2.4. Surface Concentration
The surface concentrations of piperidone derivatives (Γ) were calculated employing the following equation:
where I is the peak current; n = 2 (number of electrons involved in the electrochemical process); F is the Faraday constant 96,485 C mol
−1; A is the surface area, 0.0707cm
2 (the diameter of the working electrode, the glassy carbon electrode, is 0.3 cm, and from this, the surface area of the electrode was calculated); υ represents the scan rate; R is the gas constant, 8.314 JK
−1mol
−1; and T = 293 K. If we plot the graph of Ip versus the scan rate, which conforms to the following equation: Ip (μA) = 0.721υ + 10.223 (R
2 = 0.9153), in this equation the slope, gives the value of n
2F
2AΓ/4RT, from which we can calculate Γ. The results are presented in
Table 4.
In the electrochemical process, there is an important difference between the concentration of a species at the surface of the electrode and its concentration at some distance from it, generally known as bulk concentration. The surface concentration depends on the rate at which the reactants are brought to the electrode surface by either diffusion or flow processes, which determines the rate of the electrochemical reaction.
All compounds exhibit a surface concentration in the order of 10−7 mol/cm2, compound C4 (N-methyl-4-piperidone with trifluoromethyl in phenyl rings) has the lowest value (1.72 × 10−7), and compound C6 (N-benzyl-4-piperidone unsubstituted in phenyl rings) has the highest value (5.01 × 10−7).
2.6. Quantum Chemical Calculation
The oxidation potentials at DFT M06 and M06-2x level using 6-311+G(d,p) basis set were obtained for the ten derivatives. M06-2X functionals have been extensively used to calculate redox potentials [
33,
34,
35,
36,
37]; however, in the specific case of oxidation potential, previous work has shown that functionals with high levels of Hartree–Fock (HF) exchange percentage tend to significantly overestimate the potential [
38]. Therefore, we tested M06-2X (54% HF) and M06 functionals to obtain the potentials. The number of electrons transferred in the reactions, according to Laviron’s equation in
Section 3.2, was two. Accordingly, we considered the two possible electronic states for oxidized species (singlet and triplet dications) for the calculations.
Table 5 and
Table 6 show the values for experimental and theoretical oxidation potentials calculated with M06-2X and M06 functionals, respectively. In all cases, the mean absolute error (MAE) for M06 was lower than for M06-2X, which agrees with what has been previously reported [
38]. MAE values are still overestimated with the M06 functional, but they are in the range of previous work of calculated oxidation potentials [
39]. Based on the above, we use data obtained with the M06 functional to discuss the results.
Taking both series (N-methyl and N-benzyl) together, experimental and theoretical potentials did not correlate well, neither when considering the oxidized species as a triplet, nor as a singlet (R2 = 0.0796 for singlet, R2 = 0.1667 for triplet). However, when both series were treated separately, it was found that the series with the N-benzyl group at the piperidone ring presented a good correlation between experimental and theoretical oxidation potential.
Figure 4 presents the correlation between experimental and theoretical oxidation potential for the N-methyl (A) and N-benzyl (B) series considering the oxidized species as a singlet, and for the N-methyl (C) and N-benzyl (D) series considering the oxidized species as a triplet.
For the series with N-benzyl (compounds C6 to C10), good correlations were found considering both kinds of oxidized species, triplet or singlet (R2 = 0.8886 and R2 = 0.9846, respectively). Meanwhile, the series with the N-methyl group did not show a good correlation between experimental and theoretical oxidation potentials, neither when considering the oxidized species as triplet nor as singlet (R2 = 0.4918 and R2 = 0.3786, respectively).
Considering that the N-benzyl derivative series presented a good correlation between experimental and theoretical oxidation potentials, unlike that evidenced for the N-methyl series, it can be suggested that oxidation of this last series of compounds did not occur by a simple mechanism. The above indicates that variation of the substituent at nitrogen is a key factor that determines the redox behavior of these derivatives. In addition, based on the structural analysis mentioned above, it is reasonable to consider that the trend for both series will remain independent of the addition of more compounds for each series.
Triplet oxidized species for all compounds present a similar geometry, regardless of the substituent on the N-position or phenyl rings. However, for singlet oxidized species, the behavior varies between the N-methyl and N-benzyl series (
Figure 5). For the N-methyl series, the oxidized species of compounds
C2,
C3, and
C5 (with p-Br, p-Cl, and p-OCH3 in the phenyl rings) present a notable geometry distortion of the piperidone ring, which does not occur for
C1 and
C4 (unsubstituted and with p-CF3 in the phenyl rings). On the other hand, all oxidized species of the N-benzyl series present a notable geometrical distortion of the piperidone ring, regardless of the substituent on the phenyl rings.
To obtain insights into the behavior observed for these two series of compounds, we studied the energy of frontier molecular orbitals. In previous works, theoretical calculations of some of these compounds had been carried out to study their reactivity and the behavior of their radical anions [
16], as well as optical properties [
28]. In our previous work [
16], we obtained, at the B3LYP/6-31G(d) level, the energies of the frontier molecular orbitals of some compounds studied here (
C1,
C2,
C3,
C6,
C7, and
C8).
Table 7 shows the values for HOMO and LUMO energies (E
HOMO and E
LUMO, respectively) and the HOMO–LUMO energy gap (GAP
H-L). These values, calculated at M06/6-311+G(d,p), showed slight differences from those calculated previously for some of the compounds at the B3LYP/6-31G(d) level. The E
HOMO values calculated at M06/6-311+G(d,p) tend to be lower, while the E
LUMO values tend to be higher, which leads to the GAP
H-L being lower for M06/6-311+G(d,p). However, the tendency for the three compounds in common for each series is the same. Unsubstituted derivatives in the aromatic ring show the highest E
HOMO and E
LUMO, followed by
p-Br and p-Cl (both for N-methyl and N-benzyl series). In addition, it can be observed that the value of GAP
H-L did not show significant differences among compounds from the N-methyl and N-benzyl series.
We examined the correlation among E
HOMO and experimental E
ox pairs, considering that electrons on this orbital are those that are removed in the oxidation process.
Figure 6 shows the correlation for both the N-methyl and N-benzyl series. We found that the E
HOMO of N-benzyl derivatives correlates well with experimental E
ox, with an R
2 = 0.9524. On the other hand, N-methyl derivatives did not present a good correlation, obtaining an R
2 = 0.6342. These results also support the assumption that the oxidation of the N-methyl derivatives, unlike the N-benzyl ones, suffers a complex oxidation process that does not only involve the direct subtraction of two electrons from the HOMO.
To evaluate alternative possible mechanisms, which can imply the heterolytic cleavage of a carbon–hydrogen (C-H) bond, we study the bond dissociation enthalpies (BDEs) of the C–H bonds potentially breakable in compounds
1 and
6 (BDE1 and BDE2 in
Table 8). A significant difference between the lower BDE would be indicative of a possible differential mechanism between the compounds of both series.
Results show that for both compounds, the BDE2 was the lowest, and there are no significant differences in the values (61.50 kcal/mol for 1 vs. 60.15 kcal/mol for 6). The latter suggests that neither of the two series of compounds (N-methyl and N-benzyl derivatives) has a significant preference for the proton-coupled electron transfer (PCET) mechanism; therefore, this cannot explain the differences between both series.
A plausible explanation for the irreversible oxidation of the compounds is to consider a chemical reaction coupled to the electrochemical process. For heterocyclic tertiary amines, an electrochemical oxidation reaction (Shono oxidation) has been described that gives products with a substituent in the carbon vicinal to the nitrogen [
30,
40,
41]. The process consists of a two-electron oxidation coupled to a proton transfer to generate an iminium cation intermediate which reacts with nucleophiles to achieve a great variety of products depending on the conditions [
42,
43,
44]. In our case, it is possible that the attacking nucleophile corresponds to the same molecule generating a dimer. This hypothesis needs to be evaluated in future works that delve into mechanistic aspects of the oxidation of these compounds.

 and
and 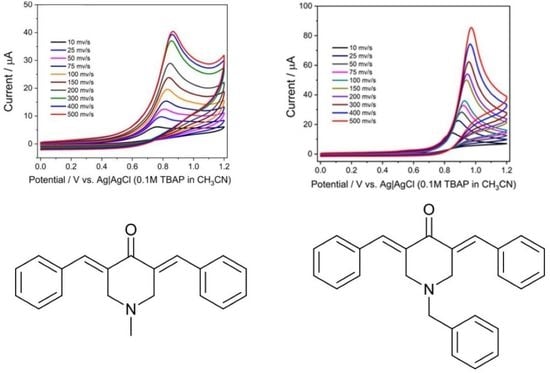







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


