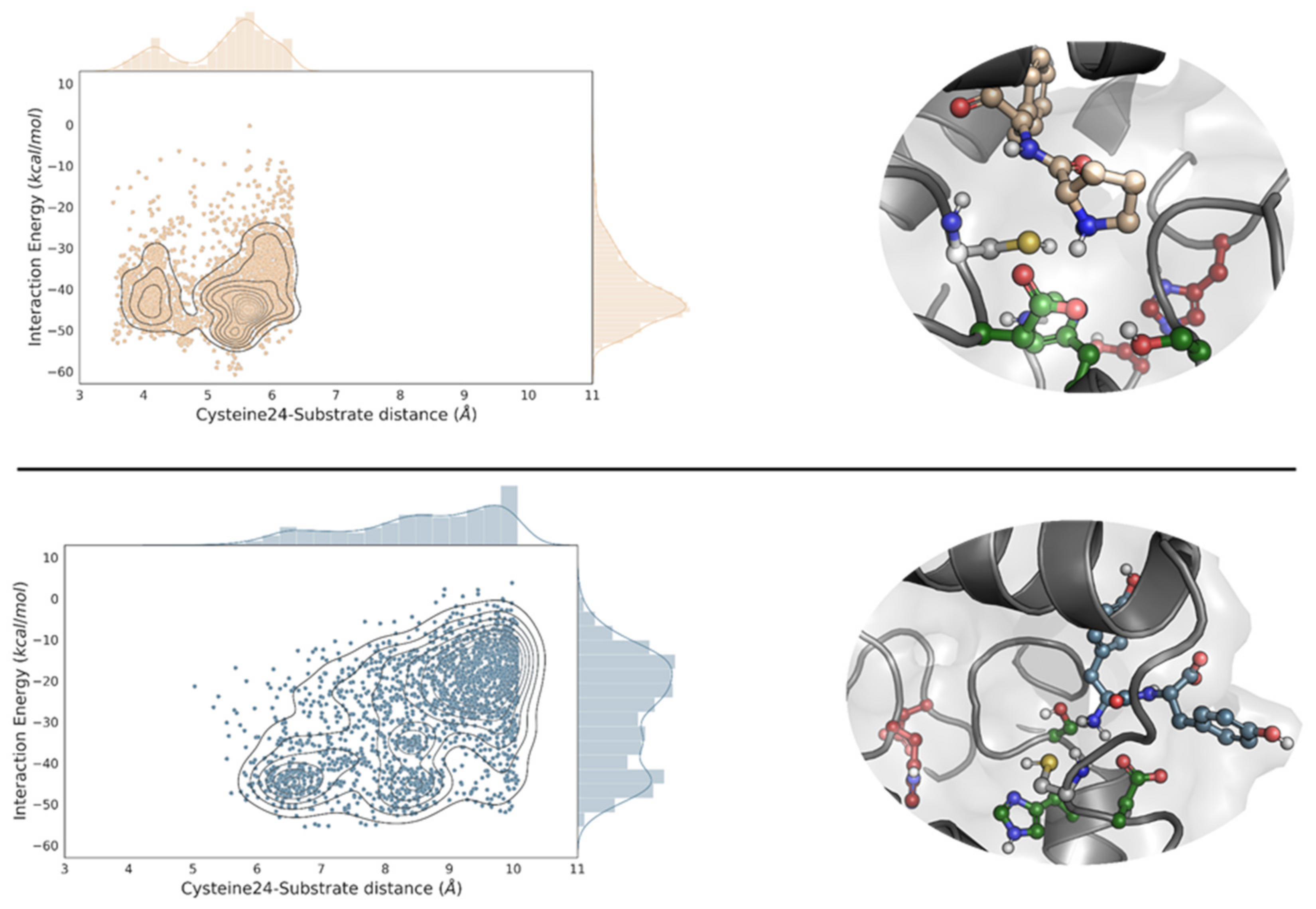
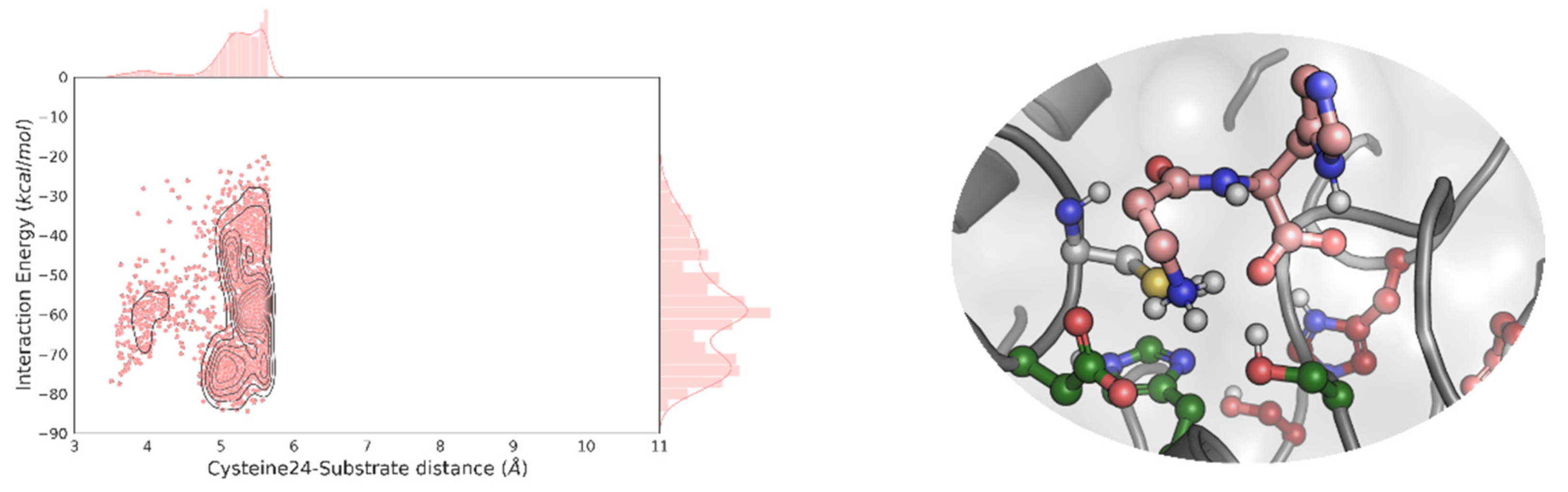
2.1. Molecular Simulations
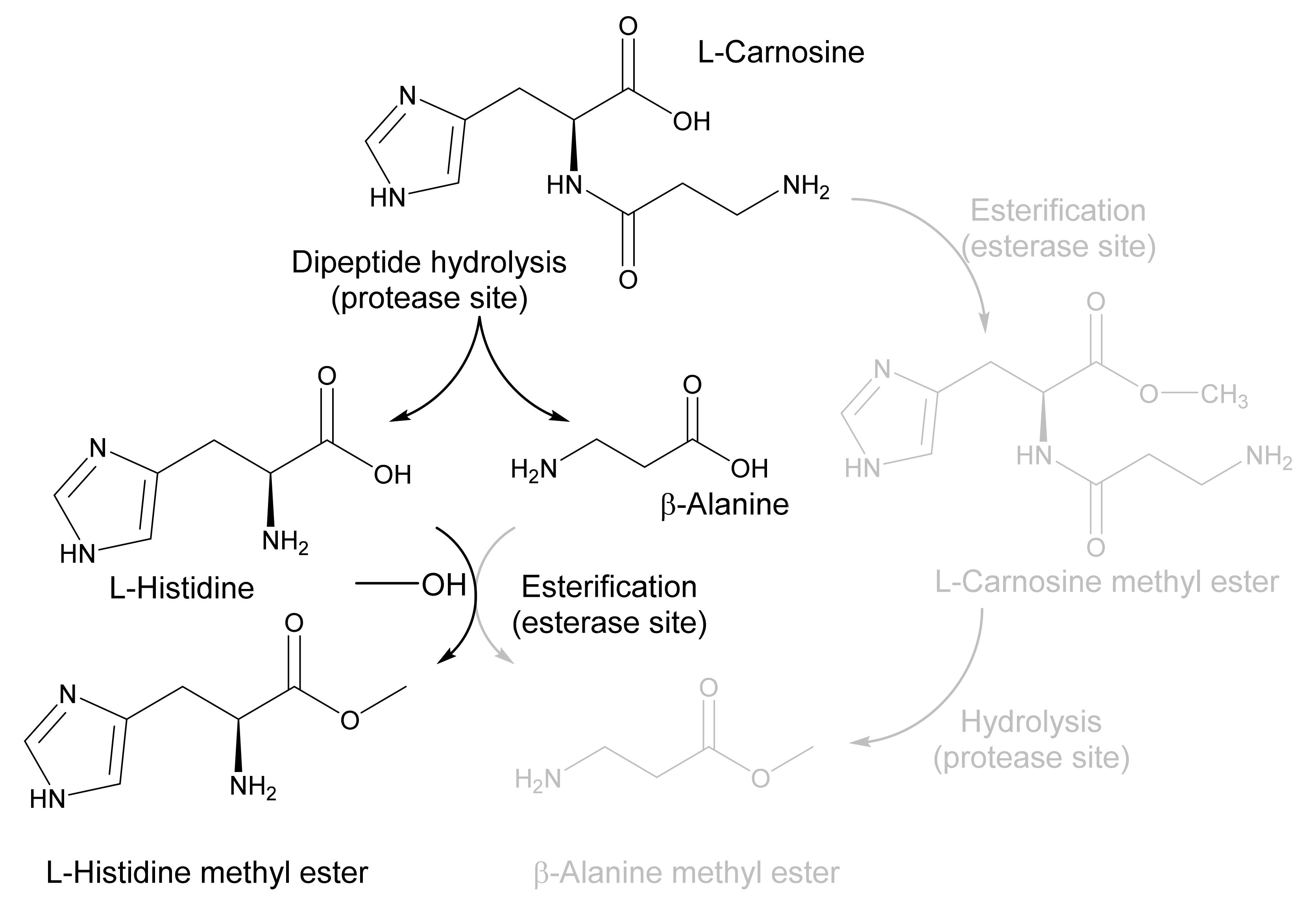
2.2. Experimental Validation: EH1AB1C Is an Efficient Protease
2.3. Application of EH1AB1C in a One-Pot Cascade Reaction
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Protein and Chemical Preparation for the In Silico Analysis
4.3. Protein Energy Landscape Exploration (PELE) Simulations
4.4. Prediction of ΔΔG in the EH1AB1 Variant
4.5. Source and Production of EH1AB1C
4.6. Activity Tests
4.7. Cascade Reaction and the HPLC Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicolaou, K.C.; Edmonds, D.J.; Bulger, P.G. Cascade Reactions in Total Synthesis. Angew. Chem. Int. Ed. Engl. 2006, 45, 7134–7186. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. Engl. 2021, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Devine, P.N.; Howard, R.M.; Kumar, R.; Thompson, M.P.; Truppo, M.D.; Turner, N.J. Extending the Application of Biocatalysis to Meet the Challenges of Drug Development. Nat. Rev. Chem. 2018, 2, 409–421. [Google Scholar] [CrossRef]
- Sperl, J.M.; Sieber, V. Multienzyme Cascade Reactions—Status and Recent Advances. ACS Catal. 2018, 8, 2385–2396. [Google Scholar] [CrossRef]
- Nazor, J.; Liu, J.; Huisman, G. Enzyme Evolution for Industrial Biocatalytic Cascades. Curr. Opin. Biotechnol. 2021, 69, 182–190. [Google Scholar] [CrossRef]
- Coelho, P.S.; Brustad, E.M.; Kannan, A.; Arnold, F.H. Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Huffman, M.A.; Fryszkowska, A.; Alvizo, O.; Borra-Garske, M.; Campos, K.R.; Canada, K.A.; Devine, P.N.; Duan, D.; Forstater, J.H.; Grosser, S.T.; et al. Design of an in Vitro Biocatalytic Cascade for the Manufacture of Islatravir. Science 2019, 366, 1255–1259. [Google Scholar] [CrossRef]
- Alonso, S.; Santiago, G.; Cea-Rama, I.; Fernandez-Lopez, L.; Coscolín, C.; Modregger, J.; Ressmann, A.K.; Martínez-Martínez, M.; Marrero, H.; Bargiela, R.; et al. Genetically Engineered Proteins with Two Active Sites for Enhanced Biocatalysis and Synergistic Chemo- and Biocatalysis. Nat. Catal. 2020, 3, 319–328. [Google Scholar] [CrossRef]
- Santiago, G.; Martínez-Martínez, M.; Alonso, S.; Bargiela, R.; Coscolín, C.; Golyshin, P.N.; Guallar, V.; Ferrer, M. Rational Engineering of Multiple Active Sites in an Ester Hydrolase. Biochemistry 2018, 57, 2245–2255. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Calvó-Tusell, C.; Zhou, A.Z.; Chen, K.; Garcia-Borràs, M.; Arnold, F.H. Dual-Function Enzyme Catalysis for Enantioselective Carbon-Nitrogen Bond Formation. Nat. Chem. 2021, 13, 1166–1172. [Google Scholar] [CrossRef]
- Díaz-Caballero, M.; Navarro, S.; Nuez-Martínez, M.; Peccati, F.; Rodríguez-Santiago, L.; Sodupe, M.; Teixidor, F.; Ventura, S. PH-Responsive Self-Assembly of Amyloid Fibrils for Dual Hydrolase-Oxidase Reactions. ACS Catal. 2021, 11, 595–607. [Google Scholar] [CrossRef]
- Zhou, Z.; Roelfes, G. Synergistic Catalysis in an Artificial Enzyme by Simultaneous Action of Two Abiological Catalytic Sites. Nat. Catal. 2020, 3, 289–294. [Google Scholar] [CrossRef]
- Filice, M.; Romero, O.; Gutiérrez-Fernández, J.; de Las Rivas, B.; Hermoso, J.A.; Palomo, J.M. Synthesis of a Heterogeneous Artificial Metallolipase with Chimeric Catalytic Activity. Chem. Commun. 2015, 51, 9324–9327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christoffel, F.; Igareta, N.V.; Pellizzoni, M.M.; Tiessler-Sala, L.; Lozhkin, B.; Spiess, D.C.; Lledós, A.; Maréchal, J.-D.; Peterson, R.L.; Ward, T.R. Design and Evolution of Chimeric Streptavidin for Protein-Enabled Dual Gold Catalysis. Nat. Catal. 2021, 4, 643–653. [Google Scholar] [CrossRef]
- Knott, B.C.; Erickson, E.; Allen, M.D.; Gado, J.E.; Graham, R.; Kearns, F.L.; Pardo, I.; Topuzlu, E.; Anderson, J.J.; Austin, H.P.; et al. Characterization and Engineering of a Two-Enzyme System for Plastics Depolymerization. Proc. Natl. Acad. Sci. USA 2020, 117, 25476–25485. [Google Scholar] [CrossRef]
- Faylo, J.L.; van Eeuwen, T.; Kim, H.J.; Gorbea Colón, J.J.; Garcia, B.A.; Murakami, K.; Christianson, D.W. Structural Insight on Assembly-Line Catalysis in Terpene Biosynthesis. Nat. Commun. 2021, 12, 3487. [Google Scholar] [CrossRef]
- Zhang, Y.; Hess, H. Toward Rational Design of High-Efficiency Enzyme Cascades. ACS Catal. 2017, 7, 6018–6027. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Arnold, F.H. Engineering New Catalytic Activities in Enzymes. Nat. Catal. 2020, 3, 203–213. [Google Scholar] [CrossRef]
- Roda, S.; Robles-Martín, A.; Xiang, R.; Kazemi, M.; Guallar, V. Structural-Based Modeling in Protein Engineering. A Must Do. J. Phys. Chem. B 2021, 125, 6491–6500. [Google Scholar] [CrossRef]
- Roda, S.; Fernandez-Lopez, L.; Benedens, M.; Bollinger, A.; Thies, S.; Schumacher, J.; Coscolín, C.; Kazemi, M.; Santiago, G.; Gertzen, C.G.W.; et al. A Plurizyme with Transaminase and Hydrolase Activity Catalyzes Cascade Reactions. Angew. Chem. Int. Ed. Engl. 2022, 61, e202207344. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D.; Bode, M.L. The Hitchhiker’s Guide to Biocatalysis: Recent Advances in the Use of Enzymes in Organic Synthesis. Chem. Sci. 2020, 11, 2587–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Moyano, A.; Diaz, Y.; Navarro, J.; Almendral, D.; Puntervoll, P.; Ferrer, M.; Bjerga, G.E.K. Two-step Functional Screen on Multiple Proteinaceous Substrates Reveals Temperature-Robust Proteases with a Broad-Substrate Range. Appl. Microbiol. Biotechnol. 2021, 105, 3195–3209. [Google Scholar] [CrossRef] [PubMed]
- Dyer, R.P.; Weiss, G.A. Making the cut with protease engineering. Cell Chem. Biol. 2022, 29, 177–190. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes; their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, N. Hydrolase-Catalyzed Promiscuous Reactions and Applications in Organic Synthesis. In Molecular Biotechnology; Sedykh, S., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Ogawa, R.; Sunatsuki, Y.; Suzuki, T. Schiff Base Ligands Derived from l-Histidine Methyl Ester: Characterization, Racemization, and Dimerization of Their Transition-Metal Complexes. Eur. J. Inorg. Chem. 2018, 1733–1742. [Google Scholar] [CrossRef]
- Roda, S.; Santiago, G.; Guallar, V. Mapping Enzyme-Substrate Interactions: Its Potential to Study the Mechanism of Enzymes. Adv. Protein Chem. Struct. Biol. 2020, 122, 1–31. [Google Scholar] [PubMed]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Busto, E.; Gotor-Fernández, V.; Gotor, V. Hydrolases: Catalytically Promiscuous Enzymes for Non-Conventional Reactions in Organic Synthesis. Chem. Soc. Rev. 2010, 39, 4504–4523. [Google Scholar] [CrossRef]
- Liljeblad, A.; Kallio, P.; Vainio, M.; Niemi, J.; Kanerva, L.T. Formation and Hydrolysis of Amide Bonds by Lipase A from Candida antarctica; Exceptional Features. Org. Biomol. Chem. 2010, 8, 886–895. [Google Scholar] [CrossRef]
- Simons, C.; van Leeuwen, J.G.E.; Stemmer, R.; Arends, I.W.C.E.; Maschmeyer, T.; Sheldon, R.A.; Hanefeld, U. Enzyme-Catalysed Deprotection of N-Acetyl and N-Formyl Amino Acids. J. Mol. Catal. B Enzym. 2008, 54, 67–71. [Google Scholar] [CrossRef]
- Henke, E.; Bornscheuer, U.T. Fluorophoric Assay for the High-Throughput Determination of Amidase Activity. Anal. Chem. 2003, 75, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Gupta, M.N. Lipase Promiscuity and Its Biochemical Applications. Process Biochem. 2012, 47, 555–569. [Google Scholar] [CrossRef]
- Richter, F.; Blomberg, R.; Khare, S.D.; Kiss, G.; Kuzin, A.P.; Smith, A.J.T.; Gallaher, J.; Pianowski, Z.; Helgeson, R.C.; Grjasnow, A.; et al. Computational Design of Catalytic Dyads and Oxyanion Holes for Ester Hydrolysis. J. Am. Chem. Soc. 2012, 134, 16197–16206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, S.; Wang, C.; Yu, K.; Kuzin, A.P.; Richter, F.; Lew, S.; Miklos, A.E.; Matthews, M.L.; Seetharaman, J.; Su, M.; et al. Design of Activated Serine-Containing Catalytic Triads with Atomic-Level Accuracy. Nat. Chem. Biol. 2014, 10, 386–391. [Google Scholar] [CrossRef]
- Moroz, Y.S.; Dunston, T.T.; Makhlynets, O.V.; Moroz, O.V.; Wu, Y.; Yoon, J.H.; Olsen, A.B.; McLaughlin, J.M.; Mack, K.L.; Gosavi, P.M.; et al. New Tricks for Old Proteins: Single Mutations in a Nonenzymatic Protein Give Rise to Various Enzymatic Activities. J. Am. Chem. Soc. 2015, 137, 14905–14911. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, T.; Minowa, H.; Miyamoto, T.; Imagawa, K.; Yanagihara, R.; Yamada, T. Resolution of Non-Protein Amino Acids Via Microbial Protease-Catalyzed Ester Hydrolysis: Marked Enhancement of Enantioselectivity by the use of Esters with Longer Alkyl Chains and at Low Temperature. Tetrahedron Asymmetry 1997, 8, 367–370. [Google Scholar] [CrossRef]
- Li, J.; Sha, Y. A Convenient Synthesis of Amino Acid Methyl Esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-T.; Wang, K.-T.; Wong, C.-H. Chirally Selective Hydrolysis of D,L-Amino Acid Esters by Alkaline Protease Amino Acid-Catalyzed Hydrolysis of Amino Acid Esters with the Free α-Amino Group. J. Chem. Soc. Chem. Commun. 1986, 20, 1514–1516. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A High-performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Borrelli, K.W.; Vitalis, A.; Alcantara, R.; Guallar, V. PELE: Protein Energy Landscape Exploration. A Novel Monte Carlo Based Technique. J. Chem. Theory Comput. 2005, 1, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Bashford, D.; Case, D.A. Generalized Born Models of Macromolecular Solvation Effects. Annu. Rev. Phys. Chem. 2000, 51, 129–152. [Google Scholar] [CrossRef]
- Sumbalova, L.; Stourac, J.; Martinek, T.; Bednar, D.; Damborsky, J. HotSpot Wizard 3.0: Web Server for Automated Design of Mutations and Smart Libraries Based on Sequence Input Information. Nucleic Acids Res. 2018, 46, W356–W362. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Lopez, L.; Roda, S.; Gonzalez-Alfonso, J.L.; Plou, F.J.; Guallar, V.; Ferrer, M. Design and Characterization of In-One Protease-Esterase PluriZyme. Int. J. Mol. Sci. 2022, 23, 13337. https://doi.org/10.3390/ijms232113337
Fernandez-Lopez L, Roda S, Gonzalez-Alfonso JL, Plou FJ, Guallar V, Ferrer M. Design and Characterization of In-One Protease-Esterase PluriZyme. International Journal of Molecular Sciences. 2022; 23(21):13337. https://doi.org/10.3390/ijms232113337
Chicago/Turabian StyleFernandez-Lopez, Laura, Sergi Roda, Jose L. Gonzalez-Alfonso, Francisco J. Plou, Víctor Guallar, and Manuel Ferrer. 2022. "Design and Characterization of In-One Protease-Esterase PluriZyme" International Journal of Molecular Sciences 23, no. 21: 13337. https://doi.org/10.3390/ijms232113337