
Acid Sphingomyelinase Inhibitor, Imipramine, Reduces Hippocampal Neuronal Death after Traumatic Brain Injury
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Imipramine Reduces Acid Sphingomyelinase and Ceramide Overexpression after Traumatic Brain Injury
2.2. Imipramine Reduces TBI-Induced Hippocampal Neuron Death
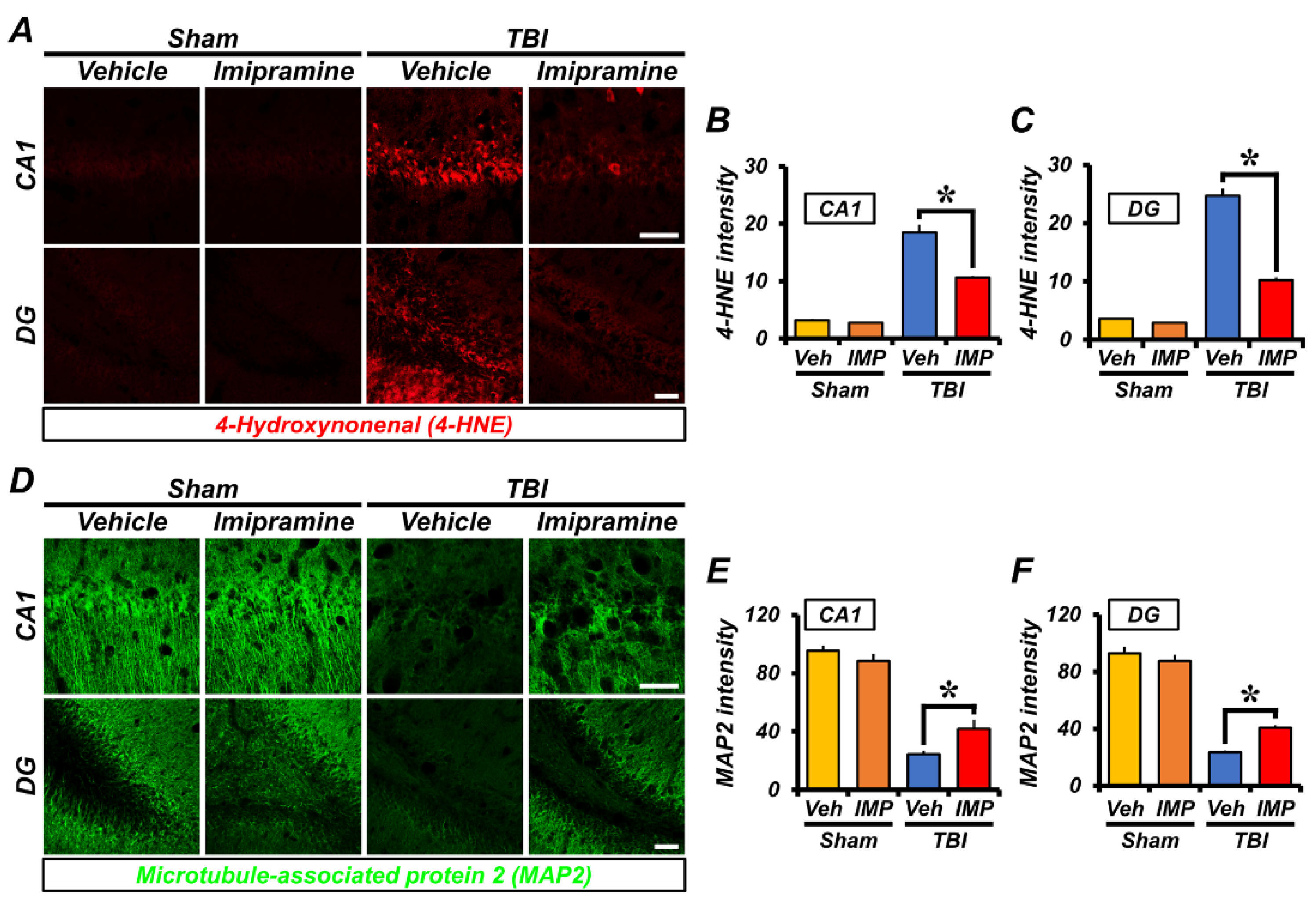
2.3. Imipramine Reduces Oxidative Damage and Dendritic Loss after TBI
2.4. Imipramine Reduces TBI-Induced Astrocyte and Microglia Activation
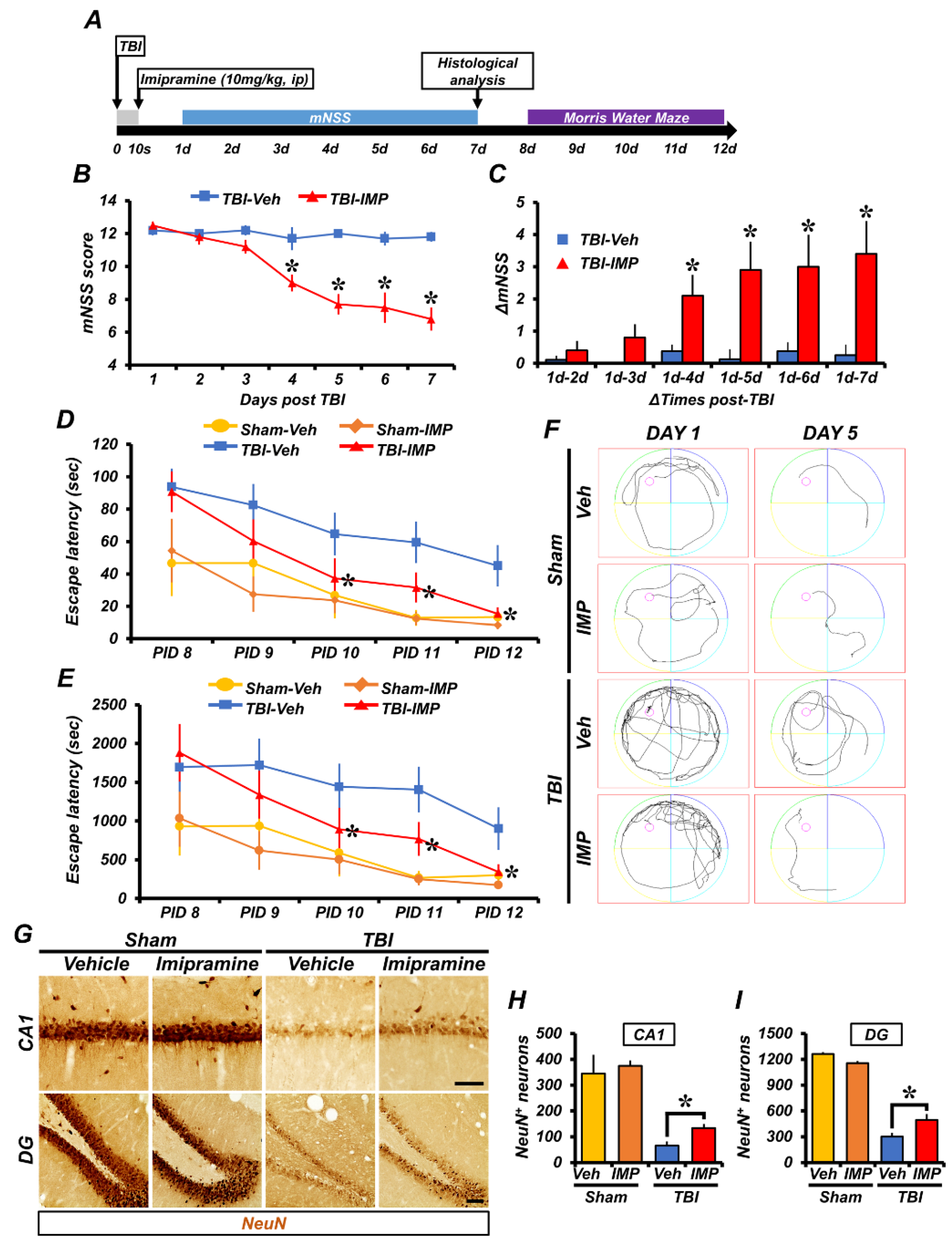
2.5. Imipramine Reduced TBI-Induced Neuronal Death and Cognitive Impairment
3. Discussion
4. Materials and Methods
4.1. Ethics Statement and Experimental Animals
4.2. Traumatic BI Surgery
4.3. Brain Sample Preparation
4.4. Evaluation of Hippocampal Neuronal Death
4.5. Evaluation of ASMase and Ceramide
4.6. Immunofluorescence Assay
4.7. Detection of Live Neurons
4.8. Behavior Test
4.9. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghajar, J. Traumatic brain injury. Lancet 2000, 356, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Najem, D.; Rennie, K.; Ribecco-Lutkiewicz, M.; Ly, D.; Haukenfrers, J.; Liu, Q.; Nzau, M.; Fraser, D.D.; Bani-Yaghoub, M. Traumatic brain injury: Classification, models, and markers. Biochem. Cell. Biol. 2018, 96, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.J. Pathophysiology of Traumatic Brain Injury. Phys. Med. Rehabil. Clin. N. Am. 2017, 28, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, Y.; Hanafy, K.A. Cell Death and Recovery in Traumatic Brain Injury. Neurotherapeutics 2020, 17, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Vella, M.A.; Crandall, M.L.; Patel, M.B. Acute Management of Traumatic Brain Injury. Surg. Clin. N. Am. 2017, 97, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Venigalla, H.; Mekala, H.M.; Dar, S.; Hassan, M.; Ayub, S. Traumatic Brain Injury and Neuropsychiatric Complications. Indian J. Psychol. Med. 2017, 39, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Van Echten-Deckert, G.; Herget, T. Sphingolipid metabolism in neural cells. Biochim. Biophys. Acta 2006, 1758, 1978–1994. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Chatterjee, S. Effects of gentamicin on sphingomyelinase activity in cultured human renal proximal tubular cells. J. Biol. Chem. 1987, 262, 12550–12556. [Google Scholar] [CrossRef]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010, 584, 1728–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnick, R. The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J. Clin. Investig. 2002, 110, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Wasserstein, M.P. Types A and B Niemann-Pick disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Luberto, C.; Argraves, K.M. Enzymes of sphingolipid metabolism: From modular to integrative signaling. Biochemistry 2001, 40, 4893–4903. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, W.J.; van der Luit, A.H.; Veldman, R.J.; Verheij, M.; Borst, J. Ceramide: Second messenger or modulator of membrane structure and dynamics? Biochem. J. 2003, 369, 199–211. [Google Scholar] [CrossRef]
- Ketteler, J.; Wittka, A.; Leonetti, D.; Roy, V.V.; Estephan, H.; Maier, P.; Reis, H.; Herskind, C.; Jendrossek, V.; Paris, F.; et al. Caveolin-1 regulates the ASMase/ceramide-mediated radiation response of endothelial cells in the context of tumor-stroma interactions. Cell. Death Dis. 2020, 11, 228. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.W.; Hojabrpour, P.; Zhang, P.; Kolesnick, R.N.; Steinbrecher, U.P.; Gomez-Munoz, A.; Duronio, V. Regulation of ceramide generation during macrophage apoptosis by ASMase and de novo synthesis. Biochim. Biophys. Acta 2015, 1851, 1482–1489. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, J. Sphingosine and ceramide signalling in apoptosis. IUBMB Life 2006, 58, 462–466. [Google Scholar] [CrossRef]
- Siskind, L.J. Mitochondrial ceramide and the induction of apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Albeituni, S.; Stiban, J. Roles of Ceramides and Other Sphingolipids in Immune Cell Function and Inflammation. Adv. Exp. Med. Biol. 2019, 1161, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.D.; Obeid, L.M. Ceramide and apoptosis: Exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer. Agents Med. Chem. 2012, 12, 340–363. [Google Scholar] [CrossRef] [PubMed]
- Truman, J.P.; Al Gadban, M.M.; Smith, K.J.; Hammad, S.M. Acid sphingomyelinase in macrophage biology. Cell. Mol. Life Sci. 2011, 68, 3293–3305. [Google Scholar] [CrossRef] [Green Version]
- Kolesnick, R.N.; Kronke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Moyano, A.L.; Li, G.; Lopez-Rosas, A.; Mansson, J.E.; van Breemen, R.B.; Givogri, M.I. Distribution of C16:0, C18:0, C24:1, and C24:0 sulfatides in central nervous system lipid rafts by quantitative ultra-high-pressure liquid chromatography tandem mass spectrometry. Anal. Biochem. 2014, 467, 31–39. [Google Scholar] [CrossRef]
- Racagni, G.; Popoli, M. The pharmacological properties of antidepressants. Int. Clin. Psychopharmacol. 2010, 25, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Gillman, P.K. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br. J. Pharmacol. 2007, 151, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Kolzer, M.; Werth, N.; Sandhoff, K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 2004, 559, 96–98. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, J.; Tripal, P.; Gulbins, E.; Muehlbacher, M. Functional inhibitors of acid sphingomyelinase (FIASMAs). Handb. Exp. Pharmacol. 2013, 215, 169–186. [Google Scholar] [CrossRef]
- Li, Y.; Lu, Z.; Zhang, L.; Kirkwood, C.L.; Kirkwood, K.L.; Lopes-Virella, M.F.; Huang, Y. Inhibition of acid sphingomyelinase by imipramine abolishes the synergy between metabolic syndrome and periodontitis on alveolar bone loss. J. Periodontal. Res. 2022, 57, 173–185. [Google Scholar] [CrossRef]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Muhle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): A novel pharmacological group of drugs with broad clinical applications. Cell. Physiol. Biochem. 2010, 26, 9–20. [Google Scholar] [CrossRef]
- Hwang, J.; Zheng, L.T.; Ock, J.; Lee, M.G.; Kim, S.H.; Lee, H.W.; Lee, W.H.; Park, H.C.; Suk, K. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology 2008, 55, 826–834. [Google Scholar] [CrossRef]
- Schiavon, A.P.; Milani, H.; Romanini, C.V.; Foresti, M.L.; Castro, O.W.; Garcia-Cairasco, N.; de Oliveira, R.M. Imipramine enhances cell proliferation and decreases neurodegeneration in the hippocampus after transient global cerebral ischemia in rats. Neurosci. Lett. 2010, 470, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Liangpunsakul, S.; Rahmini, Y.; Ross, R.A.; Zhao, Z.; Xu, Y.; Crabb, D.W. Imipramine blocks ethanol-induced ASMase activation, ceramide generation, and PP2A activation, and ameliorates hepatic steatosis in ethanol-fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G515–G523. [Google Scholar] [CrossRef] [Green Version]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Kang, B.S.; Lee, S.H.; Suh, S.W. Administration of an Acidic Sphingomyelinase (ASMase) Inhibitor, Imipramine, Reduces Hypoglycemia-Induced Hippocampal Neuronal Death. Cells 2022, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, Y.; Mahmood, A.; Chopp, M. Investigational agents for treatment of traumatic brain injury. Expert Opin. Investig. Drugs 2015, 24, 743–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyano, A.L.; Pituch, K.; Li, G.; van Breemen, R.; Mansson, J.E.; Givogri, M.I. Levels of plasma sulfatides C18:0 and C24:1 correlate with disease status in relapsing-remitting multiple sclerosis. J. Neurochem. 2013, 127, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Jamjoom, A.A.B.; Rhodes, J.; Andrews, P.J.D.; Grant, S.G.N. The synapse in traumatic brain injury. Brain 2021, 144, 18–31. [Google Scholar] [CrossRef]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflamm. 2016, 13, 264. [Google Scholar] [CrossRef] [Green Version]
- Park, M.K.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, D.H.; Kang, B.S.; Suh, S.W. Effects of Transient Receptor Potential Cation 5 (TRPC5) Inhibitor, NU6027, on Hippocampal Neuronal Death after Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 8256. [Google Scholar] [CrossRef]
- Choi, S.; Hong, D.K.; Choi, B.Y.; Suh, S.W. Zinc in the Brain: Friend or Foe? Int. J. Mol. Sci. 2020, 21, 8941. [Google Scholar] [CrossRef] [PubMed]
- Schissel, S.L.; Keesler, G.A.; Schuchman, E.H.; Williams, K.J.; Tabas, I. The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J. Biol. Chem. 1998, 273, 18250–18259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, S.A.; Voltin, J.R.; Wang, W.; Tomlinson, S.; Riley, C.L.; Gudz, T.I. Acid sphingomyelinase deficiency protects mitochondria and improves function recovery after brain injury. J. Lipid Res. 2019, 60, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Roux, A.; Muller, L.; Jackson, S.N.; Post, J.; Baldwin, K.; Hoffer, B.; Balaban, C.D.; Barbacci, D.; Schultz, J.A.; Gouty, S.; et al. Mass spectrometry imaging of rat brain lipid profile changes over time following traumatic brain injury. J. Neurosci. Methods 2016, 272, 19–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, N.T.; Shumilina, E.; Schmid, E.; Bhavsar, S.K.; Rexhepaj, R.; Gotz, F.; Gulbins, E.; Lang, F. Role of acidic sphingomyelinase in thymol-mediated dendritic cell death. Mol. Nutr. Food Res. 2010, 54, 1833–1841. [Google Scholar] [CrossRef]
- Rhein, C.; Zoicas, I.; Marx, L.M.; Zeitler, S.; Hepp, T.; von Zimmermann, C.; Muhle, C.; Richter-Schmidinger, T.; Lenz, B.; Erim, Y.; et al. mRNA Expression of SMPD1 Encoding Acid Sphingomyelinase Decreases upon Antidepressant Treatment. Int. J. Mol. Sci. 2021, 22, 5700. [Google Scholar] [CrossRef]
- Calvi, A.; Fischetti, I.; Verzicco, I.; Belvederi Murri, M.; Zanetidou, S.; Volpi, R.; Coghi, P.; Tedeschi, S.; Amore, M.; Cabassi, A. Antidepressant Drugs Effects on Blood Pressure. Front. Cardiovasc. Med. 2021, 8, 704281. [Google Scholar] [CrossRef]
- Mochizucki, D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum. Psychopharmacol. 2004, 19 (Suppl. S1), S15–S19. [Google Scholar] [CrossRef]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell. Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Czarnota, G.J. Involvement of Ceramide Signalling in Radiation-Induced Tumour Vascular Effects and Vascular-Targeted Therapy. Int. J. Mol. Sci. 2022, 23, 6671. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Acid Sphingomyelinase, a Lysosomal and Secretory Phospholipase C, Is Key for Cellular Phospholipid Catabolism. Int. J. Mol. Sci. 2021, 22, 9001. [Google Scholar] [CrossRef]
- Mizrachi, A.; Ben-Aharon, I.; Li, H.; Bar-Joseph, H.; Bodden, C.; Hikri, E.; Popovtzer, A.; Shalgi, R.; Haimovitz-Friedman, A. Chemotherapy-induced acute vascular injury involves intracellular generation of ROS via activation of the acid sphingomyelinase pathway. Cell. Signal. 2021, 82, 109969. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Lee, S.H.; Choi, H.C.; Lee, S.K.; Yoon, H.S.; Park, J.B.; Chung, W.S.; Suh, S.W. Alcohol dependence treating agent, acamprosate, prevents traumatic brain injury-induced neuron death through vesicular zinc depletion. Transl. Res. 2019, 207, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Andrieu-Abadie, N.; Gouaze, V.; Salvayre, R.; Levade, T. Ceramide in apoptosis signaling: Relationship with oxidative stress. Free Radic. Biol. Med. 2001, 31, 717–728. [Google Scholar] [CrossRef]
- He, X.; Schuchman, E.H. Ceramide and Ischemia/Reperfusion Injury. J. Lipids 2018, 2018, 3646725. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, S.; Mukherjee, A.; Sepulveda, S.; Becerra-Calixto, A.; Bravo-Vasquez, N.; Gherardelli, C.; Chavez, M.; Soto, C. Modeling Traumatic Brain Injury in Human Cerebral Organoids. Cells 2021, 10, 2683. [Google Scholar] [CrossRef]
- Inaba, T.; Murate, M.; Tomishige, N.; Lee, Y.F.; Hullin-Matsuda, F.; Pollet, B.; Humbert, N.; Mely, Y.; Sako, Y.; Greimel, P.; et al. Formation of tubules and helical ribbons by ceramide phosphoethanolamine-containing membranes. Sci. Rep. 2019, 9, 5812. [Google Scholar] [CrossRef] [Green Version]
- Hirata, H.; Hibasami, H.; Yoshida, T.; Ogawa, M.; Matsumoto, M.; Morita, A.; Uchida, A. Nerve growth factor signaling of p75 induces differentiation and ceramide-mediated apoptosis in Schwann cells cultured from degenerating nerves. Glia 2001, 36, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Barbacci, D.C.; Roux, A.; Muller, L.; Jackson, S.N.; Post, J.; Baldwin, K.; Hoffer, B.; Balaban, C.D.; Schultz, J.A.; Gouty, S.; et al. Mass Spectrometric Imaging of Ceramide Biomarkers Tracks Therapeutic Response in Traumatic Brain Injury. ACS Chem. Neurosci. 2017, 8, 2266–2274. [Google Scholar] [CrossRef]
- Li, C.; Guo, S.; Pang, W.; Zhao, Z. Crosstalk Between Acid Sphingomyelinase and Inflammasome Signaling and Their Emerging Roles in Tissue Injury and Fibrosis. Front Cell. Dev. Biol. 2019, 7, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualtierotti, R.; Guarnaccia, L.; Beretta, M.; Navone, S.E.; Campanella, R.; Riboni, L.; Rampini, P.; Marfia, G. Modulation of Neuroinflammation in the Central Nervous System: Role of Chemokines and Sphingolipids. Adv. Ther. 2017, 34, 396–420. [Google Scholar] [CrossRef] [PubMed]
- Raghupathi, R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004, 14, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.M.; Keyser, R.E.; Dsurney, J.; Chan, L. Improved cognitive performance following aerobic exercise training in people with traumatic brain injury. Arch. Phys. Med. Rehabil. 2015, 96, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Reus, G.Z.; Stringari, R.B.; Ribeiro, K.F.; Ferraro, A.K.; Vitto, M.F.; Cesconetto, P.; Souza, C.T.; Quevedo, J. Ketamine plus imipramine treatment induces antidepressant-like behavior and increases CREB and BDNF protein levels and PKA and PKC phosphorylation in rat brain. Behav. Brain Res. 2011, 221, 166–171. [Google Scholar] [CrossRef]
- Wainwright, S.R.; Workman, J.L.; Tehrani, A.; Hamson, D.K.; Chow, C.; Lieblich, S.E.; Galea, L.A. Testosterone has antidepressant-like efficacy and facilitates imipramine-induced neuroplasticity in male rats exposed to chronic unpredictable stress. Horm. Behav. 2016, 79, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Glombik, K.; Slusarczyk, J.; Trojan, E.; Chamera, K.; Budziszewska, B.; Lason, W.; Basta-Kaim, A. Regulation of insulin receptor phosphorylation in the brains of prenatally stressed rats: New insight into the benefits of antidepressant drug treatment. Eur. Neuropsychopharmacol. 2017, 27, 120–131. [Google Scholar] [CrossRef] [PubMed]
- El Kaffas, A.; Al-Mahrouki, A.; Hashim, A.; Law, N.; Giles, A.; Czarnota, G.J. Role of Acid Sphingomyelinase and Ceramide in Mechano-Acoustic Enhancement of Tumor Radiation Responses. J. Natl. Cancer Inst. 2018, 110, 1009–1018. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Kang, B.S.; Park, M.K.; Lee, C.J.; Yang, H.W.; Woo, S.Y.; Park, S.W.; et al. Acid Sphingomyelinase Inhibitor, Imipramine, Reduces Hippocampal Neuronal Death after Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 14749. https://doi.org/10.3390/ijms232314749
Lee SH, Kho AR, Lee SH, Hong DK, Kang BS, Park MK, Lee CJ, Yang HW, Woo SY, Park SW, et al. Acid Sphingomyelinase Inhibitor, Imipramine, Reduces Hippocampal Neuronal Death after Traumatic Brain Injury. International Journal of Molecular Sciences. 2022; 23(23):14749. https://doi.org/10.3390/ijms232314749
Chicago/Turabian StyleLee, Si Hyun, A Ra Kho, Song Hee Lee, Dae Ki Hong, Beom Seok Kang, Min Kyu Park, Chang Juhn Lee, Hyun Wook Yang, Seo Young Woo, Se Wan Park, and et al. 2022. "Acid Sphingomyelinase Inhibitor, Imipramine, Reduces Hippocampal Neuronal Death after Traumatic Brain Injury" International Journal of Molecular Sciences 23, no. 23: 14749. https://doi.org/10.3390/ijms232314749