

Chronic Trazodone and Citalopram Treatments Increase Trophic Factor and Circadian Rhythm Gene Expression in Rat Brain Regions Relevant for Antidepressant Efficacy
 , , and
, , and
Abstract
:1. Introduction
2. Results
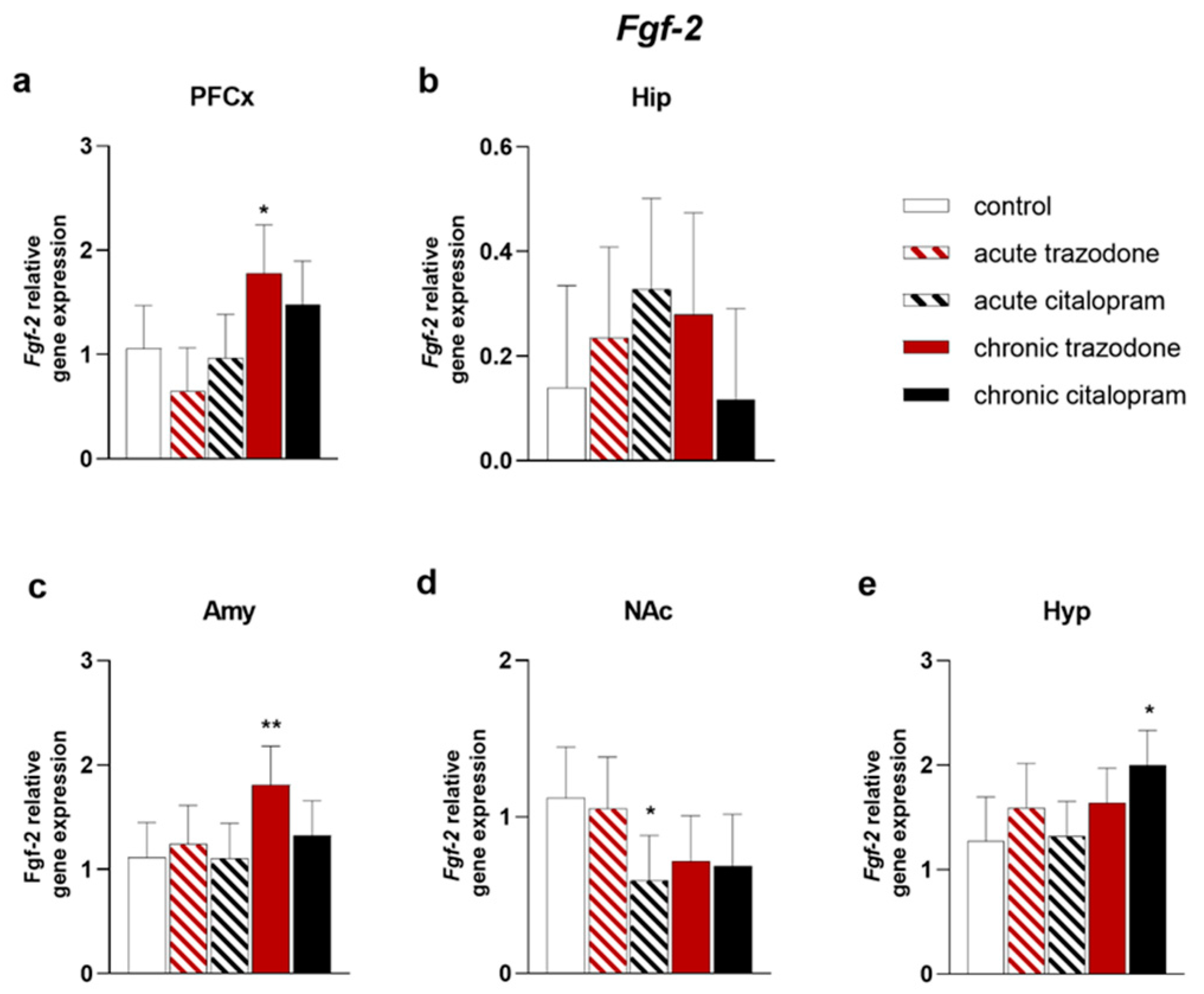
2.1. Trophic Factors
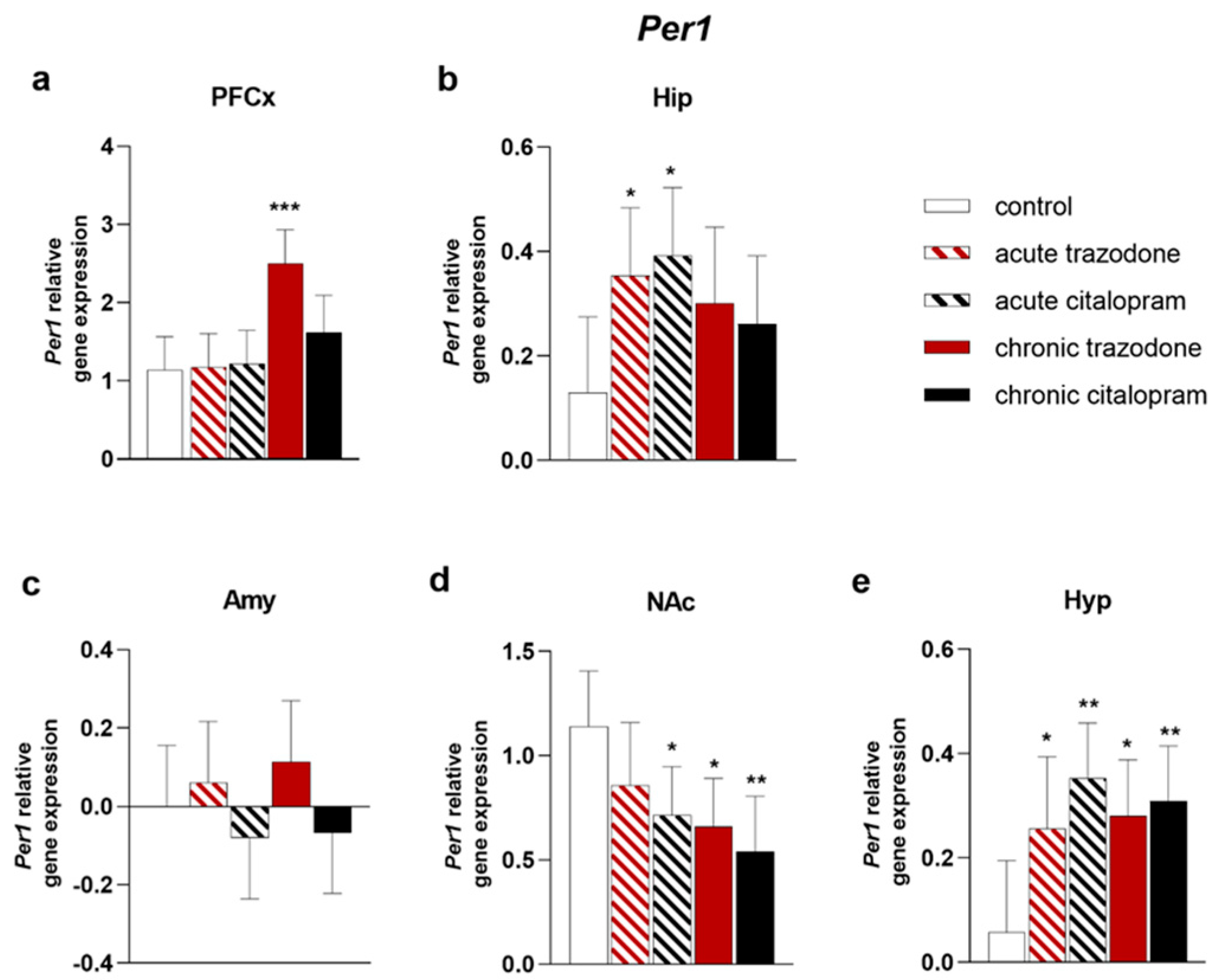
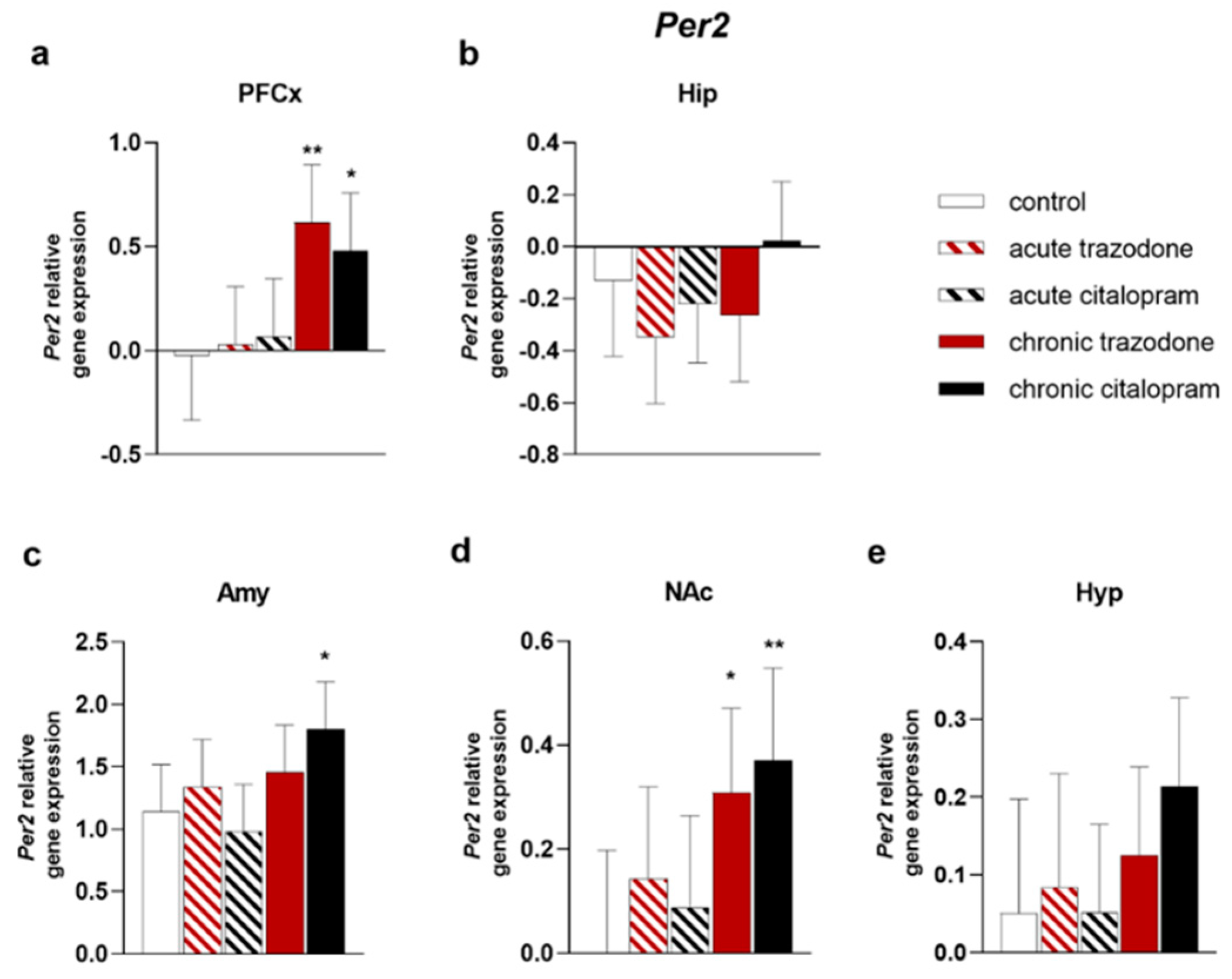
2.2. Circadian Rhythm Genes
2.3. Multivariate Analysis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Treatments
4.3. Gene Expression
4.4. Protein Levels
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stahl, S.M. Mechanism of action of trazodone: A multifunctional drug. CNS Spectr. 2009, 14, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Khouzam, H.R. A review of trazodone use in psychiatric and medical conditions. Postgrad. Med. 2017, 129, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, A.; Bianchetti, A.; Cagnin, A.; De Berardis, D.; Di Fazio, I.; Incalzi, R.A.; Marra, C.; Neviani, F.; Laurenzi, P.F.; Nicoletti, F. Trazodone: A multifunctional antidepressant. evaluation of its properties and real-world use. J. Gerontol. Geriatr. 2021, 69, 120–129. [Google Scholar] [CrossRef]
- Fagiolini, A.; Comandini, A.; Dell’Osso, M.C.; Kasper, S. Rediscovering trazodone for the treatment of major depressive disorder. CNS Drugs 2012, 26, 1033–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossini, L.; Coluccia, A.; Casolaro, I.; Benbow, J.; Amodeo, G.; De Giorgi, R.; Fagiolini, A. Off-Label Trazodone Prescription: Evidence, Benefits and Risks. Curr. Pharm. Des. 2015, 21, 3343–3351. [Google Scholar] [CrossRef]
- Goracci, A.; Forgione, R.N.; De Giorgi, R.; Coluccia, A.; Cuomo, A.; Fagiolini, A. Practical guidance for prescribing trazodone extended-release in major depression. Expert Opin. Pharmacother. 2016, 17, 433–441. [Google Scholar] [CrossRef]
- Opal, M.D.; Klenotich, S.C.; Morais, M.; Bessa, J.; Winkle, J.; Doukas, D.; Kay, L.J.; Sousa, N.; Dulawa, S.M. Serotonin 2C receptor antagonists induce fast-onset antidepressant effects. Mol. Psychiatry 2014, 19, 1106–1114. [Google Scholar] [CrossRef]
- Sheehan, D.V.; Croft, H.A.; Gossen, E.R.; Levitt, R.J.; Brullé, C.; Bouchard, S.; Rozova, A. Extended-release trazodone in major depressive disorder: A randomized, double-blind, placebo-controlled study. Psychiatry 2009, 6, 20–33. [Google Scholar]
- Bossini, L.; Casolaro, I.; Koukouna, D.; Cecchini, F.; Fagiolini, A. Off-label uses of trazodone: A review. Expert Opin. Pharmacother. 2012, 13, 1707–1717. [Google Scholar] [CrossRef]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Saletu-Zyhlarz, G.M.; Anderer, P.; Arnold, O.; Saletu, B. Confirmation of the neurophysiologically predicted therapeutic effects of trazodone on its target symptoms depression, anxiety and insomnia by postmarketing clinical studies with a controlled-release formulation in depressed outpatients. Neuropsychobiology 2003, 48, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lv, T.; Wu, J.; Lyu, Y. Trazodone changed the polysomnographic sleep architecture in insomnia disorder: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 14453. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, K.Y.; Chang, T.; Vanle, B.; Dang, J.; Steiner, A.J.; Loera, N.; Abdelmesseh, M.; Danovitch, I.; Ishak, W.W. Trazodone for Insomnia: A Systematic Review. Innov. Clin. Neurosci. 2017, 14, 24–34. [Google Scholar] [PubMed]
- Camargos, E.F.; Louzada, L.L.; Quintas, J.L.; Naves, J.O.S.; Louzada, F.M.; Nóbrega, O.T. Trazodone improves sleep parameters in Alzheimer disease patients: A randomized, double-blind, and placebo-controlled study. Am. J. Geriatr. Psychiatry 2014, 22, 1565–1574. [Google Scholar] [CrossRef]
- La, A.L.; Walsh, C.M.; Neylan, T.C.; Vossel, K.A.; Yaffe, K.; Krystal, A.D.; Miller, B.L.; Karageorgiou, E. Long-Term Trazodone Use and Cognition: A Potential Therapeutic Role for Slow-Wave Sleep Enhancers. J. Alzheimer’s Dis. 2019, 67, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, J.E.; Hur, K.; Kane, C.; Au, A.; Bishop, T.M.; Pigeon, W.R. Prescription Medications for the Treatment of Insomnia and Risk of Suicide Attempt: A Comparative Safety Study. J. Gen. Intern. Med. 2019, 34, 1554–1563. [Google Scholar] [CrossRef]
- Pyke, R.E. Trazodone in Sexual Medicine: Underused and Overdosed? Sex. Med. Rev. 2020, 8, 206–216. [Google Scholar] [CrossRef]
- Harmer, C.J.; Duman, R.S.; Cowen, P.J. How do antidepressants work? New perspectives for refining future treatment approaches. Lancet Psychiatry 2017, 4, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the pathophysiology and treatment of depression: Activity-dependent effects distinguish rapid-acting antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef]
- Castrén, E.; Kojima, M. Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol. Dis. 2017, 97, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Tardito, D.; Perez, J.; Tiraboschi, E.; Musazzi, L.; Racagni, G.; Popoli, M. Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: A critical overview. Pharmacol. Rev. 2006, 58, 115–134. [Google Scholar] [CrossRef]
- Carboni, L.; Nguyen, T.-P.; Caberlotto, L. Systems biology integration of proteomic data in rodent models of depression reveals involvement of the immune response and glutamatergic signaling. Proteom. Clin. Appl. 2016, 10, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Monteggia, L.M. A Neurotrophic Model for Stress-Related Mood Disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef]
- Caputi, F.F.; Rullo, L.; Stamatakos, S.; Candeletti, S.; Romualdi, P. Modulation of the Negative Affective Dimension of Pain: Focus on Selected Neuropeptidergic System Contributions. Int. J. Mol. Sci. 2019, 20, 4010. [Google Scholar] [CrossRef] [Green Version]
- Caputi, F.F.; Stopponi, S.; Rullo, L.; Palmisano, M.; Ubaldi, M.; Candeletti, S.; Ciccocioppo, R.; Romualdi, P. Dysregulation of Nociceptin/Orphanin FQ and Dynorphin Systems in the Extended Amygdala of Alcohol Preferring Marchigian Sardinian (msP) Rats. Int. J. Mol. Sci. 2021, 22, 2448. [Google Scholar] [CrossRef]
- Carboni, L.; Lattanzio, F.; Candeletti, S.; Porcellini, E.; Raschi, E.; Licastro, F.; Romualdi, P. Peripheral leukocyte expression of the potential biomarker proteins Bdnf, Sirt1, and Psen1 is not regulated by promoter methylation in Alzheimer’s disease patients. Neurosci. Lett. 2015, 605, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 2021, 90, 128–136. [Google Scholar] [CrossRef]
- Björkholm, C.; Monteggia, L.M. BDNF—A key transducer of antidepressant effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Aurbach, E.L.; Inui, E.G.; Turner, C.A.; Hagenauer, M.H.; Prater, K.E.; Li, J.Z.; Absher, D.; Shah, N.; Blandino, P.; Bunney, W.E.; et al. Fibroblast growth factor 9 is a novel modulator of negative affect. Proc. Natl. Acad. Sci. USA 2015, 112, 11953–11958. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.A.; Watson, S.J.; Akil, H. The Fibroblast Growth Factor Family: Neuromodulation of Affective Behavior. Neuron 2012, 76, 160–174. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.A.; Eren-Koçak, E.; Inui, E.G.; Watson, S.J.; Akil, H. Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Semin. Cell Dev. Biol. 2016, 53, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Tseng, P.; Chen, Y.; Tu, K.; Lin, P. Significantly higher peripheral fibroblast growth factor-2 levels in patients with major depressive disorder: A preliminary meta-analysis under MOOSE guidelines. Medicine 2016, 95, e4563. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Hisaoka-Nakashima, K.; Morioka, N.; Okada-Tsuchioka, M.; Kaneko, M.; Kasai, M.; Shibasaki, C.; Nakata, Y.; Takebayashi, M. Antidepressant acts on astrocytes leading to an increase in the expression of neurotrophic/growth factors: Differential regulation of FGF-2 by noradrenaline. PLoS ONE 2012, 7, e51197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallei, A.; Shi, B.; Mocchetti, I. Antidepressant treatments induce the expression of basic fibroblast growth factor in cortical and hippocampal neurons. Mol. Pharmacol. 2002, 61, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- Walker, W.H.; Walton, J.C.; DeVries, A.C.; Nelson, R.J. Circadian rhythm disruption and mental health. Transl. Psychiatry 2020, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Logan, R.W.; McClung, C.A. Rhythms of life: Circadian disruption and brain disorders across the lifespan. Nat. Rev. Neurosci. 2019, 20, 49–65. [Google Scholar] [CrossRef]
- McClung, C.A. How might circadian rhythms control mood? Let me count the ways. Biol. Psychiatry 2013, 74, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Bunney, B.; Mendoza-Viveros, L.; Bunney, W.; Borrelli, E.; Sassone-Corsi, P.; Orozco-Solis, R. Rapid-acting antidepressants and the circadian clock. Neuropsychopharmacology 2022, 47, 805–816. [Google Scholar] [CrossRef]
- Bunney, B.G.; Li, J.Z.; Walsh, D.M.; Stein, R.; Vawter, M.P.; Cartagena, P.; Barchas, J.D.; Schatzberg, A.F.; Myers, R.M.; Watson, S.J.; et al. Circadian dysregulation of clock genes: Clues to rapid treatments in major depressive disorder. Mol. Psychiatry 2015, 20, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Li, S.X.; Liu, L.J.; Xu, L.Z.; Gao, L.; Wang, X.F.; Zhang, J.T.; Lu, L. Diurnal alterations in circadian genes and peptides in major depressive disorder before and after escitalopram treatment. Psychoneuroendocrinology 2013, 38, 2789–2799. [Google Scholar] [CrossRef]
- Gray, J.D.; Milner, T.A.; McEwen, B.S. Dynamic plasticity: The role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience 2013, 239, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Benatti, C.; Capone, G.; Corsini, D.; Caggia, F.; Tascedda, F.; Mendlewicz, J.; Brunello, N. Time-dependent effects of escitalopram on brain derived neurotrophic factor (BDNF) and neuroplasticity related targets in the central nervous system of rats. Eur. J. Pharmacol. 2010, 643, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Burstein, O.; Franko, M.; Gale, E.; Handelsman, A.; Barak, S.; Motsan, S.; Shamir, A.; Toledano, R.; Simhon, O.; Hirshler, Y.; et al. Escitalopram and NHT normalized stress-induced anhedonia and molecular neuroadaptations in a mouse model of depression. PLoS ONE 2017, 12, e0188043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, J.P.R.; Mørk, A. The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain-derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5-HT and 5-HIAA levels. Brain Res. 2004, 1024, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Russo-Neustadt, A.A.; Alejandre, H.; Garcia, C.; Ivy, A.S.; Chen, M.J. Hippocampal brain-derived neurotrophic factor expression following treatment with reboxetine, citalopram, and physical exercise. Neuropsychopharmacology 2004, 29, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Saadati, N.; Bananej, M.; Khakpai, F.; Zarrindast, M.R.; Alibeik, H. Synergistic antidepressant effects of citalopram and SB-334867 in the REM sleep-deprived mice: Possible role of BDNF. Pharmacol. Biochem. Behav. 2022, 219, 173449. [Google Scholar] [CrossRef]
- Rogóż, Z.; Kamińska, K.; Pańczyszyn-Trzewik, P.; Sowa-Kućma, M. Repeated co-treatment with antidepressants and risperidone increases BDNF mRNA and protein levels in rats. Pharmacol. Rep. 2017, 69, 885–893. [Google Scholar] [CrossRef]
- Kozisek, M.E.; Middlemas, D.; Bylund, D.B. The differential regulation of BDNF and TrkB levels in juvenile rats after four days of escitalopram and desipramine treatment. Neuropharmacology 2008, 54, 251–257. [Google Scholar] [CrossRef]
- Seo, M.K.; Lee, J.G.; Park, S.W. Effects of escitalopram and ibuprofen on a depression-like phenotype induced by chronic stress in rats. Neurosci. Lett. 2019, 696, 168–173. [Google Scholar] [CrossRef]
- Arunrut, T.; Alejandre, H.; Chen, M.; Cha, J.; Russo-Neustadt, A. Differential behavioral and neurochemical effects of exercise, reboxetine and citalopram with the forced swim test. Life Sci. 2009, 84, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Dionisie, V.; Ciobanu, A.M.; Toma, V.A.; Manea, M.C.; Baldea, I.; Olteanu, D.; Sevastre-Berghian, A.; Clichici, S.; Manea, M.; Riga, S.; et al. Escitalopram targets oxidative stress, caspase-3, bdnf and mecp2 in the hippocampus and frontal cortex of a rat model of depression induced by chronic unpredictable mild stress. Int. J. Mol. Sci. 2021, 22, 7483. [Google Scholar] [CrossRef]
- Gong, W.G.; Wang, Y.J.; Zhou, H.; Li, X.L.; Bai, F.; Ren, Q.G.; Zhang, Z.J. Citalopram Ameliorates Synaptic Plasticity Deficits in Different Cognition-Associated Brain Regions Induced by Social Isolation in Middle-Aged Rats. Mol. Neurobiol. 2017, 54, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Doron, R.; Lotan, D.; Versano, Z.; Benatav, L.; Franko, M.; Armoza, S.; Kately, N.; Rehavi, M. Escitalopram or novel herbal mixture treatments during or following exposure to stress reduce anxiety-like behavior through corticosterone and BDNF modifications. PLoS ONE 2014, 9, e91455. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.D.; Grover, L.M.; Spangler, P.R. Venlafaxine treatment stimulates expression of brain-derived neurotrophic factor protein in frontal cortex and inhibits long-term potentiation in hippocampus. Neuroscience 2009, 162, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Da Pozzo, E.; Zappelli, E.; Martini, C. Trazodone treatment protects neuronal-like cells from inflammatory insult by inhibiting NF-κB, p38 and JNK. Cell. Signal. 2015, 27, 1609–1629. [Google Scholar] [CrossRef] [PubMed]
- Rantamäki, T.; Hendolin, P.; Kankaanpää, A.; Mijatovic, J.; Piepponen, P.; Domenici, E.; Chao, M.V.; Männistö, P.T.; Castrén, E. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cγ signaling pathways in mouse brain. Neuropsychopharmacology 2007, 32, 2152–2162. [Google Scholar] [CrossRef] [Green Version]
- Rantamäki, T. TrkB neurotrophin receptor at the core of antidepressant effects, but how? Cell Tissue Res. 2019, 377, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e19. [Google Scholar] [CrossRef]
- Kusmider, M.; Faron-Górecka, A.; Solich, J.; Pabian, P.; Dziedzicka-Wasylewska, M. Time-course of changes in key catecholaminergic receptors and trophic systems in rat brain after antidepressant administration. Neurochem. Int. 2020, 141, 104885. [Google Scholar] [CrossRef]
- Xu, Y.; Wei, H.; Zhu, Y.; Zhu, Y.; Zhang, N.; Qin, J.; Zhu, X.; Yu, M.; Li, Y. Potential serum biomarkers for the prediction of the efficacy of escitalopram for treating depression. J. Affect. Disord. 2019, 250, 307–312. [Google Scholar] [CrossRef]
- Bachis, A.; Mallei, A.; Cruz, M.I.; Wellstein, A.; Mocchetti, I. Chronic antidepressant treatments increase basic fibroblast growth factor and fibroblast growth factor-binding protein in neurons. Neuropharmacology 2008, 55, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Molteni, R.; Gabriel, C.; Mocaer, E.; Racagni, G.; Riva, M.A. Modulation of neuroplastic molecules in selected brain regions after chronic administration of the novel antidepressant agomelatine. Psychopharmacology 2011, 215, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, X.X.; Chen, X.; Qin, X.Y.; Kardami, E.; Cheng, Y. Antidepressant-Like Effects of Low- And High-Molecular Weight FGF-2 on Chronic Unpredictable Mild Stress Mice. Front. Mol. Neurosci. 2018, 11, 377. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, M.; Banasr, M.; Duric, V.; Fournier, N.M.; Licznerski, P.; Duman, R.S. Antidepressant Effects of Fibroblast Growth Factor-2 in Behavioral and Cellular Models of Depression. Biol. Psychiatry 2012, 72, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlezon, W.A.; Duman, R.S.; Nestler, E.J. The many faces of CREB. Trends Neurosci. 2005, 28, 436–445. [Google Scholar] [CrossRef]
- Malberg, J.E.; Blendy, J.A. Antidepressant action: To the nucleus and beyond. Trends Pharmacol. Sci. 2005, 26, 631–638. [Google Scholar] [CrossRef]
- Satyanarayanan, S.K.; Chien, Y.C.; Chang, J.P.C.; Huang, S.Y.; Guu, T.W.; Su, H.; Su, K.P. Melatonergic agonist regulates circadian clock genes and peripheral inflammatory and neuroplasticity markers in patients with depression and anxiety. Brain. Behav. Immun. 2020, 85, 142–151. [Google Scholar] [CrossRef]
- Christiansen, S.L.; Bouzinova, E.V.; Fahrenkrug, J.; Wiborg, O. Altered expression pattern of clock genes in a rat model of depression. Int. J. Neuropsychopharmacol. 2016, 19, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Erburu, M.; Cajaleon, L.; Guruceaga, E.; Venzala, E.; Muñoz-Cobo, I.; Beltrán, E.; Puerta, E.; Tordera, R.M. Chronic mild stress and imipramine treatment elicit opposite changes in behavior and in gene expression in the mouse prefrontal cortex. Pharmacol. Biochem. Behav. 2015, 135, 227–236. [Google Scholar] [CrossRef]
- Savalli, G.; Diao, W.; Schulz, S.; Todtova, K.; Pollak, D.D. Diurnal oscillation of Amygdala clock gene expression and loss of synchrony in a mouse model of depression. Int. J. Neuropsychopharmacol. 2015, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nagy, A.D.; Iwamoto, A.; Kawai, M.; Goda, R.; Matsuo, H.; Otsuka, T.; Nagasawa, M.; Furuse, M.; Yasuo, S. Melatonin adjusts the expression pattern of clock genes in the suprachiasmatic nucleus and induces antidepressant-like effect in a mouse model of seasonal affective disorder. Chronobiol. Int. 2015, 32, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Moriya, S.; Tahara, Y.; Sasaki, H.; Hamaguchi, Y.; Kuriki, D.; Ishikawa, R.; Ishigooka, J.; Shibata, S. Effect of quetiapine on Per1, Per2, and Bmal1 clock gene expression in the mouse amygdala and hippocampus. J. Pharmacol. Sci. 2014, 125, 329–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grippe, T.C.; Gonçalves, B.S.B.; Louzada, L.L.; Quintas, J.L.; Naves, J.O.S.; Camargos, E.F.; Nóbrega, O.T. Circadian rhythm in Alzheimer disease after trazodone use. Chronobiol. Int. 2015, 32, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Du Sert, N.P.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting Animal Research: Explanation and Elaboration for the Arrive Guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef]
- FDA Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers (accessed on 1 March 2018).
- Carboni, L.; Pischedda, F.; Piccoli, G.; Lauria, M.; Musazzi, L.; Popoli, M.; Mathé, A.A.; Domenici, E. Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment. Cells 2020, 9, 1818. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bate, S.T.; Clark, R.A. The Design and Statistical Analysis of Animal Experiments; Cambridge University Press: Cambridge, UK, 2014; ISBN 978-1-107-69094-3. [Google Scholar]
- Clark, R.A.; Shoaib, M.; Hewitt, K.N.; Stanford, S.C.; Bate, S.T. A comparison of InVivoStat with other statistical software packages for analysis of data generated from animal experiments. J. Psychopharmacol. 2012, 26, 1136–1142. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer | Tm | Product Size |
|---|---|---|---|---|
| Bmal1 | TGCCACTGACTACCAAGAAAGT | TGTCCCGACGCCTCTTTTCA | 59.56 61.75 | 134 |
| Bdnf | AAGTCTGCATTACATTCCTCGA | GTTTTCTGAAAGAGGGACAGTTTAT | 57.53 57.49 | 138 [25] |
| Creb | CAGACAACCAGCAGAGTGGA | CTGGACTGTCTGCCCATTG | 59.61 58.14 | 176 |
| Fgf-2 | GCGAACCGGTACCTGGCTAT | GCCCAGTTCGTTTCAGTGCC | 62.02 62.13 | 160 |
| Fgfr2 | CCGGCCCTCCTTCAGTTTAG | TTCAACATGCAGCGCAACTC | 60.11 60.04 | 132 |
| Gapdh | AGACAGCCGCATCTTCTTGT | CTTGCCGTGGGTAGAGTCAT | 59.68 59.46 | 207 [25] |
| Per1 | GCGTTGCAAACGGGATGT | GCAGGCGAGATGGTGTAGTAGA | 59.36 61.59 | 101 |
| Per2 | TGACGGGTCGAGCAAAGGAC | CCACGTCTTCCTGGAGCACA | 62.43 62.10 | 163 |
| TrkB | AAGTTCTACGGTGTCTGTGTG | TTCTCTCCTACCAAGCAGTTC | 57.91 57.04 | 257 [25] |
| Antibody | Dilution | Cat. Number | RRID | Reactivity | Manufacturer |
|---|---|---|---|---|---|
| anti-phospho CREB Ser 133 | 1:1000 | 06-519 | AB_310153 | mouse, rat, hamster, human | Millipore, Burlington, MA, USA |
| anti-CREB | 1:1000 | 9197 | AB_331277 | human, mouse, rat, monkey, D. melanogaster | Cell Signaling, Danvers, MA, USA |
| anti-Gapdh | 1:1000 | MA5-15738 | AB_10977387 | bacteria, canine, chicken, hamster, human, insect, mouse, rabbit, rat, yeast | Thermo Fisher Scientific, Waltham, MA, USA |
| Donkey Anti-rabbit IgG | 1:3000 | NA934 | AB_772206 | rabbit | GE Healthcare, Chicago, IL, USA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carboni, L.; Rullo, L.; Caputi, F.F.; Stamatakos, S.; Candeletti, S.; Romualdi, P. Chronic Trazodone and Citalopram Treatments Increase Trophic Factor and Circadian Rhythm Gene Expression in Rat Brain Regions Relevant for Antidepressant Efficacy. Int. J. Mol. Sci. 2022, 23, 14041. https://doi.org/10.3390/ijms232214041
Carboni L, Rullo L, Caputi FF, Stamatakos S, Candeletti S, Romualdi P. Chronic Trazodone and Citalopram Treatments Increase Trophic Factor and Circadian Rhythm Gene Expression in Rat Brain Regions Relevant for Antidepressant Efficacy. International Journal of Molecular Sciences. 2022; 23(22):14041. https://doi.org/10.3390/ijms232214041
Chicago/Turabian StyleCarboni, Lucia, Laura Rullo, Francesca Felicia Caputi, Serena Stamatakos, Sanzio Candeletti, and Patrizia Romualdi. 2022. "Chronic Trazodone and Citalopram Treatments Increase Trophic Factor and Circadian Rhythm Gene Expression in Rat Brain Regions Relevant for Antidepressant Efficacy" International Journal of Molecular Sciences 23, no. 22: 14041. https://doi.org/10.3390/ijms232214041