
Inflammaging: Implications in Sarcopenia
 , , , and
, , , and {kind=link}
Abstract
:1. Introduction
2. Aging Factors in Skeletal Muscle (When You Are Old and Gray)
3. Decline of Muscle Stem Cells (Running out the Gold Mine of Skeletal Muscle Regeneration)
4. Impairment of Damage Sensors and Increase in Alarm Signaling (When the Enemy Is Within)
5. Cytokines/Myokines (Friends or Foes)
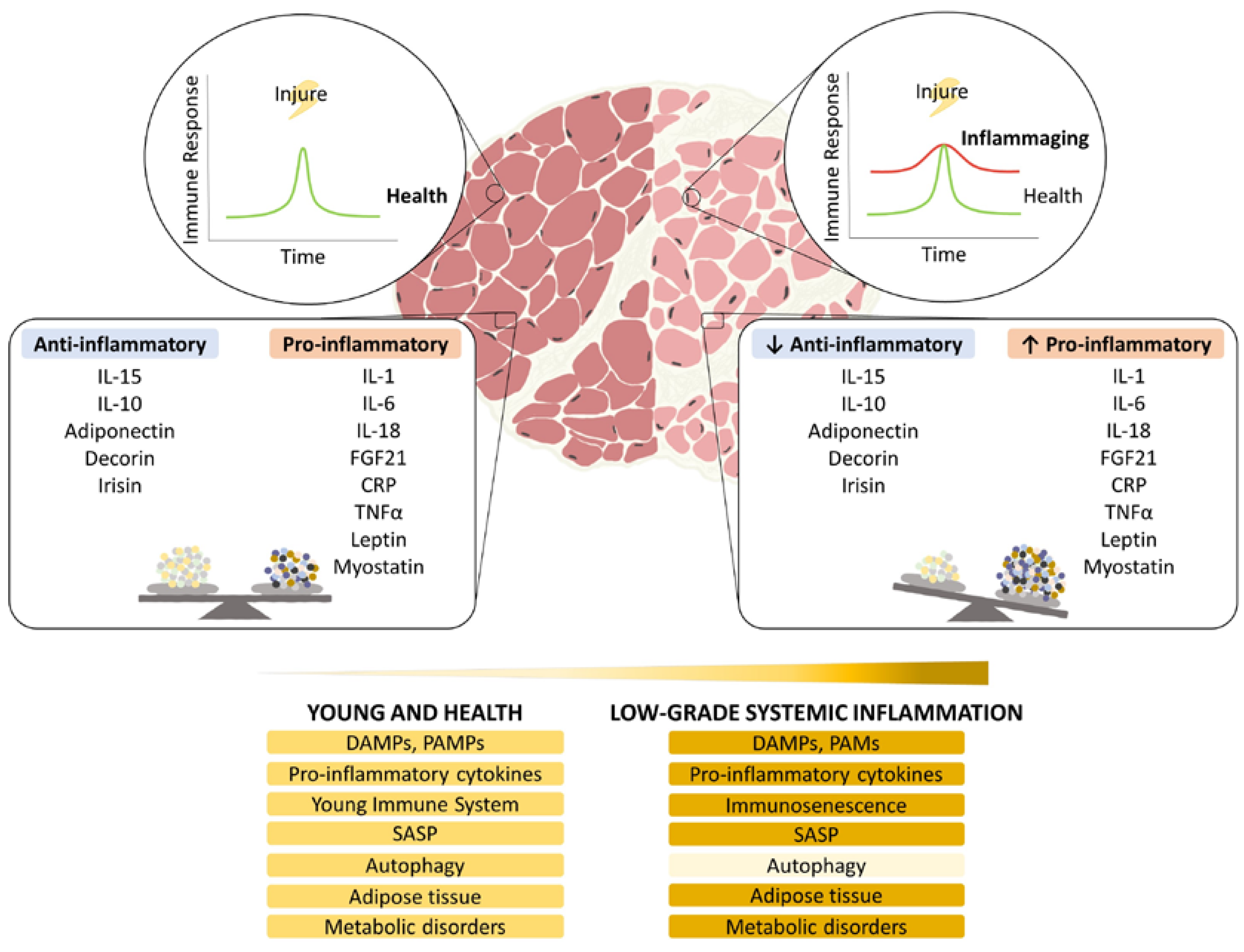
6. Immunosenescence (I Guess I Must Be Getting Old)
7. Gut and Metabolic Disorders: Metabolic Syndrome (Toxic Sugar)
8. Adipose Tissue Implications (Toxic Energy)
9. Autophagy and Oxidative Stress Implications (Destroy Everything You Touch)
10. Interventions
10.1. Senolytics (Eliminate the Wrong)
10.2. Melatonin (The Many Roads of Melatonin: Anti-Inflammatory Effect)
11. Conclusions (Aging Is the Only Way of Living)
12. Future Directions (Save Myself)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crimmins, E.M. Lifespan and Healthspan: Past, Present, and Promise. Gerontologist 2015, 55, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.C. Living too long: The current focus of medical research on increasing the quantity, rather than the quality, of life is damaging our health and harming the economy. EMBO Rep. 2015, 16, 137–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. World Report on Ageing and Health; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Rudnicka, E.; Napierała, P.; Podfigurna, A.; Męczekalski, B.; Smolarczyk, R.; Grymowicz, M. The World Health Organization (WHO) approach to healthy ageing. Maturitas 2020, 139, 6–11. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Q.L. The frailty syndrome: Definition and natural history. Clin. Geriatr. Med. 2011, 27, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fried, L.P.; Tangen, C.M.; Walston, J.; Newman, A.B.; Hirsch, C.; Gottdiener, J.; Seeman, T.; Tracy, R.; Kop, W.J.; Burke, G.; et al. Frailty in older adults: Evidence for a phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, M146–M156. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [Green Version]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Malmstrom, T.K.; Miller, D.K.; Simonsick, E.M.; Ferrucci, L.; Morley, J.E. SARC-F: A symptom score to predict persons with sarcopenia at risk for poor functional outcomes. J. Cachexia Sarcopenia Muscle 2016, 7, 28–36. [Google Scholar] [CrossRef]
- Bertschi, D.; Kiss, C.M.; Beerli, N.; Mauthner, O.; Kressig, R.W. Impact of sarcopenia on daily functioning: A cross-sectional study among older inpatients. Aging Clin. Exp. Res. 2022, 34, 2041–2046. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Wakabayashi, H.; Bise, T.; Nagano, F.; Shimazu, S.; Shiraishi, A.; Yamaga, M.; Koga, H. Sarcopenia is associated with worse recovery of physical function and dysphagia and a lower rate of home discharge in Japanese hospitalized adults undergoing convalescent rehabilitation. Nutrition 2019, 61, 111–118. [Google Scholar] [CrossRef]
- Lang, T.; Streeper, T.; Cawthon, P.; Baldwin, K.; Taaffe, D.R.; Harris, T.B. Sarcopenia: Etiology, clinical consequences, intervention, and assessment. Osteoporos. Int. 2010, 21, 543–559. [Google Scholar] [CrossRef] [Green Version]
- Rezuş, E.; Burlui, A.; Cardoneanu, A.; Rezuş, C.; Codreanu, C.; Pârvu, M.; Rusu Zota, G.; Tamba, B.I. Inactivity and Skeletal Muscle Metabolism: A Vicious Cycle in Old Age. Int. J. Mol. Sci. 2020, 21, 592. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Yakabe, M.; Akishita, M. Age-related sarcopenia and its pathophysiological bases. Inflamm. Regen. 2016, 36, 17. [Google Scholar] [CrossRef] [Green Version]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef]
- Hilton, T.N.; Tuttle, L.J.; Bohnert, K.L.; Mueller, M.J.; Sinacore, D.R. Excessive adipose tissue infiltration in skeletal muscle in individuals with obesity, diabetes mellitus, and peripheral neuropathy: Association with performance and function. Phys. Ther. 2008, 88, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef]
- Meng, S.J.; Yu, L.J. Oxidative stress, molecular inflammation and sarcopenia. Int. J. Mol. Sci. 2010, 11, 1509–1526. [Google Scholar] [CrossRef] [Green Version]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The Role of Inflammation in Age-Related Sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef]
- Aluganti Narasimhulu, C.; Singla, D.K. Amelioration of diabetes-induced inflammation mediated pyroptosis, sarcopenia, and adverse muscle remodelling by bone morphogenetic protein-7. J. Cachexia Sarcopenia Muscle 2021, 12, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Nardone, O.M.; de Sire, R.; Petito, V.; Testa, A.; Villani, G.; Scaldaferri, F.; Castiglione, F. Inflammatory Bowel Diseases and Sarcopenia: The Role of Inflammation and Gut Microbiota in the Development of Muscle Failure. Front. Immunol. 2021, 12, 694217. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. New York Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo-Calvo, D.; Fernández-García, B.; de Luxán-Delgado, B.; Rodríguez-González, S.; García-Macia, M.; Suárez, F.M.; Solano, J.J.; Rodríguez-Colunga, M.J.; Coto-Montes, A. Chronic training increases blood oxidative damage but promotes health in elderly men. Age 2013, 35, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020, 11, 932. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; Spiga, R.; Mancuso, E.; La Sala, L.; Antonicelli, R.; Testa, R.; Procopio, A.D.; Olivieri, F.; Ceriello, A. Inflammageing and metaflammation: The yin and yang of type 2 diabetes. Ageing Res. Rev. 2018, 41, 1–17. [Google Scholar] [CrossRef]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef] [Green Version]
- Gorelick, P.B. Role of inflammation in cognitive impairment: Results of observational epidemiological studies and clinical trials. Ann. NY Acad. Sci. 2010, 1207, 155–162. [Google Scholar] [CrossRef]
- Ginaldi, L.; Di Benedetto, M.C.; De Martinis, M. Osteoporosis, inflammation and ageing. Immun. Ageing 2005, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Calvani, R. Molecular Mechanism and Pathogenesis of Sarcopenia: An Overview. Int. J. Mol. Sci. 2021, 22, 3032. [Google Scholar] [CrossRef]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar] [CrossRef] [Green Version]
- Cros, D.; Harnden, P.; Pellissier, J.F.; Serratrice, G. Muscle hypertrophy in Duchenne muscular dystrophy. A pathological and morphometric study. J. Neurol. 1989, 236, 43–47. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [Green Version]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef]
- El Assar, M.; Álvarez-Bustos, A.; Sosa, P.; Angulo, J.; Rodríguez-Mañas, L. Effect of Physical Activity/Exercise on Oxidative Stress and Inflammation in Muscle and Vascular Aging. Int. J. Mol. Sci. 2022, 23, 8713. [Google Scholar] [CrossRef]
- Ziemkiewicz, N.; Hilliard, G.; Pullen, N.A.; Garg, K. The Role of Innate and Adaptive Immune Cells in Skeletal Muscle Regeneration. Int. J. Mol. Sci. 2021, 22, 3265. [Google Scholar] [CrossRef]
- Dhawan, J.; Rando, T.A. Stem cells in postnatal myogenesis: Molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005, 15, 666–673. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [Green Version]
- Barberi, L.; Scicchitano, B.M.; De Rossi, M.; Bigot, A.; Duguez, S.; Wielgosik, A.; Stewart, C.; McPhee, J.; Conte, M.; Narici, M.; et al. Age-dependent alteration in muscle regeneration: The critical role of tissue niche. Biogerontology 2013, 14, 273–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montarras, D.; L’Honoré, A.; Buckingham, M. Lying low but ready for action: The quiescent muscle satellite cell. FEBS J. 2013, 280, 4036–4050. [Google Scholar] [CrossRef] [PubMed]
- Scharner, J.; Zammit, P.S. The muscle satellite cell at 50: The formative years. Skelet. Muscle 2011, 1, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perandini, L.A.; Chimin, P.; Lutkemeyer, D.D.S.; Câmara, N.O.S. Chronic inflammation in skeletal muscle impairs satellite cells function during regeneration: Can physical exercise restore the satellite cell niche? FEBS J. 2018, 285, 1973–1984. [Google Scholar] [CrossRef] [Green Version]
- Bouché, M.; Muñoz-Cánoves, P.; Rossi, F.; Coletti, D. Inflammation in muscle repair, aging, and myopathies. BioMed Res. Int. 2014, 2014, 821950. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol. 2007, 578, 327–336. [Google Scholar] [CrossRef]
- Reidy, P.T.; McKenzie, A.I.; Mahmassani, Z.S.; Petrocelli, J.J.; Nelson, D.B.; Lindsay, C.C.; Gardner, J.E.; Morrow, V.R.; Keefe, A.C.; Huffaker, T.B.; et al. Aging impairs mouse skeletal muscle macrophage polarization and muscle-specific abundance during recovery from disuse. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E85–E98. [Google Scholar] [CrossRef]
- Cui, C.Y.; Driscoll, R.K.; Piao, Y.; Chia, C.W.; Gorospe, M.; Ferrucci, L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell 2019, 18, e13032. [Google Scholar] [CrossRef] [Green Version]
- Villalta, S.A.; Rinaldi, C.; Deng, B.; Liu, G.; Fedor, B.; Tidball, J.G. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet. 2011, 20, 790–805. [Google Scholar] [CrossRef]
- Madaro, L.; Torcinaro, A.; De Bardi, M.; Contino, F.F.; Pelizzola, M.; Diaferia, G.R.; Imeneo, G.; Bouchè, M.; Puri, P.L.; De Santa, F. Macrophages fine tune satellite cell fate in dystrophic skeletal muscle of mdx mice. PLoS Genet. 2019, 15, e1008408. [Google Scholar] [CrossRef]
- Segawa, M.; Fukada, S.; Yamamoto, Y.; Yahagi, H.; Kanematsu, M.; Sato, M.; Ito, T.; Uezumi, A.; Hayashi, S.; Miyagoe-Suzuki, Y.; et al. Suppression of macrophage functions impairs skeletal muscle regeneration with severe fibrosis. Exp. Cell Res. 2008, 314, 3232–3244. [Google Scholar] [CrossRef]
- Zhang, C.; Cheng, N.; Qiao, B.; Zhang, F.; Wu, J.; Liu, C.; Li, Y.; Du, J. Age-related decline of interferon-gamma responses in macrophage impairs satellite cell proliferation and regeneration. J. Cachexia Sarcopenia Muscle 2020, 11, 1291–1305. [Google Scholar] [CrossRef]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Datzkiw, D.; Rudnicki, M.A. Satellite cells in ageing: Use it or lose it. Open Biol. 2020, 10, 200048. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Chakkalakal, J.V.; Jones, K.M.; Basson, M.A.; Brack, A.S. The aged niche disrupts muscle stem cell quiescence. Nature 2012, 490, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef]
- Palla, A.R.; Ravichandran, M.; Wang, Y.X.; Alexandrova, L.; Yang, A.V.; Kraft, P.; Holbrook, C.A.; Schürch, C.M.; Ho, A.T.V.; Blau, H.M. Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science 2021, 371, eabc8059. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, N.P.; Misyak, S.A.; Robertson, J.L.; Bassaganya-Riera, J.; Grange, R.W. Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil. 2009, 88, 502–522. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Li, Q.; Ding, J.; Liang, F.; Gusev, E.; Lapohos, O.; Fonseca, G.J.; Kaufmann, E.; Divangahi, M.; Petrof, B.J. TLR4 is a regulator of trained immunity in a murine model of Duchenne muscular dystrophy. Nat. Commun. 2022, 13, 879. [Google Scholar] [CrossRef]
- De Paepe, B. Progressive Skeletal Muscle Atrophy in Muscular Dystrophies: A Role for Toll-like Receptor-Signaling in Disease Pathogenesis. Int. J. Mol. Sci. 2020, 21, 4440. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Yang, J.Y.; Guo, Y.; Liu, Y.X.; Zhong, X.Y. Altered levels of circulating mitochondrial DNA in elderly people with sarcopenia: Association with mitochondrial impairment. Exp. Gerontol. 2022, 163, 111802. [Google Scholar] [CrossRef]
- Hahn, A.; Kny, M.; Pablo-Tortola, C.; Todiras, M.; Willenbrock, M.; Schmidt, S.; Schmoeckel, K.; Jorde, I.; Nowak, M.; Jarosch, E.; et al. Serum amyloid A1 mediates myotube atrophy via Toll-like receptors. J. Cachexia Sarcopenia Muscle 2020, 11, 103–119. [Google Scholar] [CrossRef] [Green Version]
- Verzola, D.; Bonanni, A.; Sofia, A.; Montecucco, F.; D’Amato, E.; Cademartori, V.; Parodi, E.L.; Viazzi, F.; Venturelli, C.; Brunori, G.; et al. Toll-like receptor 4 signalling mediates inflammation in skeletal muscle of patients with chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 131–144. [Google Scholar] [CrossRef]
- Parveen, A.; Bohnert, K.R.; Tomaz da Silva, M.; Wen, Y.; Bhat, R.; Roy, A.; Kumar, A. MyD88-mediated signaling intercedes in neurogenic muscle atrophy through multiple mechanisms. FASEB J. 2021, 35, e21821. [Google Scholar] [CrossRef]
- Wang, J.; Leung, K.S.; Chow, S.K.; Cheung, W.H. Inflammation and age-associated skeletal muscle deterioration (sarcopaenia). J. Orthop. Translat. 2017, 10, 94–101. [Google Scholar] [CrossRef]
- Torrente, Y.; El Fahime, E.; Caron, N.J.; Del Bo, R.; Belicchi, M.; Pisati, F.; Tremblay, J.P.; Bresolin, N. Tumor necrosis factor-alpha (TNF-alpha) stimulates chemotactic response in mouse myogenic cells. Cell Transplant. 2003, 12, 91–100. [Google Scholar] [CrossRef]
- Chen, S.E.; Jin, B.; Li, Y.P. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am. J. Physiol. Cell Physiol. 2007, 292, C1660–C1671. [Google Scholar] [CrossRef]
- Reid, M.B.; Li, Y.P. Tumor necrosis factor-alpha and muscle wasting: A cellular perspective. Respir. Res. 2001, 2, 269–272. [Google Scholar] [CrossRef]
- Bian, A.L.; Hu, H.Y.; Rong, Y.D.; Wang, J.; Wang, J.X.; Zhou, X.Z. A study on relationship between elderly sarcopenia and inflammatory factors IL-6 and TNF-α. Eur. J. Med. Res. 2017, 22, 25. [Google Scholar] [CrossRef] [Green Version]
- Li, C.W.; Yu, K.; Shyh-Chang, N.; Li, G.X.; Jiang, L.J.; Yu, S.L.; Xu, L.Y.; Liu, R.J.; Guo, Z.J.; Xie, H.Y.; et al. Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J. Cachexia Sarcopenia Muscle 2019, 10, 586–600. [Google Scholar] [CrossRef] [Green Version]
- de Gonzalo-Calvo, D.; de Luxán-Delgado, B.; Martínez-Camblor, P.; Rodríguez-González, S.; García-Macia, M.; Suárez, F.M.; Solano, J.J.; Rodríguez-Colunga, M.J.; Coto-Montes, A. Chronic inflammation as predictor of 1-year hospitalization and mortality in elderly population. Eur. J. Clin. Investig. 2012, 42, 1037–1046. [Google Scholar] [CrossRef]
- Cantini, M.; Massimino, M.L.; Rapizzi, E.; Rossini, K.; Catani, C.; Dalla Libera, L.; Carraro, U. Human satellite cell proliferation in vitro is regulated by autocrine secretion of IL-6 stimulated by a soluble factor(s) released by activated monocytes. Biochem. Biophys. Res. Commun. 1995, 216, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Lee, J.; Baz, M.; Wells, C.E.; Bloch, S.; Lewis, A.; Donaldson, A.V.; Garfield, B.E.; Hopkinson, N.S.; Natanek, A.; et al. Growth differentiation factor-15 is associated with muscle mass in chronic obstructive pulmonary disease and promotes muscle wasting in vivo. J. Cachexia Sarcopenia Muscle 2016, 7, 436–448. [Google Scholar] [CrossRef] [Green Version]
- Schaap, L.A.; Pluijm, S.M.; Deeg, D.J.; Visser, M. Inflammatory markers and loss of muscle mass (sarcopenia) and strength. Am. J. Med. 2006, 119, 526.e9–526.e17. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, J.L.; He, L.; Virecoulon Giudici, K.; Guyonnet, S.; Parini, A.; Dray, C.; Valet, P.; Pereira, O.; Vellas, B.; Rolland, Y.; et al. Circulating Levels of Apelin, GDF-15 and Sarcopenia: Lack of Association in the MAPT Study. J. Nutr. Health Aging 2022, 26, 564–570. [Google Scholar] [CrossRef]
- Kim, H.; Kim, K.M.; Kang, M.J.; Lim, S. Growth differentiation factor-15 as a biomarker for sarcopenia in aging humans and mice. Exp. Gerontol. 2020, 142, 111115. [Google Scholar] [CrossRef]
- Shokri-Mashhadi, N.; Moradi, S.; Heidari, Z.; Saadat, S. Association of circulating C-reactive protein and high-sensitivity C-reactive protein with components of sarcopenia: A systematic review and meta-analysis of observational studies. Exp. Gerontol. 2021, 150, 111330. [Google Scholar] [CrossRef]
- Tuttle, C.S.L.; Thang, L.A.N.; Maier, A.B. Markers of inflammation and their association with muscle strength and mass: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 64, 101185. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Baylis, D.; Bartlett, D.B.; Syddall, H.E.; Ntani, G.; Gale, C.R.; Cooper, C.; Lord, J.M.; Sayer, A.A. Immune-endocrine biomarkers as predictors of frailty and mortality: A 10-year longitudinal study in community-dwelling older people. Age 2013, 35, 963–971. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 2006, 83, 447s–455s. [Google Scholar] [CrossRef] [Green Version]
- Schrager, M.A.; Metter, E.J.; Simonsick, E.; Ble, A.; Bandinelli, S.; Lauretani, F.; Ferrucci, L. Sarcopenic obesity and inflammation in the InCHIANTI study. J. Appl. Physiol. 2007, 102, 919–925. [Google Scholar] [CrossRef]
- O’Leary, M.F.; Wallace, G.R.; Bennett, A.J.; Tsintzas, K.; Jones, S.W. IL-15 promotes human myogenesis and mitigates the detrimental effects of TNFα on myotube development. Sci. Rep. 2017, 7, 12997. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, A.; Silay, K.; Balik, A.R.; Avcioğlu, G.; Aydin, A.S. The relationship between plasma interleukin-15 levels and sarcopenia in outpatient older people. Aging Clin. Exp. Res. 2018, 30, 783–790. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [Green Version]
- Steen, E.H.; Wang, X.; Balaji, S.; Butte, M.J.; Bollyky, P.L.; Keswani, S.G. The Role of the Anti-Inflammatory Cytokine Interleukin-10 in Tissue Fibrosis. Adv. Wound Care 2020, 9, 184–198. [Google Scholar] [CrossRef] [Green Version]
- Ko, F.; Abadir, P.; Marx, R.; Westbrook, R.; Cooke, C.; Yang, H.; Walston, J. Impaired mitochondrial degradation by autophagy in the skeletal muscle of the aged female interleukin 10 null mouse. Exp. Gerontol. 2016, 73, 23–27. [Google Scholar] [CrossRef] [Green Version]
- Dagdeviren, S.; Jung, D.Y.; Friedline, R.H.; Noh, H.L.; Kim, J.H.; Patel, P.R.; Tsitsilianos, N.; Inashima, K.; Tran, D.A.; Hu, X.; et al. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 2017, 31, 701–710. [Google Scholar] [CrossRef]
- Rong, Y.D.; Bian, A.L.; Hu, H.Y.; Ma, Y.; Zhou, X.Z. Study on relationship between elderly sarcopenia and inflammatory cytokine IL-6, anti-inflammatory cytokine IL-10. BMC Geriatr. 2018, 18, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Bai, L.; Wang, S.; Lin, H. The Association of Systemic Interleukin 6 and Interleukin 10 Levels with Sarcopenia in Elderly Patients with Chronic Obstructive Pulmonary Disease. Int. J. Gen. Med. 2021, 14, 5893–5902. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Kishioka, Y.; Wakamatsu, J.; Hattori, A.; Hennebry, A.; Berry, C.J.; Sharma, M.; Kambadur, R.; Nishimura, T. Decorin binds myostatin and modulates its activity to muscle cells. Biochem. Biophys. Res. Commun. 2006, 340, 675–680. [Google Scholar] [CrossRef] [PubMed]
- McCroskery, S.; Thomas, M.; Maxwell, L.; Sharma, M.; Kambadur, R. Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell Biol. 2003, 162, 1135–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanzleiter, T.; Rath, M.; Görgens, S.W.; Jensen, J.; Tangen, D.S.; Kolnes, A.J.; Kolnes, K.J.; Lee, S.; Eckel, J.; Schürmann, A.; et al. The myokine decorin is regulated by contraction and involved in muscle hypertrophy. Biochem. Biophys. Res. Commun. 2014, 450, 1089–1094. [Google Scholar] [CrossRef]
- Kwon, J.H.; Moon, K.M.; Min, K.W. Exercise-Induced Myokines can Explain the Importance of Physical Activity in the Elderly: An Overview. Healthcare 2020, 8, 378. [Google Scholar] [CrossRef]
- Bekki, M.; Hashida, R.; Kawaguchi, T.; Goshima, N.; Yoshiyama, T.; Otsuka, T.; Koya, S.; Hirota, K.; Matsuse, H.; Niizeki, T.; et al. The association between sarcopenia and decorin, an exercise-induced myokine, in patients with liver cirrhosis: A pilot study. JCSM Rapid Commun. 2018, 1, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Baczek, J.; Silkiewicz, M.; Wojszel, Z.B. Myostatin as a Biomarker of Muscle Wasting and other Pathologies-State of the Art and Knowledge Gaps. Nutrients 2020, 12, 2401. [Google Scholar] [CrossRef]
- Peng, L.N.; Lee, W.J.; Liu, L.K.; Lin, M.H.; Chen, L.K. Healthy community-living older men differ from women in associations between myostatin levels and skeletal muscle mass. J. Cachexia Sarcopenia Muscle 2018, 9, 635–642. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Zhou, X.; Yuan, C.; Li, R.; Ma, Y.; Tang, X. Association between serum irisin concentrations and sarcopenia in patients with liver cirrhosis: A cross-sectional study. Sci. Rep. 2020, 10, 16093. [Google Scholar] [CrossRef]
- Chang, J.S.; Kim, T.H.; Nguyen, T.T.; Park, K.S.; Kim, N.; Kong, I.D. Circulating irisin levels as a predictive biomarker for sarcopenia: A cross-sectional community-based study. Geriatr. Gerontol. Int. 2017, 17, 2266–2273. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Dong, Y.; Dong, Y.; Chen, F.; Mitch, W.E.; Zhang, L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int. J. Obes. 2016, 40, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [Green Version]
- Mancinelli, R.; Checcaglini, F.; Coscia, F.; Gigliotti, P.; Fulle, S.; Fanò-Illic, G. Biological Aspects of Selected Myokines in Skeletal Muscle: Focus on Aging. Int. J. Mol. Sci. 2021, 22, 8520. [Google Scholar] [CrossRef]
- Alberro, A.; Iribarren-Lopez, A.; Sáenz-Cuesta, M.; Matheu, A.; Vergara, I.; Otaegui, D. Inflammaging markers characteristic of advanced age show similar levels with frailty and dependency. Sci. Rep. 2021, 11, 4358. [Google Scholar] [CrossRef]
- Pawelec, G. Immunosenescence: Impact in the young as well as the old? Mech. Ageing Dev. 1999, 108, 1–7. [Google Scholar] [CrossRef]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- Chargé, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef]
- Saclier, M.; Lapi, M.; Bonfanti, C.; Rossi, G.; Antonini, S.; Messina, G. The Transcription Factor Nfix Requires RhoA-ROCK1 Dependent Phagocytosis to Mediate Macrophage Skewing during Skeletal Muscle Regeneration. Cells 2020, 9, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounier, R.; Théret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, N.R.; Rumbaut, R.E. Age-related responses of the microcirculation to ischemia-reperfusion and inflammation. Pathophysiology 2001, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.F.; Zamunér, S.R.; Zuliani, J.P.; Fernandes, C.M.; Cruz-Hofling, M.A.; Fernandes, I.; Chaves, F.; Gutiérrez, J.M. Neutrophils do not contribute to local tissue damage, but play a key role in skeletal muscle regeneration, in mice injected with Bothrops asper snake venom. Muscle Nerve 2003, 28, 449–459. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Modesti, M.; Ginaldi, L. Phenotypic and functional changes of circulating monocytes and polymorphonuclear leucocytes from elderly persons. Immunol. Cell Biol. 2004, 82, 415–420. [Google Scholar] [CrossRef]
- Saclier, M.; Cuvellier, S.; Magnan, M.; Mounier, R.; Chazaud, B. Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J. 2013, 280, 4118–4130. [Google Scholar] [CrossRef]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musarò, A.; Rosenthal, N. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol. Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef] [Green Version]
- Przybyla, B.; Gurley, C.; Harvey, J.F.; Bearden, E.; Kortebein, P.; Evans, W.J.; Sullivan, D.H.; Peterson, C.A.; Dennis, R.A. Aging alters macrophage properties in human skeletal muscle both at rest and in response to acute resistance exercise. Exp. Gerontol. 2006, 41, 320–327. [Google Scholar] [CrossRef]
- Della Gatta, P.A.; Garnham, A.P.; Peake, J.M.; Cameron-Smith, D. Effect of exercise training on skeletal muscle cytokine expression in the elderly. Brain Behav. Immun. 2014, 39, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Pereira, M.G.; Ciciliot, S.; Rovere-Querini, P. Regulatory T cells and skeletal muscle regeneration. FEBS J. 2017, 284, 517–524. [Google Scholar] [CrossRef]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Lee, J.; d’Aigle, J.; Atadja, L.; Quaicoe, V.; Honarpisheh, P.; Ganesh, B.P.; Hassan, A.; Graf, J.; Petrosino, J.; Putluri, N.; et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Poststroke Recovery in Aged Mice. Circ. Res. 2020, 127, 453–465. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Nagpal, R.; Mainali, R.; Ahmadi, S.; Wang, S.; Singh, R.; Kavanagh, K.; Kitzman, D.W.; Kushugulova, A.; Marotta, F.; Yadav, H. Gut microbiome and aging: Physiological and mechanistic insights. Nutr. Healthy Aging 2018, 4, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jazwinski, S.M. The Gut Microbiota and Healthy Aging: A Mini-Review. Gerontology 2018, 64, 513–520. [Google Scholar] [CrossRef]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 2017, 21, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Ryan, E.; McNicholas, D.; Creavin, B.; Kelly, M.E.; Walsh, T.; Beddy, D. Sarcopenia and Inflammatory Bowel Disease: A Systematic Review. Inflamm. Bowel Dis. 2019, 25, 67–73. [Google Scholar] [CrossRef]
- Puleo, F.; Meirelles, K.; Navaratnarajah, M.; Fitzpatrick, L.; Shumate, M.L.; Cooney, R.N.; Lang, C.H. Skeletal muscle catabolism in trinitrobenzene sulfonic acid-induced murine colitis. Metabolism 2010, 59, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- van Langenberg, D.R.; Della Gatta, P.; Hill, B.; Zacharewicz, E.; Gibson, P.R.; Russell, A.P. Delving into disability in Crohn’s disease: Dysregulation of molecular pathways may explain skeletal muscle loss in Crohn’s disease. J. Crohn’s Colitis 2014, 8, 626–634. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.B.; Kang, J.; Ahn, D.W.; Kim, J.W.; Kim, B.G.; Lee, K.L.; Oh, S.; Yoon, S.H.; Park, S.J.; et al. Association between sarcopenia level and metabolic syndrome. PLoS ONE 2021, 16, e0248856. [Google Scholar] [CrossRef] [PubMed]
- Cleasby, M.E.; Jamieson, P.M.; Atherton, P.J. Insulin resistance and sarcopenia: Mechanistic links between common co-morbidities. J. Endocrinol. 2016, 229, R67–R81. [Google Scholar] [CrossRef] [PubMed]
- Gluvic, Z.; Zaric, B.; Resanovic, I.; Obradovic, M.; Mitrovic, A.; Radak, D.; Isenovic, E.R. Link between Metabolic Syndrome and Insulin Resistance. Curr. Vasc. Pharmacol. 2017, 15, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Cline, G.W.; Petersen, K.F.; Krssak, M.; Shen, J.; Hundal, R.S.; Trajanoski, Z.; Inzucchi, S.; Dresner, A.; Rothman, D.L.; Shulman, G.I. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N. Engl. J. Med. 1999, 341, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, Z.; Hu, J.; Du, J.; Mitch, W.E. Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 2006, 147, 4160–4168. [Google Scholar] [CrossRef] [Green Version]
- Mirza, S.; Hossain, M.; Mathews, C.; Martinez, P.; Pino, P.; Gay, J.L.; Rentfro, A.; McCormick, J.B.; Fisher-Hoch, S.P. Type 2-diabetes is associated with elevated levels of TNF-alpha, IL-6 and adiponectin and low levels of leptin in a population of Mexican Americans: A cross-sectional study. Cytokine 2012, 57, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Fève, B.; Bastard, J.P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 305–311. [Google Scholar] [CrossRef]
- Brown, A.E.; Palsgaard, J.; Borup, R.; Avery, P.; Gunn, D.A.; De Meyts, P.; Yeaman, S.J.; Walker, M. p38 MAPK activation upregulates proinflammatory pathways in skeletal muscle cells from insulin-resistant type 2 diabetic patients. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E63–E70. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., III; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- He, M.; Chiang, H.H.; Luo, H.; Zheng, Z.; Qiao, Q.; Wang, L.; Tan, M.; Ohkubo, R.; Mu, W.C.; Zhao, S.; et al. An Acetylation Switch of the NLRP3 Inflammasome Regulates Aging-Associated Chronic Inflammation and Insulin Resistance. Cell Metab. 2020, 31, 580–591. [Google Scholar] [CrossRef]
- Stout, M.B.; Justice, J.N.; Nicklas, B.J.; Kirkland, J.L. Physiological Aging: Links Among Adipose Tissue Dysfunction, Diabetes, and Frailty. Physiology 2017, 32, 9–19. [Google Scholar] [CrossRef]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E745–E751. [Google Scholar] [CrossRef]
- Starr, M.E.; Evers, B.M.; Saito, H. Age-associated increase in cytokine production during systemic inflammation: Adipose tissue as a major source of IL-6. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 723–730. [Google Scholar] [CrossRef]
- Morin, C.L.; Pagliassotti, M.J.; Windmiller, D.; Eckel, R.H. Adipose tissue-derived tumor necrosis factor-alpha activity is elevated in older rats. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, B190–B195. [Google Scholar] [CrossRef] [Green Version]
- Jerschow, E.; Anwar, S.; Barzilai, N.; Rosenstreich, D. Macrophages accumulation in visceral and subcutaneous adipose tissue correlates with age. J. Allergy Clin. Immunol. 2007, 119, S179. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Liu, J.; Geletka, L.; Delaney, C.; Delproposto, J.; Desai, A.; Oatmen, K.; Martinez-Santibanez, G.; Julius, A.; Garg, S.; et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J. Immunol. 2011, 187, 6208–6216. [Google Scholar] [CrossRef] [Green Version]
- Paris, M.T.; Bell, K.E.; Mourtzakis, M. Myokines and adipokines in sarcopenia: Understanding cross-talk between skeletal muscle and adipose tissue and the role of exercise. Curr. Opin. Pharmacol. 2020, 52, 61–66. [Google Scholar] [CrossRef]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Kalinkovich, A.; Livshits, G. Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res. Rev. 2017, 35, 200–221. [Google Scholar] [CrossRef] [PubMed]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilski, J.; Pierzchalski, P.; Szczepanik, M.; Bonior, J.; Zoladz, J.A. Multifactorial Mechanism of Sarcopenia and Sarcopenic Obesity. Role of Physical Exercise, Microbiota and Myokines. Cells 2022, 11, 160. [Google Scholar] [CrossRef] [PubMed]
- Farup, J.; Just, J.; de Paoli, F.; Lin, L.; Jensen, J.B.; Billeskov, T.; Roman, I.S.; Cömert, C.; Møller, A.B.; Madaro, L.; et al. Human skeletal muscle CD90(+) fibro-adipogenic progenitors are associated with muscle degeneration in type 2 diabetic patients. Cell Metab. 2021, 33, 2201–2214. [Google Scholar] [CrossRef]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Yang, M.Y.; Shi, Y.X.; Xie, M.M.; Zhu, M.; Zheng, X.L.; Zhang, C.K.; Ge, Z.L.; Bian, X.T.; Lv, J.T.; et al. Interleukin-15 facilitates muscle regeneration through modulation of fibro/adipogenic progenitors. Cell Commun. Signal. 2018, 16, 42. [Google Scholar] [CrossRef] [Green Version]
- Biferali, B.; Proietti, D.; Mozzetta, C.; Madaro, L. Fibro-Adipogenic Progenitors Cross-Talk in Skeletal Muscle: The Social Network. Front. Physiol. 2019, 10, 1074. [Google Scholar] [CrossRef]
- Batsis, J.A.; Villareal, D.T. Sarcopenic obesity in older adults: Aetiology, epidemiology and treatment strategies. Nat. Rev. Endocrinol. 2018, 14, 513–537. [Google Scholar] [CrossRef]
- Tyrovolas, S.; Koyanagi, A.; Olaya, B.; Ayuso-Mateos, J.L.; Miret, M.; Chatterji, S.; Tobiasz-Adamczyk, B.; Koskinen, S.; Leonardi, M.; Haro, J.M. Factors associated with skeletal muscle mass, sarcopenia, and sarcopenic obesity in older adults: A multi-continent study. J. Cachexia Sarcopenia Muscle 2016, 7, 312–321. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Speakman, J.; Hambly, C.; Mitchell, S.; Król, E. Animal models of obesity. Obes. Rev. 2007, 8 (Suppl. 1), 55–61. [Google Scholar] [CrossRef]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436. [Google Scholar] [CrossRef] [Green Version]
- de Luxán-Delgado, B.; Potes, Y.; Rubio-González, A.; Caballero, B.; Solano, J.J.; Fernández-Fernández, M.; Bermúdez, M.; Rodrigues Moreira Guimarães, M.; Vega-Naredo, I.; Boga, J.A.; et al. Melatonin reduces endoplasmic reticulum stress and autophagy in liver of leptin-deficient mice. J. Pineal Res. 2016, 61, 108–123. [Google Scholar] [CrossRef]
- de Luxán-Delgado, B.; Caballero, B.; Potes, Y.; Rubio-González, A.; Rodríguez, I.; Gutiérrez-Rodríguez, J.; Solano, J.J.; Coto-Montes, A. Melatonin administration decreases adipogenesis in the liver of ob/ob mice through autophagy modulation. J. Pineal. Res. 2014, 56, 126–133. [Google Scholar] [CrossRef]
- Rubio-González, A.; Bermejo-Millo, J.C.; de Luxán-Delgado, B.; Potes, Y.; Pérez-Martínez, Z.; Boga, J.A.; Vega-Naredo, I.; Caballero, B.; Solano, J.J.; Coto-Montes, A. Melatonin Prevents the Harmful Effects of Obesity on the Brain, Including at the Behavioral Level. Mol. Neurobiol. 2018, 55, 5830–5846. [Google Scholar] [CrossRef]
- Rubio-González, A.; Reiter, R.J.; de Luxán-Delgado, B.; Potes, Y.; Caballero, B.; Boga, J.A.; Solano, J.J.; Vega-Naredo, I.; Coto-Montes, A. Pleiotropic role of melatonin in brain mitochondria of obese mice. Melatonin Res. 2020, 3, 538–557. [Google Scholar] [CrossRef]
- Grumati, P.; Bonaldo, P. Autophagy in skeletal muscle homeostasis and in muscular dystrophies. Cells 2012, 1, 325–345. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal muscle mitochondria and aging: A review. J. Aging Res. 2012, 2012, 194821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canty, T.G., Jr.; Boyle, E.M., Jr.; Farr, A.; Morgan, E.N.; Verrier, E.D.; Pohlman, T.H. Oxidative stress induces NF-kappaB nuclear translocation without degradation of IkappaBalpha. Circulation 1999, 100 (Suppl. 19), II-361–Ii–364. [Google Scholar] [CrossRef]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Blanco, L.; Bermúdez, M.; Bermejo-Millo, J.C.; Gutiérrez-Rodríguez, J.; Solano, J.J.; Antuña, E.; Menéndez-Valle, I.; Caballero, B.; Vega-Naredo, I.; Potes, Y.; et al. Cell interactome in sarcopenia during aging. J. Cachexia Sarcopenia Muscle 2022, 13, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Riffelmacher, T.; Richter, F.C.; Simon, A.K. Autophagy dictates metabolism and differentiation of inflammatory immune cells. Autophagy 2018, 14, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Cui, J.; Sun, Y.; Xu, W.; Wang, Z.; Wu, M.; Dong, H.; Yang, C.; Hong, S.; Yin, S.; et al. Autophagy deficiency promotes M1 macrophage polarization to exacerbate acute liver injury via ATG5 repression during aging. Cell Death Discov. 2021, 7, 397. [Google Scholar] [CrossRef]
- Stranks, A.J.; Hansen, A.L.; Panse, I.; Mortensen, M.; Ferguson, D.J.; Puleston, D.J.; Shenderov, K.; Watson, A.S.; Veldhoen, M.; Phadwal, K.; et al. Autophagy Controls Acquisition of Aging Features in Macrophages. J. Innate Immun. 2015, 7, 375–391. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef]
- Saitoh, T.; Akira, S. Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 2010, 189, 925–935. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Furman, D.; Chang, J.; Lartigue, L.; Bolen, C.R.; Haddad, F.; Gaudilliere, B.; Ganio, E.A.; Fragiadakis, G.K.; Spitzer, M.H.; Douchet, I.; et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 2017, 23, 174–184. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Conley, K.E.; Jubrias, S.A.; Esselman, P.C. Oxidative capacity and ageing in human muscle. J. Physiol. 2000, 526, 203–210. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Potes, Y.; de Luxán-Delgado, B.; Rodriguez-González, S.; Guimarães, M.R.M.; Solano, J.J.; Fernández-Fernández, M.; Bermúdez, M.; Boga, J.A.; Vega-Naredo, I.; Coto-Montes, A. Overweight in elderly people induces impaired autophagy in skeletal muscle. Free Radic. Biol. Med. 2017, 110, 31–41. [Google Scholar] [CrossRef]
- García-Macia, M.; Sierra, V.; Palanca, A.; Vega-Naredo, I.; de Gonzalo-Calvo, D.; Rodríguez-González, S.; Oliván, M.; Coto-Montes, A. Autophagy during beef aging. Autophagy 2014, 10, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Fry, C.S.; Lee, J.D.; Mula, J.; Kirby, T.J.; Jackson, J.R.; Liu, F.; Yang, L.; Mendias, C.L.; Dupont-Versteegden, E.E.; McCarthy, J.J.; et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 2015, 21, 76–80. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biran, A.; Perelmutter, M.; Gal, H.; Burton, D.G.; Ovadya, Y.; Vadai, E.; Geiger, T.; Krizhanovsky, V. Senescent cells communicate via intercellular protein transfer. Genes Dev. 2015, 29, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J. Pineal melatonin: Cell biology of its synthesis and of its physiological interactions. Endocr. Rev. 1991, 12, 151–180. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Guerrero, J.M.; Reiter, R.J. Melatonin-immune system relationships. Curr. Top. Med. Chem. 2002, 2, 167–179. [Google Scholar] [CrossRef]
- Wu, Y.H.; Swaab, D.F. The human pineal gland and melatonin in aging and Alzheimer’s disease. J. Pineal Res. 2005, 38, 145–152. [Google Scholar] [CrossRef]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef] [Green Version]
- Bubenik, G.A.; Konturek, S.J. Melatonin and aging: Prospects for human treatment. J. Physiol. Pharmacol. 2011, 62, 13–19. [Google Scholar]
- Luo, F.; Sandhu, A.F.; Rungratanawanich, W.; Williams, G.E.; Akbar, M.; Zhou, S.; Song, B.J.; Wang, X. Melatonin and Autophagy in Aging-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 7174. [Google Scholar] [CrossRef]
- Hardeland, R.; Madrid, J.A.; Tan, D.X.; Reiter, R.J. Melatonin, the circadian multioscillator system and health: The need for detailed analyses of peripheral melatonin signaling. J. Pineal Res. 2012, 52, 139–166. [Google Scholar] [CrossRef]
- McMullan, C.J.; Schernhammer, E.S.; Rimm, E.B.; Hu, F.B.; Forman, J.P. Melatonin secretion and the incidence of type 2 diabetes. JAMA 2013, 309, 1388–1396. [Google Scholar] [CrossRef] [Green Version]
- Cardinali, D.P.; Hardeland, R. Inflammaging, Metabolic Syndrome and Melatonin: A Call for Treatment Studies. Neuroendocrinology 2017, 104, 382–397. [Google Scholar] [CrossRef] [Green Version]
- Santos-Ledo, A.; Luxán-Delgado, B.; Caballero, B.; Potes, Y.; Rodríguez-González, S.; Boga, J.A.; Coto-Montes, A.; García-Macia, M. Melatonin Ameliorates Autophagy Impairment in a Metabolic Syndrome Model. Antioxidants 2021, 10, 796. [Google Scholar] [CrossRef]
- Boga, J.A.; Caballero, B.; Potes, Y.; Perez-Martinez, Z.; Reiter, R.J.; Vega-Naredo, I.; Coto-Montes, A. Therapeutic potential of melatonin related to its role as an autophagy regulator: A review. J. Pineal Res. 2019, 66, e12534. [Google Scholar] [CrossRef] [Green Version]
- Coto-Montes, A.; Boga, J.A.; Rosales-Corral, S.; Fuentes-Broto, L.; Tan, D.X.; Reiter, R.J. Role of melatonin in the regulation of autophagy and mitophagy: A review. Mol. Cell Endocrinol. 2012, 361, 12–23. [Google Scholar] [CrossRef]
- Szewczyk-Golec, K.; Woźniak, A.; Reiter, R.J. Inter-relationships of the chronobiotic, melatonin, with leptin and adiponectin: Implications for obesity. J. Pineal Res. 2015, 59, 277–291. [Google Scholar] [CrossRef]
- Celinski, K.; Konturek, P.C.; Slomka, M.; Cichoz-Lach, H.; Gonciarz, M.; Bielanski, W.; Reiter, R.J.; Konturek, S.J. Altered basal and postprandial plasma melatonin, gastrin, ghrelin, leptin and insulin in patients with liver cirrhosis and portal hypertension without and with oral administration of melatonin or tryptophan. J. Pineal Res. 2009, 46, 408–414. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Zhang, T.; Ren, Q.; Sun, C. Melatonin alleviates adipose inflammation through elevating α-ketoglutarate and diverting adipose-derived exosomes to macrophages in mice. J. Pineal Res. 2018, 64, e12455. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and inflammation-Story of a double-edged blade. J. Pineal Res. 2018, 65, e12525. [Google Scholar] [CrossRef] [PubMed]
- Coto-Montes, A.; Boga, J.A.; Tan, D.X.; Reiter, R.J. Melatonin as a Potential Agent in the Treatment of Sarcopenia. Int. J. Mol. Sci. 2016, 17, 1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Rusanova, I.; Rahim, I.; Escames, G.; López, L.C.; Mokhtar, D.M.; Acuña-Castroviejo, D. The Protective Effect of Melatonin Against Age-Associated, Sarcopenia-Dependent Tubular Aggregate Formation, Lactate Depletion, and Mitochondrial Changes. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1330–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Aranda-Martínez, P.; Fernández-Martínez, J.; Guerra-Librero, A.; Escames, G.; López, L.C.; Alsaadawy, R.M.; Acuña-Castroviejo, D. Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef]
- Ortiz, F.; García, J.A.; Acuña-Castroviejo, D.; Doerrier, C.; López, A.; Venegas, C.; Volt, H.; Luna-Sánchez, M.; López, L.C.; Escames, G. The beneficial effects of melatonin against heart mitochondrial impairment during sepsis: Inhibition of iNOS and preservation of nNOS. J. Pineal Res. 2014, 56, 71–81. [Google Scholar] [CrossRef]
- Vural, E.M.; van Munster, B.C.; de Rooij, S.E. Optimal dosages for melatonin supplementation therapy in older adults: A systematic review of current literature. Drugs Aging 2014, 31, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Stacchiotti, A.; Favero, G.; Rodella, L.F. Impact of Melatonin on Skeletal Muscle and Exercise. Cells 2020, 9, 288. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Minciullo, P.L.; Catalano, A.; Mandraffino, G.; Casciaro, M.; Crucitti, A.; Maltese, G.; Morabito, N.; Lasco, A.; Gangemi, S.; Basile, G. Inflammaging and Anti-Inflammaging: The Role of Cytokines in Extreme Longevity. Arch. Immunol. Ther. Exp. 2016, 64, 111–126. [Google Scholar] [CrossRef]
- Zhou, L.; Ge, M.; Zhang, Y.; Wu, X.; Leng, M.; Gan, C.; Mou, Y.; Zhou, J.; Valencia, C.A.; Hao, Q.; et al. Centenarians Alleviate Inflammaging by Changing the Ratio and Secretory Phenotypes of T Helper 17 and Regulatory T Cells. Front. Pharmacol. 2022, 13, 877709. [Google Scholar] [CrossRef]
- Csete, M.; Doyle, J. Bow ties, metabolism and disease. Trends Biotechnol. 2004, 22, 446–450. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antuña, E.; Cachán-Vega, C.; Bermejo-Millo, J.C.; Potes, Y.; Caballero, B.; Vega-Naredo, I.; Coto-Montes, A.; Garcia-Gonzalez, C. Inflammaging: Implications in Sarcopenia. Int. J. Mol. Sci. 2022, 23, 15039. https://doi.org/10.3390/ijms232315039
Antuña E, Cachán-Vega C, Bermejo-Millo JC, Potes Y, Caballero B, Vega-Naredo I, Coto-Montes A, Garcia-Gonzalez C. Inflammaging: Implications in Sarcopenia. International Journal of Molecular Sciences. 2022; 23(23):15039. https://doi.org/10.3390/ijms232315039
Chicago/Turabian StyleAntuña, Eduardo, Cristina Cachán-Vega, Juan Carlos Bermejo-Millo, Yaiza Potes, Beatriz Caballero, Ignacio Vega-Naredo, Ana Coto-Montes, and Claudia Garcia-Gonzalez. 2022. "Inflammaging: Implications in Sarcopenia" International Journal of Molecular Sciences 23, no. 23: 15039. https://doi.org/10.3390/ijms232315039