
Interaction of Influenza A Nucleoprotein with Host hnRNP-C Is Implicated in Viral Replication
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Host Heterogeneous Nuclear Ribonucleoprotein C (hnRNP-C) Was Identified as an Interacting Partner of NP
2.2. NP and hnRNP-C Directly Bind to One Another
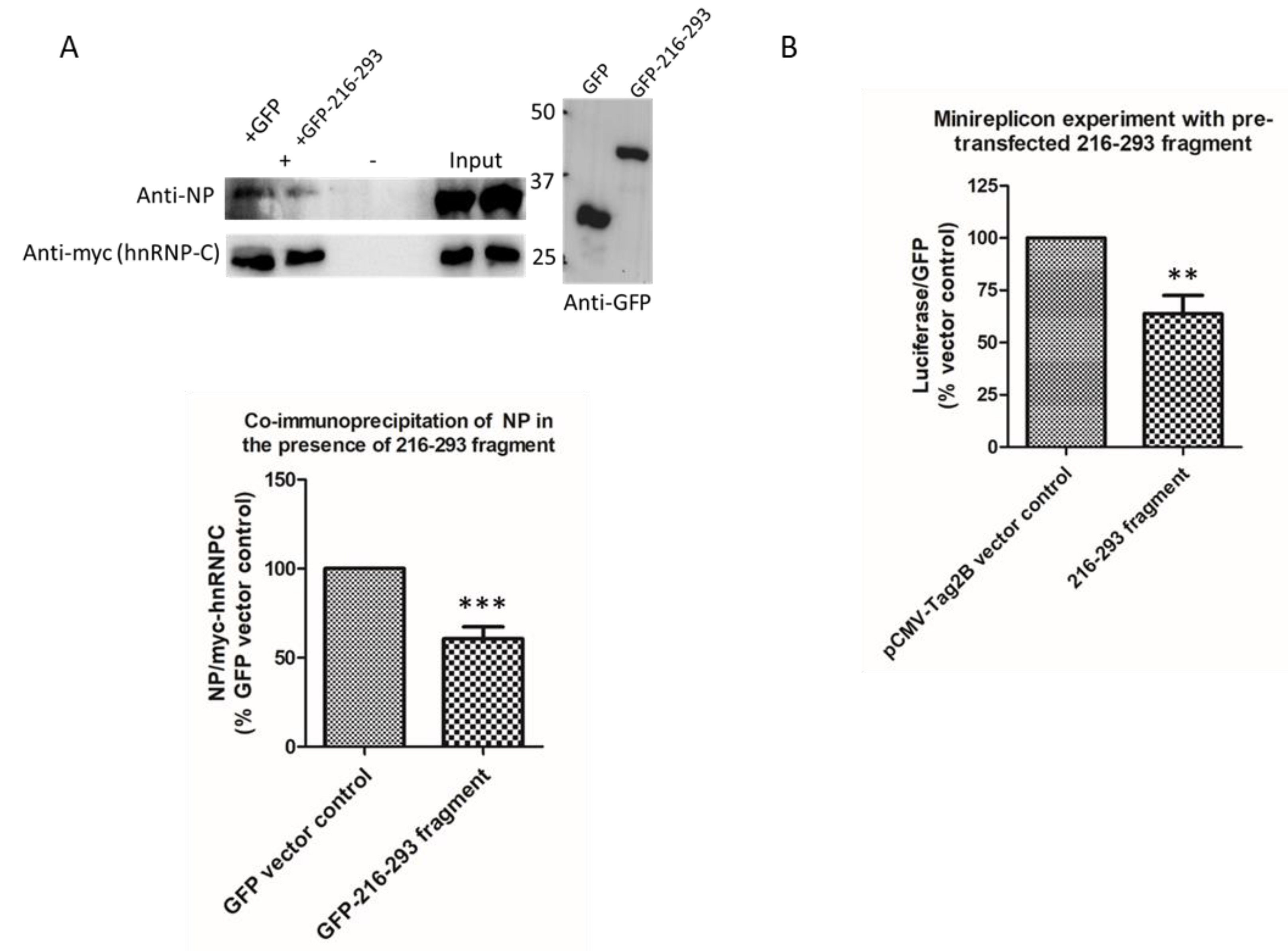
2.3. Characterization of NP–hnRNP-C Interaction
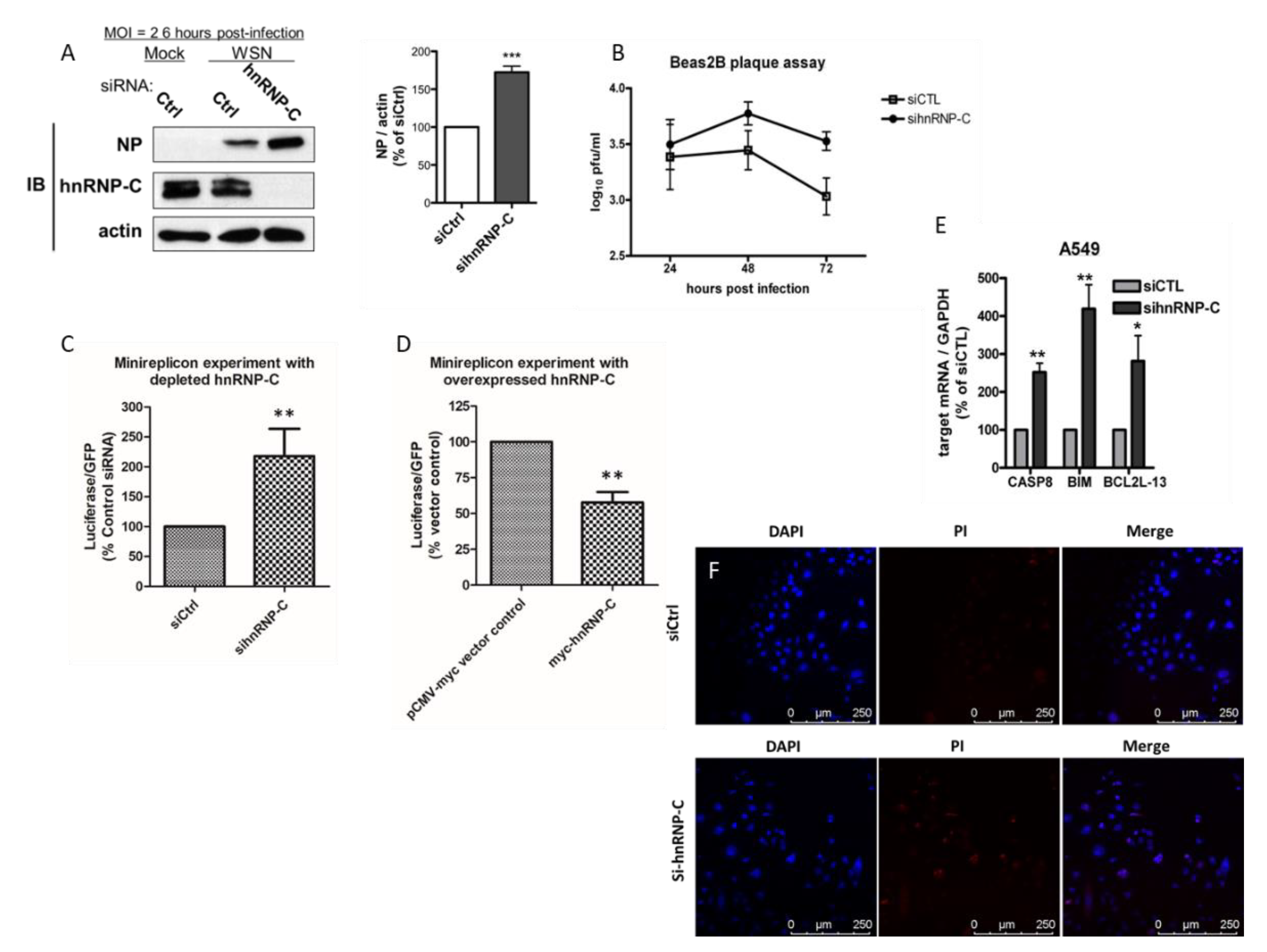
2.4. hnRNP-C Is Implicated in Influenza Virus Replication
2.5. Perturbation of NP–hnRNP-C Interaction Reduces Ribonucleoprotein (RNP) Activity
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cells
4.3. Antibodies
4.4. Expression of NP, Pull-Down, and Mass Spectrometric Analysis
4.5. Virus Infection
4.6. Co-Immunoprecipitation
4.7. In Vitro NHS Pull-Down
4.8. Reverse Transcription qPCR (RT-qPCR)
4.9. Knockdown of hnRNP-C
4.10. Minigenome Luciferase Reporter Assay
4.11. Fluorescence Staining
4.12. Western Blot
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vreede, F.T.; Ng, A.K.-L.; Shaw, P.C.; Fodor, E. Stabilization of Influenza Virus Replication Intermediates Is Dependent on the RNA-Binding but Not the Homo-Oligomerization Activity of the Viral Nucleoprotein. J. Virol. 2011, 85, 12073–12078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, M.; Wills, E.G.; Helenius, A.; Whittaker, G.R. Role of the Influenza Virus M1 Protein in Nuclear Export of Viral Ribonucleoproteins. J. Virol. 2000, 74, 1781–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, K.; Tripathi, S.; Ranjan, P.; Kumar, P.; Garten, R.; Deyde, V.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sambhara, S.; et al. Influenza A Virus Nucleoprotein Exploits Hsp40 to Inhibit PKR Activation. PLoS ONE 2011, 6, e20215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Mayank, A.K.; Nailwal, H.; Tripathi, S.; Patel, J.R.; Bowzard, J.B.; Gaur, P.; Donis, R.O.; Katz, J.M.; Cox, N.J.; et al. Influenza A Viral Nucleoprotein Interacts with Cytoskeleton Scaffolding Protein α-Actinin-4 for Viral Replication. FEBS J. 2014, 281, 2899–2914. [Google Scholar] [CrossRef]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 Inhibits Influenza A Virus Infection by Targeting the Viral Nucleoprotein for Degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.; Batra, J.; Cao, W.; Sharma, K.; Patel, J.R.; Ranjan, P.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; et al. Influenza A Virus Nucleoprotein Induces Apoptosis in Human Airway Epithelial Cells: Implications of a Novel Interaction between Nucleoprotein and Host Protein Clusterin. Cell Death Dis. 2013, 4, e562. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Song, W.; Mok, B.W.Y.; Zhao, P.; Qin, K.; Lai, A.; Smith, G.J.D.; Zhang, J.; Lin, T.; Guan, Y.; et al. Nuclear Factor 90 Negatively Regulates Influenza Virus Replication by Interacting with Viral Nucleoprotein. J. Virol. 2009, 83, 7850–7861. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Sankhala, R.S.; Florio, T.J.; Zhou, L.; Nguyen, N.L.T.; Lokareddy, R.K.; Cingolani, G.; Panté, N. Synergy of Two Low-Affinity NLSs Determines the High Avidity of Influenza A Virus Nucleoprotein NP for Human Importin α Isoforms. Sci. Rep. 2017, 7, 11381. [Google Scholar] [CrossRef] [Green Version]
- Nakada, R.; Hirano, H.; Matsuura, Y. Structure of Importin-α Bound to a Non-Classical Nuclear Localization Signal of the Influenza A Virus Nucleoprotein. Sci. Rep. 2015, 5, 15055. [Google Scholar] [CrossRef]
- Ye, Q.; Krug, R.M.; Tao, Y.J. The Mechanism by Which Influenza A Virus Nucleoprotein Forms Oligomers and Binds RNA. Nature 2006, 444, 1078–1082. [Google Scholar] [CrossRef]
- Ng, A.K.L.; Zhang, H.; Tan, K.; Li, Z.; Liu, J.H.; Chan, P.K.S.; Li, S.M.; Chan, W.Y.; Au, S.W.N.; Joachimiak, A.; et al. Structure of the Influenza Virus A H5N1 Nucleoprotein: Implications for RNA Binding, Oligomerization, and Vaccine Design. FASEB J. 2008, 22, 3638–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.S.; Xu, S.; Chen, Y.W.; Wang, J.H.; Shaw, P.C. Crystal Structures of Influenza Nucleoprotein Complexed with Nucleic Acid Provide Insights into the Mechanism of RNA Interaction. Nucleic Acids Res. 2021, 49, 4144–4154. [Google Scholar] [CrossRef]
- Lewis, S.M.; Veyrier, A.; Hosszu Ungureanu, N.; Bonnal, S.; Vagner, S.; Holcik, M. Subcellular Relocalization of a Trans-Acting Factor Regulates XIAP IRES-Dependent Translation. Mol. Biol. Cell 2007, 18, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holcík, M.; Gordon, B.W.; Korneluk, R.G. The Internal Ribosome Entry Site-Mediated Translation of Antiapoptotic Protein XIAP Is Modulated by the Heterogeneous Nuclear Ribonucleoproteins C1 and C2. Mol. Cell. Biol. 2003, 23, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Burd, C.G.; Swanson, M.S.; Görlach, M.; Dreyfuss, G. Primary Structures of the Heterogeneous Nuclear Ribonucleoprotein A2, B1, and C2 Proteins: A Diversity of RNA Binding Proteins Is Generated by Small Peptide Inserts. Proc. Natl. Acad. Sci. USA 1989, 86, 9788–9792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechtawewat, T.; Songprakhon, P.; Limjindaporn, T.; Puttikhunt, C.; Kasinrerk, W.; Saitornuang, S.; Yenchitsomanus, P.; Noisakran, S. Role of Human Heterogeneous Nuclear Ribonucleoprotein C1/C2 in Dengue Virus Replication. Virol. J. 2015, 12, 14. [Google Scholar] [CrossRef] [Green Version]
- Gustin, K.E.; Sarnow, P. Effects of Poliovirus Infection on Nucleo-Cytoplasmic Trafficking and Nuclear Pore Complex Composition. EMBO J. 2001, 20, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Casaca, A.; Fardilha, M.; da Cruz e Silva, E.; Cunha, C. The Heterogeneous Ribonuclear Protein C Interacts with the Hepatitis Delta Virus Small Antigen. Virol. J. 2011, 8, 358. [Google Scholar] [CrossRef] [Green Version]
- Shabman, R.S.; Gulcicek, E.E.; Stone, K.L.; Basler, C.F. The Ebola Virus VP24 Protein Prevents HnRNP C1/C2 Binding to Karyopherin A1 and Partially Alters Its Nuclear Import. J. Infect. Dis. 2011, 204 (Suppl. 3), S904–S910. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chu, H.; Chik, K.K.H.; Wen, L.; Shuai, H.; Yang, D.; Wang, Y.; Hou, Y.; Yuen, T.T.T.; Cai, J.P.; et al. HnRNP C Modulates MERS-CoV and SARS-CoV-2 Replication by Governing the Expression of a Subset of CircRNAs and Cognitive MRNAs. Emerg. Microbes Infect. 2022, 11, 519. [Google Scholar] [CrossRef]
- Chang, C.-K.; Chen, C.-J.; Wu, C.-C.; Chen, S.-W.; Shih, S.-R.; Kuo, R.L. Cellular HnRNP A2/B1 Interacts with the NP of Influenza A Virus and Impacts Viral Replication. PLoS ONE 2017, 12, e0188214. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lin, L.; Zhong, Y.; Feng, M.; Yu, T.; Yan, Y.; Zhou, J.; Liao, M. Cellular HnRNPAB Binding to Viral Nucleoprotein Inhibits Flu Virus Replication by Blocking Nuclear Export of Viral MRNA. iScience 2021, 24, 102160. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Batra, J.; Stuchlik, O.; Reed, M.S.; Pohl, J.; Sambhara, S.; Lal, S.K. Heterogeneous Ribonucleoprotein A1 (HnRNPA1) Interacts with the Nucleoprotein of the Influenza a Virus and Impedes Virus Replication. Viruses 2022, 14, 199. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, S.; Yu, Y.; Zhang, J.; Sun, Z.; Yan, Y.; Zhou, J. Subcellular Proteomic Analysis of Human Host Cells Infected with H3N2 Swine Influenza Virus. Proteomics 2013, 13, 3309–3326. [Google Scholar] [CrossRef] [PubMed]
- Mayer, D.; Molawi, K.; Martínez-Sobrido, L.; Ghanem, A.; Thomas, S.; Baginsky, S.; Grossmann, J.; García-Sastre, A.; Schwemmle, M. Identification of Cellular Interaction Partners of the Influenza Virus Ribonucleoprotein Complex and Polymerase Complex Using Proteomic-Based Approaches. J. Proteome Res. 2007, 6, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portela, A.; Digard, P. The Influenza Virus Nucleoprotein: A Multifunctional RNA-Binding Protein Pivotal to Virus Replication. J. Gen. Virol. 2002, 83, 723–734. [Google Scholar] [CrossRef]
- Cieniková, Z.; Damberger, F.F.; Hall, J.; Allain, F.H.T.; Maris, C. Structural and Mechanistic Insights into Poly(Uridine) Tract Recognition by the HnRNP C RNA Recognition Motif. J. Am. Chem. Soc. 2014, 136, 14536–14544. [Google Scholar] [CrossRef]
- Brunner, J.E.; Nguyen, J.H.C.; Roehl, H.H.; Ho, T.V.; Swiderek, K.M.; Semler, B.L. Functional Interaction of Heterogeneous Nuclear Ribonucleoprotein C with Poliovirus RNA Synthesis Initiation Complexes. J. Virol. 2005, 79, 3254–3266. [Google Scholar] [CrossRef] [Green Version]
- Dhanjal, S.; Kajitani, N.; Glahder, J.; Mossberg, A.K.; Johansson, C.; Schwartz, S. Heterogeneous Nuclear Ribonucleoprotein C Proteins Interact with the Human Papillomavirus Type 16 (HPV16) Early 3’-Untranslated Region and Alleviate Suppression of HPV16 Late L1 MRNA Splicing. J. Biol. Chem. 2015, 290, 13354–13371. [Google Scholar] [CrossRef] [Green Version]
- Mühlbauer, D.; Dzieciolowski, J.; Hardt, M.; Hocke, A.; Schierhorn, K.L.; Mostafa, A.; Müller, C.; Wisskirchen, C.; Herold, S.; Wolff, T.; et al. Influenza Virus-Induced Caspase-Dependent Enlargement of Nuclear Pores Promotes Nuclear Export of Viral Ribonucleoprotein Complexes. J. Virol. 2015, 89, 6009–6021. [Google Scholar] [CrossRef]
- Yu, K.; Ren, Y.; Zhang, X.; Qiao, T.; Liu, Z.; Shi, J.; Wang, Y. ShRNA-Mediated NP Knockdown Inhibits the Apoptosis of Cardiomyocytes Induced by H1N1pdm2009 Influenza Virus. Mol. Med. Rep. 2017, 16, 1376–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deveraux, Q.L.; Leo, E.; Stennicke, H.R.; Welsh, K.; Salvesen, G.S.; Reed, J.C. Cleavage of Human Inhibitor of Apoptosis Protein XIAP Results in Fragments with Distinct Specificities for Caspases. EMBO J. 1999, 18, 5242–5251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, C.Y.; Li, O.T.W.; Tang, W.P.; Hu, C.; Wang, G.X.; Ngo, J.C.K.; Wan, D.C.C.; Poon, L.L.M.; Shaw, P.C. Identification of Influenza Polymerase Inhibitors Targeting C-Terminal Domain of PA through Surface Plasmon Resonance Screening. Sci. Rep. 2018, 8, 2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, W.C.; Hsia, H.-P.; Tang, Y.S.; Shaw, P.-C. Interaction between Influenza A Virus Nucleoprotein and PB2 Cap-Binding Domain Is Mediated by RNA. PLoS ONE 2020, 15, e0239899. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.-S.; So, W.-K.; Ng, K.-L.A.; Mok, K.-P.C.; Shaw, P.-C. Interaction of Influenza A Nucleoprotein with Host hnRNP-C Is Implicated in Viral Replication. Int. J. Mol. Sci. 2022, 23, 13613. https://doi.org/10.3390/ijms232113613
Tang Y-S, So W-K, Ng K-LA, Mok K-PC, Shaw P-C. Interaction of Influenza A Nucleoprotein with Host hnRNP-C Is Implicated in Viral Replication. International Journal of Molecular Sciences. 2022; 23(21):13613. https://doi.org/10.3390/ijms232113613
Chicago/Turabian StyleTang, Yun-Sang, Wai-Kin So, Ka-Leung Andy Ng, Ka-Pun Chris Mok, and Pang-Chui Shaw. 2022. "Interaction of Influenza A Nucleoprotein with Host hnRNP-C Is Implicated in Viral Replication" International Journal of Molecular Sciences 23, no. 21: 13613. https://doi.org/10.3390/ijms232113613