
Loss of the Nuclear Envelope Protein LAP1B Disrupts the Myogenic Differentiation of Patient-Derived Fibroblasts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Myogenic Differentiation and Fusion Defects in LAP1B-Deficient Cells
2.2. Impaired Differentiation of Tor1aip1-Mutant C2C12 Myoblasts
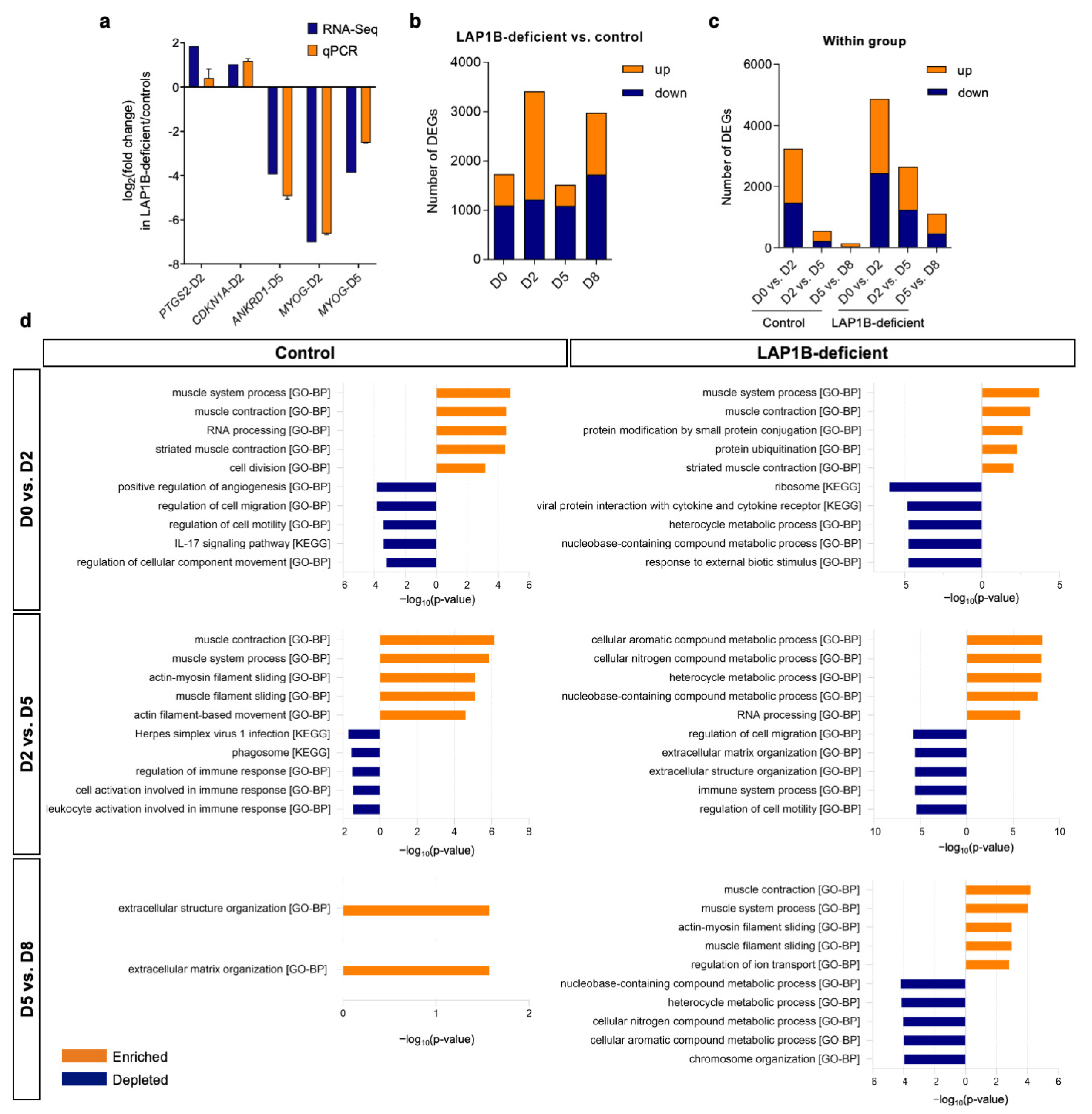
2.3. Overview of the Transcriptome Profile of Transdifferentiating Fibroblasts
2.4. Transcriptome Profiling Reflects Incomplete Reprogramming of LAP1B-Deficient Cells
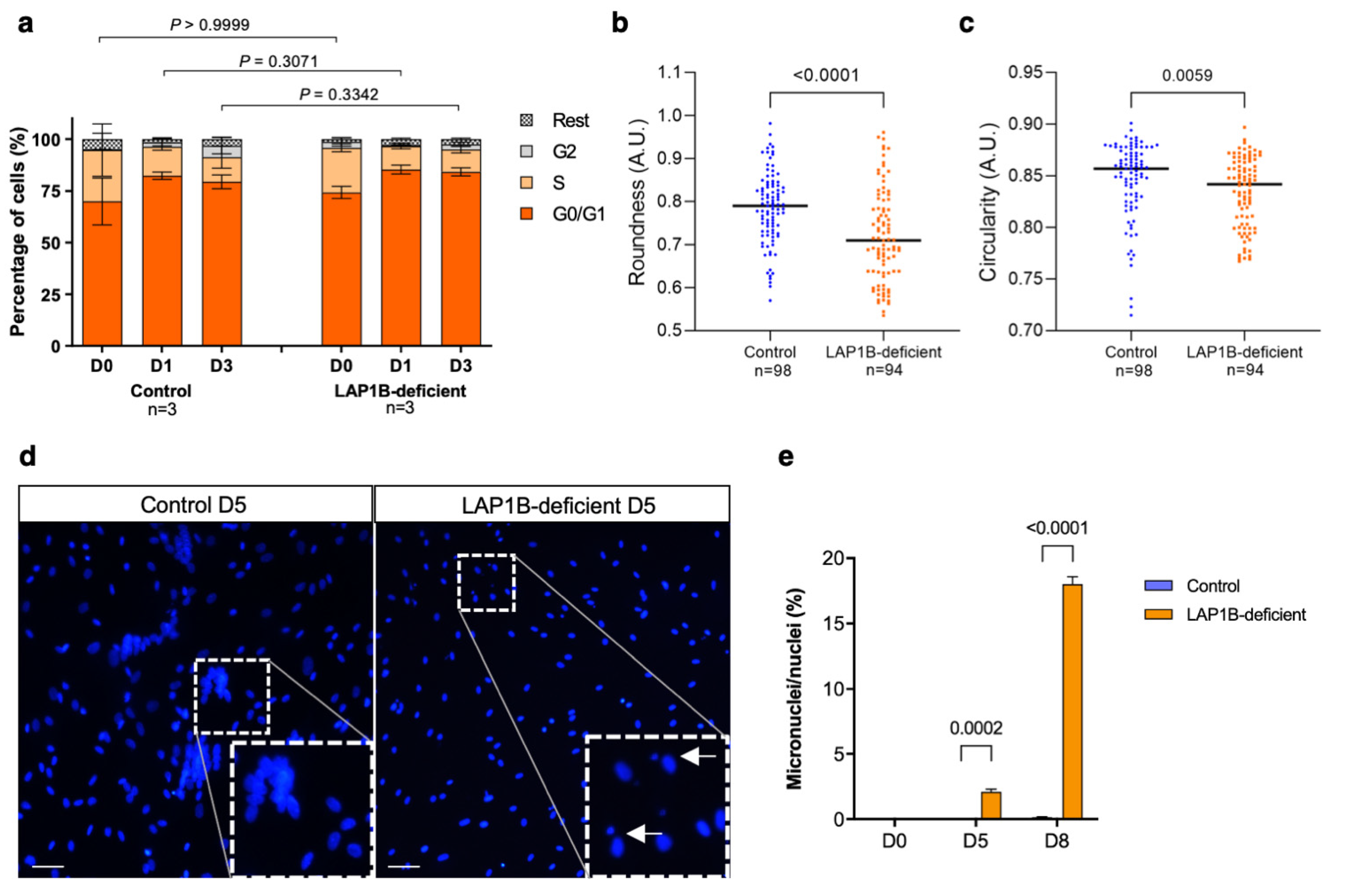
2.5. LAP1B-Deficient Fibroblasts Efficiently Withdraw from the Cell Cycle upon MyoD Expression
2.6. LAP1B-Deficient Fibroblasts Exhibit Nuclear Morphological Defects
3. Discussion
4. Materials and Methods
4.1. Immortalized MyoD-Converted Human Fibroblasts
4.2. Tor1aip1-Mutant C2C12 Myoblasts
4.3. Immunofluorescence Staining
4.4. Quantification of Myogenic Differentiation
4.5. RNA Sequencing
4.6. Pathway Analysis
4.7. Quantitative Real-Time PCR
4.8. Protein Extraction and Immunoblotting
4.9. Cell Cycle Assay
4.10. Assessment of Nuclear Morphologic and Morphometric Parameters
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worman, H.J.; Schirmer, E.C. Nuclear membrane diversity: Underlying tissue-specific pathologies in disease? Curr. Opin. Cell Biol. 2015, 34, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonne, G.; Quijano-Roy, S. Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb. Clin. Neurol. 2013, 113, 1367–1376. [Google Scholar] [PubMed] [Green Version]
- Shin, J.Y.; Dauer, W.T.; Worman, H.J. Lamina-associated polypeptide 1: Protein interactions and tissue-selective functions. Semin. Cell Dev. Biol. 2014, 29, 164–168. [Google Scholar] [CrossRef] [Green Version]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8, 335ra58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, A.; Gerace, L. Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J. Cell Biol. 1988, 107, 2029–2036. [Google Scholar] [CrossRef]
- Goodchild, R.E.; Dauer, W.T. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J. Cell Biol. 2005, 168, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Foisner, R.; Gerace, L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 1993, 73, 1267–1279. [Google Scholar] [CrossRef]
- Brown, R.S.H.; Zhao, C.; Chase, A.R.; Wang, J.; Schlieker, C. The mechanism of Torsin ATPase activation. Proc. Natl. Acad. Sci. USA 2014, 111, E4822–E4831. [Google Scholar] [CrossRef] [Green Version]
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. How lamina-associated polypeptide 1 (LAP1) activates Torsin. eLife 2014, 3, e03239. [Google Scholar] [CrossRef] [Green Version]
- Maison, C.; Pyrpasopoulou, A.; Theodoropoulos, P.A.; Georgatos, S.D. The inner nuclear membrane protein LAP1 forms a native complex with B-type lamins and partitions with spindle-associated mitotic vesicles. EMBO J. 1997, 16, 4839–4850. [Google Scholar] [CrossRef]
- Kondo, Y.; Kondoh, J.; Hayashi, D.; Ban, T.; Takagi, M.; Kamei, Y.; Tsuji, L.; Kim, J.; Yoneda, Y. Molecular cloning of one isotype of human lamina-associated polypeptide 1s and a topological analysis using its deletion mutants. Biochem. Biophys. Res. Commun. 2002, 294, 770–778. [Google Scholar] [CrossRef]
- Santos, M.; Domingues, S.C.; Costa, P.; Muller, T.; Galozzi, S.; Marcus, K.; da Cruz E Silva, E.F.; da Cruz E Silva, O.A.; Rebelo, S. Identification of a Novel Human LAP1 Isoform That Is Regulated by Protein Phosphorylation. PLoS ONE 2014, 9, e113732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.; Costa, P.; Martins, F.; da Cruz E Silva, E.F.; da Cruz E Silva, O.A.B.; Rebelo, S. LAP1 is a crucial protein for the maintenance of the nuclear envelope structure and cell cycle progression. Mol. Cell. Biochem. 2014, 399, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Walter, T.; Hériché, J.K.; Bulkescher, J.; Erfle, H.; Conrad, C.; Rogers, P.; Poser, I.; Held, M.; Liebel, U.; et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 2010, 464, 721–727. [Google Scholar] [CrossRef]
- Luithle, N.; uit de Bos, J.; Hovius, R.; Maslennikova, D.; Lewis, R.; Ungricht, R.; Fierz, B.; Kutay, U. Torsin ATPases influence chromatin interaction of the Torsin regulator LAP1. eLife 2020, 9, e63614. [Google Scholar] [CrossRef]
- Pereira, C.D.; Martins, F.; Santos, M.; Müeller, T.; da Cruz E Silva, O.A.B.; Rebelo, S. Nuclear Accumulation of LAP1:TRF2 Complex during DNA Damage Response Uncovers a Novel Role for LAP1. Cells 2020, 9, 1804. [Google Scholar] [CrossRef]
- Kayman-Kurekci, G.; Talim, B.; Korkusuz, P.; Sayar, N.; Sarioglu, T.; Oncel, I.; Sharafi, P.; Gundesli, H.; Balci-Hayta, B.; Purali, N.; et al. Mutation in TOR1AIP1 encoding LAP1B in a form of muscular dystrophy: A novel gene related to nuclear envelopathies. Neuromuscul. Disord. 2014, 24, 624–633. [Google Scholar] [CrossRef]
- Feng, X.; Wu, J.; Xian, W.; Liao, B.; Liao, S.; Yao, X.; Zhang, W. Muscular involvement and tendon contracture in limb-girdle muscular dystrophy 2Y: A mild adult phenotype and literature review. BMC Musculoskelet. Disord. 2020, 21, 588. [Google Scholar] [CrossRef]
- Ghaoui, R.; Benavides, T.; Lek, M.; Waddell, L.B.; Kaur, S.; North, K.N.; MacArthur, D.G.; Clarke, N.F.; Cooper, S.T. TOR1AIP1 as a cause of cardiac failure and recessive limb-girdle muscular dystrophy. Neuromuscul. Disord. 2016, 26, 500–503. [Google Scholar] [CrossRef]
- Cossins, J.; Webster, R.; Maxwell, S.; Rodríguez Cruz, P.M.; Knight, R.; Llewelyn, J.G.; Shin, J.-Y.; Palace, J.; Beeson, D. Congenital myasthenic syndrome due to a TOR1AIP1 mutation: A new disease pathway for impaired synaptic transmission. Brain Commun. 2020, 2, fcaa174. [Google Scholar] [CrossRef]
- Dorboz, I.; Coutelier, M.; Bertrand, A.T.; Caberg, J.-H.; Elmaleh-Bergès, M.; Lainé, J.; Stevanin, G.; Bonne, G.; Boespflug-Tanguy, O.; Servais, L. Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in TOR1AIP1. Orphanet J. Rare Dis. 2014, 9, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichtman, B.; Zagairy, F.; Biran, N.; Barsheshet, Y.; Chervinsky, E.; Ben Neriah, Z.; Shaag, A.; Assa, M.; Elpeleg, O.; Harel, A.; et al. Combined loss of LAP1B and LAP1C results in an early onset multisystemic nuclear envelopathy. Nat. Commun. 2019, 10, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessel, I.; Chen, M.J.; Lüttgen, S.; Arndt, F.; Fuchs, S.; Meien, S.; Thiele, H.; Jones, J.R.; Shaw, B.R.; Crossman, D.K.; et al. Two novel cases further expand the phenotype of TOR1AIP1-associated nuclear envelopathies. Hum. Genet. 2020, 139, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.-Y.; Méndez-López, I.; Hong, M.; Wang, Y.; Tanji, K.; Wu, W.; Shugol, L.; Krauss, R.S.; Dauer, W.T.; Worman, H.J. Lamina-associated polypeptide 1 is dispensable for embryonic myogenesis but required for postnatal skeletal muscle growth. Hum. Mol. Genet. 2016, 26, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.-Y.; Méndez-López, I.; Wang, Y.; Hays, A.P.; Tanji, K.; Lefkowitch, J.H.; Schulze, P.C.; Worman, H.J.; Dauer, W.T. Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev. Cell 2013, 26, 591–603. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.E.; Perez, A.; Perkins, G.; Ellisman, M.H.; Dauer, W.T. A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9861–9866. [Google Scholar] [CrossRef] [Green Version]
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Dev. Camb. 2017, 144, 2104–2122. [Google Scholar]
- Singh, K.; Dilworth, F.J. Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS J. 2013, 280, 3991–4003. [Google Scholar] [CrossRef]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Weintraub, H.; Tapscott, S.J.; Davis, R.L.; Thayer, M.J.; Adam, M.A.; Lassar, A.B.; Miller, A.D. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. USA 1989, 86, 5434–5438. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Costa, M.L.; Mermelstein, C.S.; Chagas, C.; Holtzer, S.; Holtzer, H. MyoD converts primary dermal fibroblasts, chondroblasts, smooth muscle, and retinal pigmented epithelial cells into striated mononucleated myoblasts and multinucleated myotubes. Proc. Natl. Acad. Sci. USA 1990, 87, 7988–7992. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, S.; Mouly, V.; Goyenvalle, A.; Vulin, A.; Mamchaoui, K.; Negroni, E.; Di Santo, J.; Butler-Browne, G.; Torrente, Y.; Garcia, L.; et al. Immortalized skin fibroblasts expressing conditional MyoD as a renewable and reliable source of converted human muscle cells to assess therapeutic strategies for muscular dystrophies: Validation of an exon-skipping approach to restore dystrophin in Duchen. Hum. Gene Ther. 2009, 20, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, L.; Salvatori, G.; Coletta, M.; Sonnino, C.; De Angelis, M.G.C.; Gioglio, L.; Murry, C.E.; Kelly, R.; Ferrari, G.; Molinaro, M.; et al. High efficiency myogenic conversion of human fibroblasts by adenoviral vector-mediated MyoD gene transfer. An alternative strategy for ex vivo gene therapy of primary myopathies. J. Clin. Investig. 1998, 101, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.; Pettersson, O.J.; Jakobsen, M.; Thomsen, R.; Pedersen, C.B.; Hertz, J.M.; Gregersen, N.; Corydon, T.J.; Jensen, T.G. Myoblasts generated by lentiviral mediated MyoD transduction of myotonic dystrophy type 1 (DM1) fibroblasts can be used for assays of therapeutic molecules. BMC Res. Notes 2011, 4, 490. [Google Scholar] [CrossRef] [Green Version]
- Muir, L.A.; Nguyen, Q.G.; Hauschka, S.D.; Chamberlain, J.S. Engraftment potential of dermal fibroblasts following in vivo myogenic conversion in immunocompetent dystrophic skeletal muscle. Mol. Ther.-Methods Clin. Dev. 2014, 1, 14025. [Google Scholar] [CrossRef]
- Faralli, H.; Dilworth, F.J. Turning on myogenin in muscle: A paradigm for understanding mechanisms of tissue-specific gene expression. Comp. Funct. Genom. 2012, 2012, 836374. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Blais, A.; Tsikitis, M.; Acosta-Alvear, D.; Sharan, R.; Kluger, Y.; Dynlacht, B.D. An initial blueprint for myogenic differentiation. Genes Dev. 2005, 19, 553–569. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Kumar, R.M.; Penn, B.H.; Berkes, C.A.; Kooperberg, C.; Boyer, L.A.; Young, R.A.; Tapscott, S.J. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J. 2006, 25, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, A.; Kim, W.; Davie, J. Myogenin is required for assembly of the transcription machinery on muscle genes during skeletal muscle differentiation. PLoS ONE 2021, 16, e0245618. [Google Scholar] [CrossRef]
- Zhang, H.; Wen, J.; Bigot, A.; Chen, J.; Shang, R.; Mouly, V.; Bi, P. Human myotube formation is determined by MyoD–Myomixer/Myomaker axis. Sci. Adv. 2020, 6, 4062–4080. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.C.; Zha, X.H.; Faralli, H.; Yin, H.; Louis-Jeune, C.; Perdiguero, E.; Pranckeviciene, E.; Muñoz-Cànoves, P.; Rudnicki, M.A.; Brand, M.; et al. Comparative expression profiling identifies differential roles for Myogenin and p38α MAPK signaling in myogenesis. J. Mol. Cell Biol. 2012, 4, 386–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, A.; Koch, A.J.; Holaska, J.M. Expression Profiling of Differentiating Emerin-Null Myogenic Progenitor Identifies Molecular Pathways Implicated in Their Impaired Differentiation. Cells 2017, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laguri, C.; Gilquin, B.; Wolff, N.; Romi-Lebrun, R.; Courchay, K.; Callebaut, I.; Worman, H.J.; Zinn-Justin, S. Structural characterization of the LEM motif common to three human inner nuclear membrane proteins. Structure 2001, 9, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Huang, Y.; Ghirlando, R.; Wilson, K.L.; Craigie, R.; Clore, G.M. Solution structure of the constant region of nuclear envelope protein LAP2 reveals two LEM-domain structures: One binds BAF and the other binds DNA. EMBO J. 2001, 20, 4399. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar] [CrossRef]
- Makatsori, D.; Kourmouli, N.; Polioudaki, H.; Shultz, L.; McLean, K.; Theodoropoulos, P.; Singh, P.; Georgatos, S. The Inner Nuclear Membrane Protein Lamin B Receptor Forms Distinct Microdomains and Links Epigenetically Marked Chromatin to the Nuclear Envelope. J. Biol. Chem. 2004, 279, P25567–P25573. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.J.P.; Kenzelmann Broz, D.; Noderer, W.L.; Ferreira, J.P.; Overton, K.W.; Spencer, S.L.; Meyer, T.; Tapscott, S.J.; Attardi, L.D.; Wang, C.L. P53 Suppresses Muscle Differentiation At the Myogenin Step in Response To Genotoxic Stress. Cell Death Differ. 2015, 22, 560–573. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2015, 31, 1120–1123. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.P.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Lowe, M.; Lage, J.; Paatela, E.; Munson, D.; Hostager, R.; Yuan, C.; Katoku-Kikyo, N.; Ruiz-Estevez, M.; Asakura, Y.; Staats, J.; et al. Cry2 Is Critical for Circadian Regulation of Myogenic Differentiation by Bclaf1-Mediated mRNA Stabilization of Cyclin D1 and Tmem176b. Cell Rep. 2018, 22, 2118–2132. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrew, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 November 2021).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Aken, B.; Achuthan, P.; Akanni, W.; Amode, M.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2016, D1, D635–D642. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stöckel, D.; Kehl, T.; Trampert, P.; Schneider, L.; Backes, C.; Ludwig, N.; Gerasch, A.; Kaufmann, M.; Gessler, M.; Graf, N.; et al. Multi-omics enrichment analysis using the GeneTrail2 web service. Bioinformatics 2016, 32, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.; Jones, M.; Rouillard, A.; Fernandez, N.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.; Jagodnik, K.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayman Kürekçi, G.; Acar, A.C.; Dinçer, P.R. Loss of the Nuclear Envelope Protein LAP1B Disrupts the Myogenic Differentiation of Patient-Derived Fibroblasts. Int. J. Mol. Sci. 2022, 23, 13615. https://doi.org/10.3390/ijms232113615
Kayman Kürekçi G, Acar AC, Dinçer PR. Loss of the Nuclear Envelope Protein LAP1B Disrupts the Myogenic Differentiation of Patient-Derived Fibroblasts. International Journal of Molecular Sciences. 2022; 23(21):13615. https://doi.org/10.3390/ijms232113615
Chicago/Turabian StyleKayman Kürekçi, Gülsüm, Aybar C. Acar, and Pervin R. Dinçer. 2022. "Loss of the Nuclear Envelope Protein LAP1B Disrupts the Myogenic Differentiation of Patient-Derived Fibroblasts" International Journal of Molecular Sciences 23, no. 21: 13615. https://doi.org/10.3390/ijms232113615