
A First-Principles Investigation on the Structural, Optoelectronic, and Thermoelectric Properties of Pyrochlore Oxides (La2Tm2O7 (Tm = Hf, Zr)) for Energy Applications
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
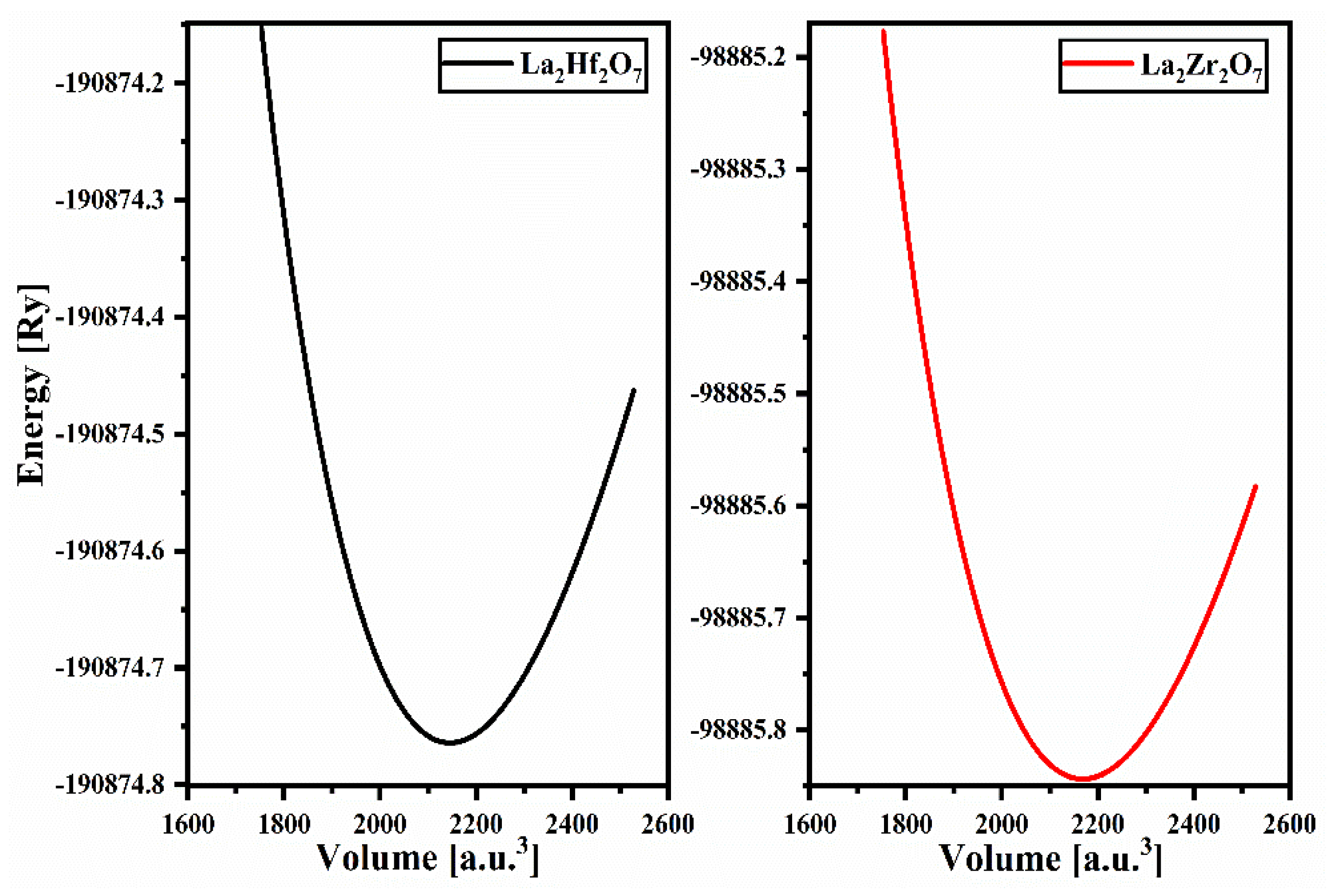
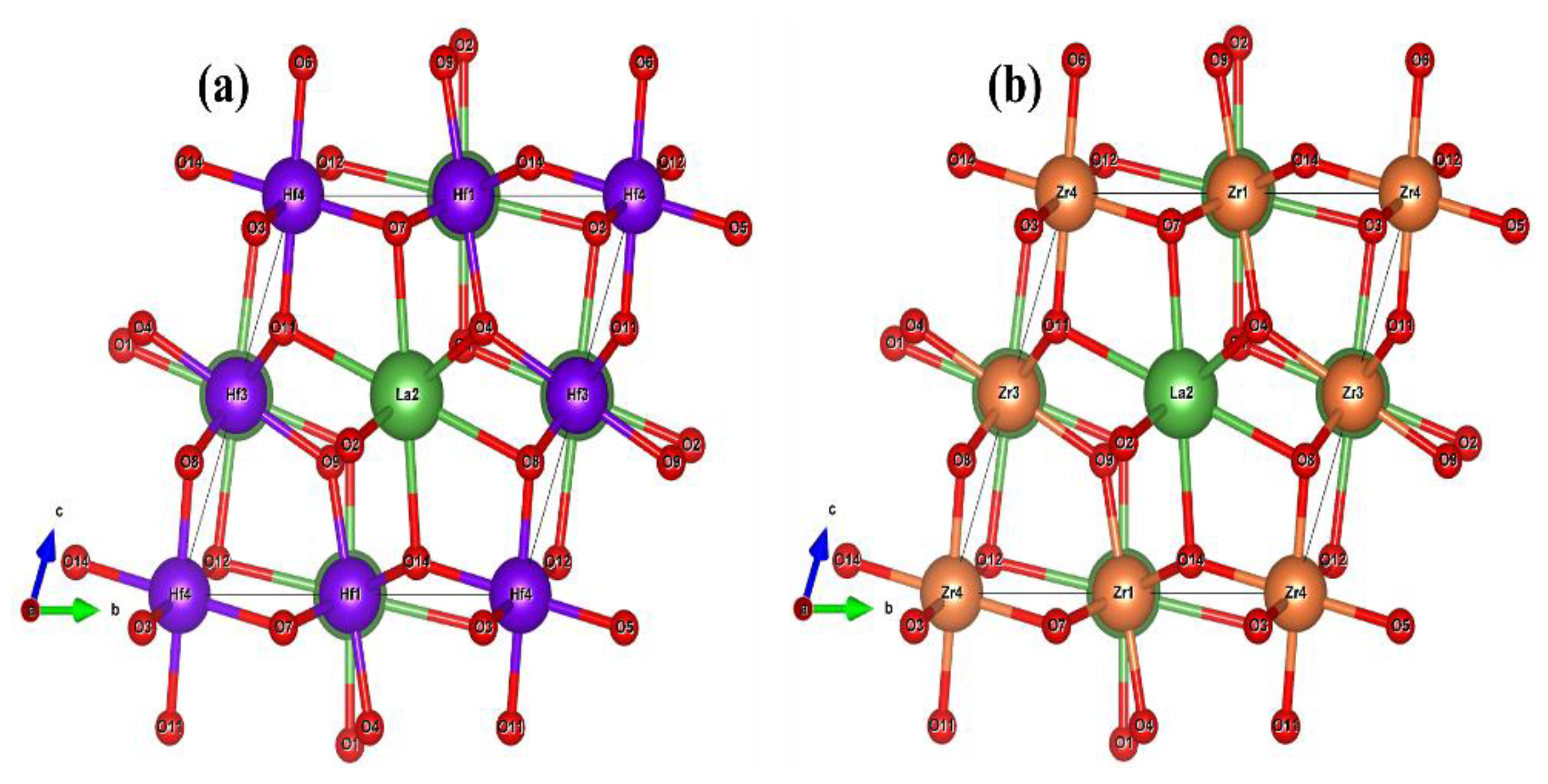
2.1. Structural Properties
2.2. Electronic Properties
2.2.1. Energy Band Structures
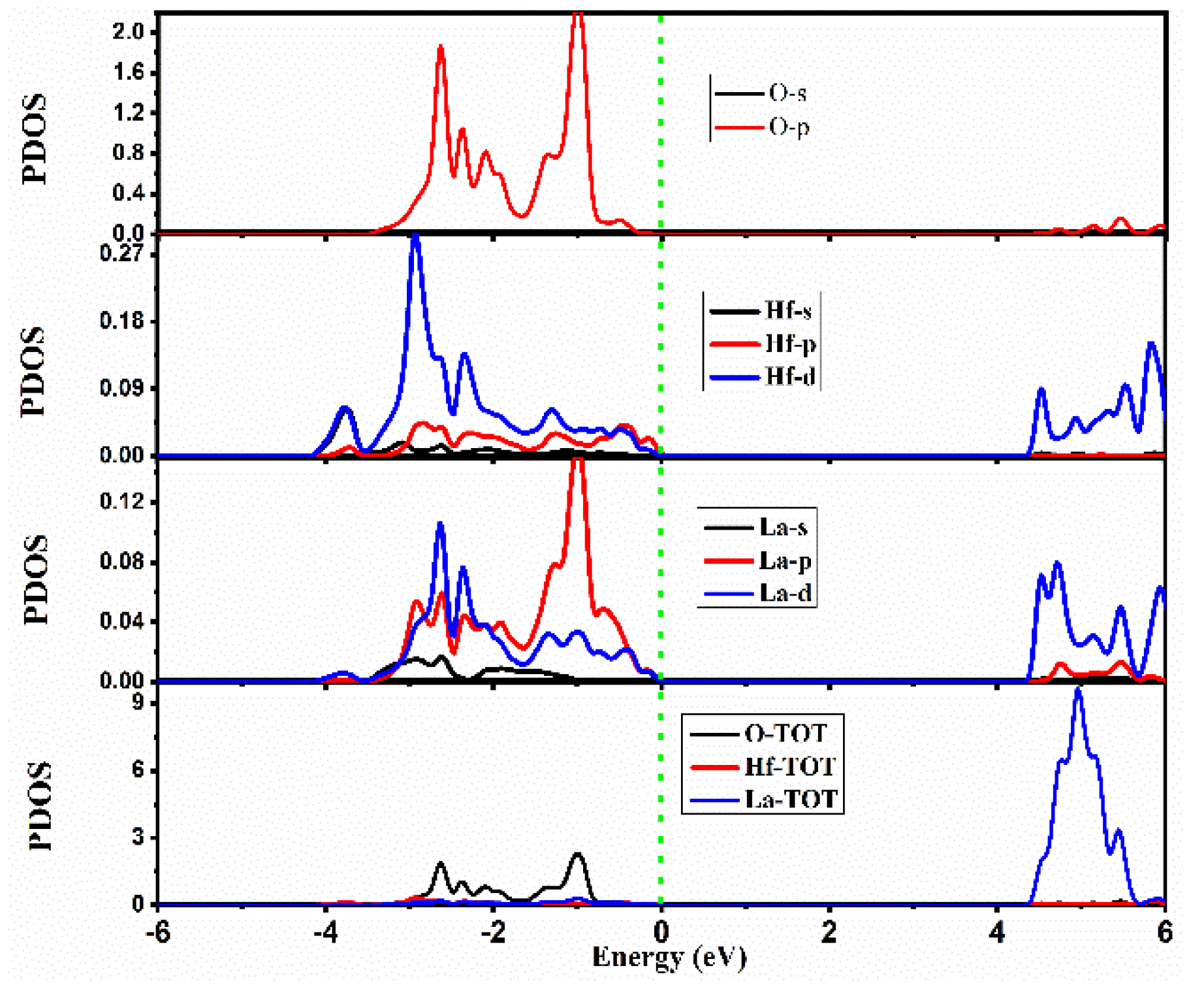
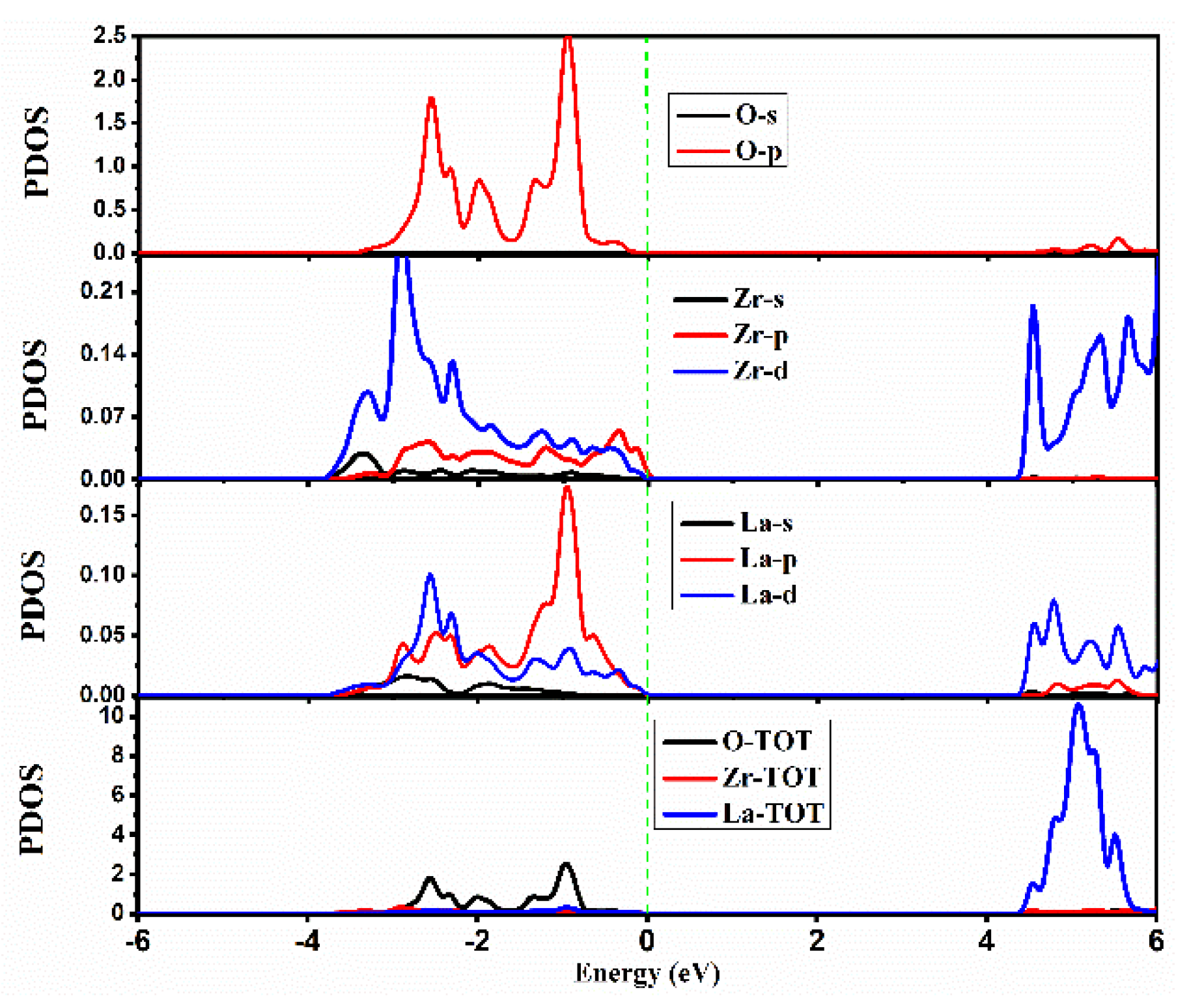
2.2.2. Density of States
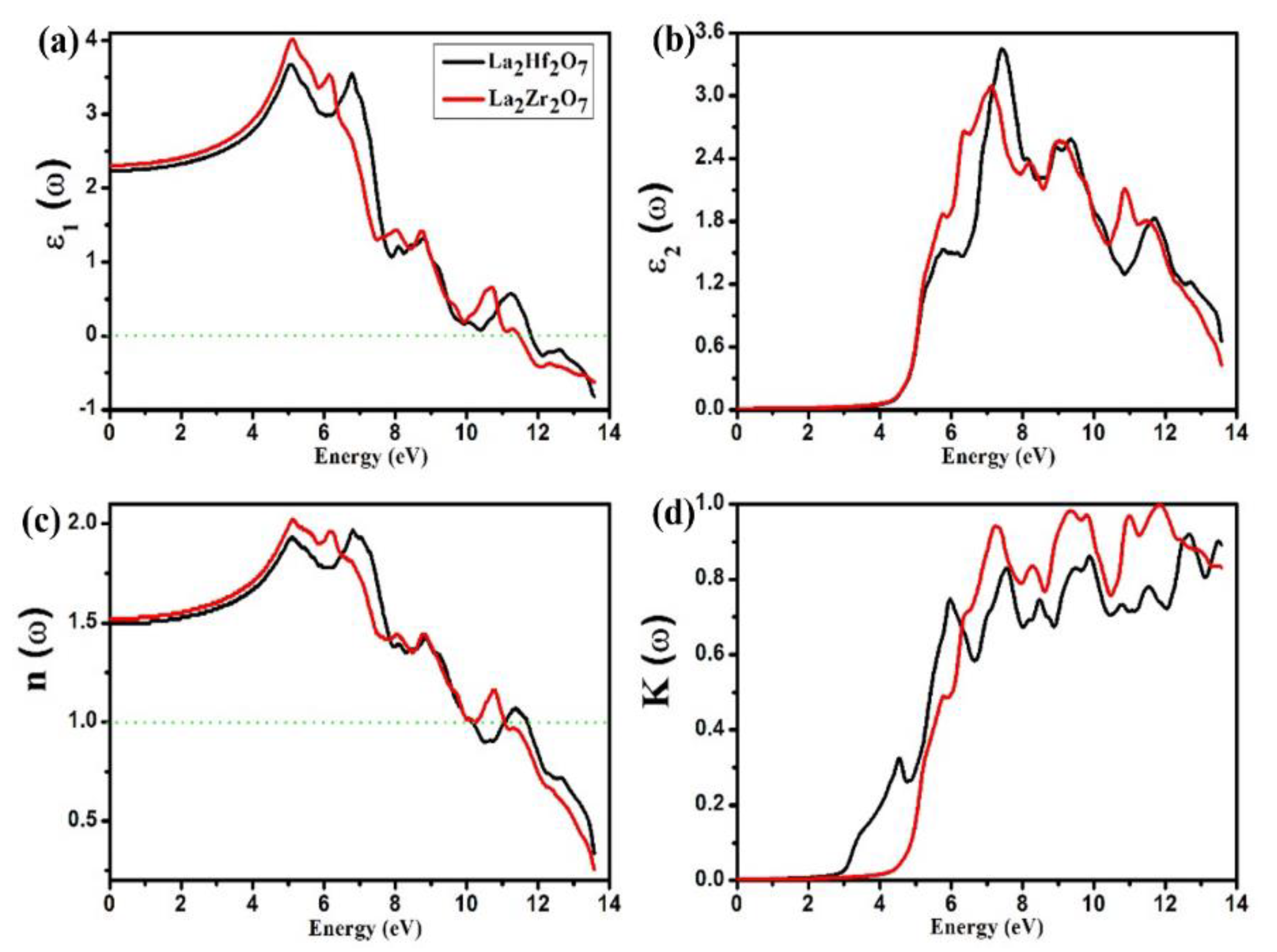
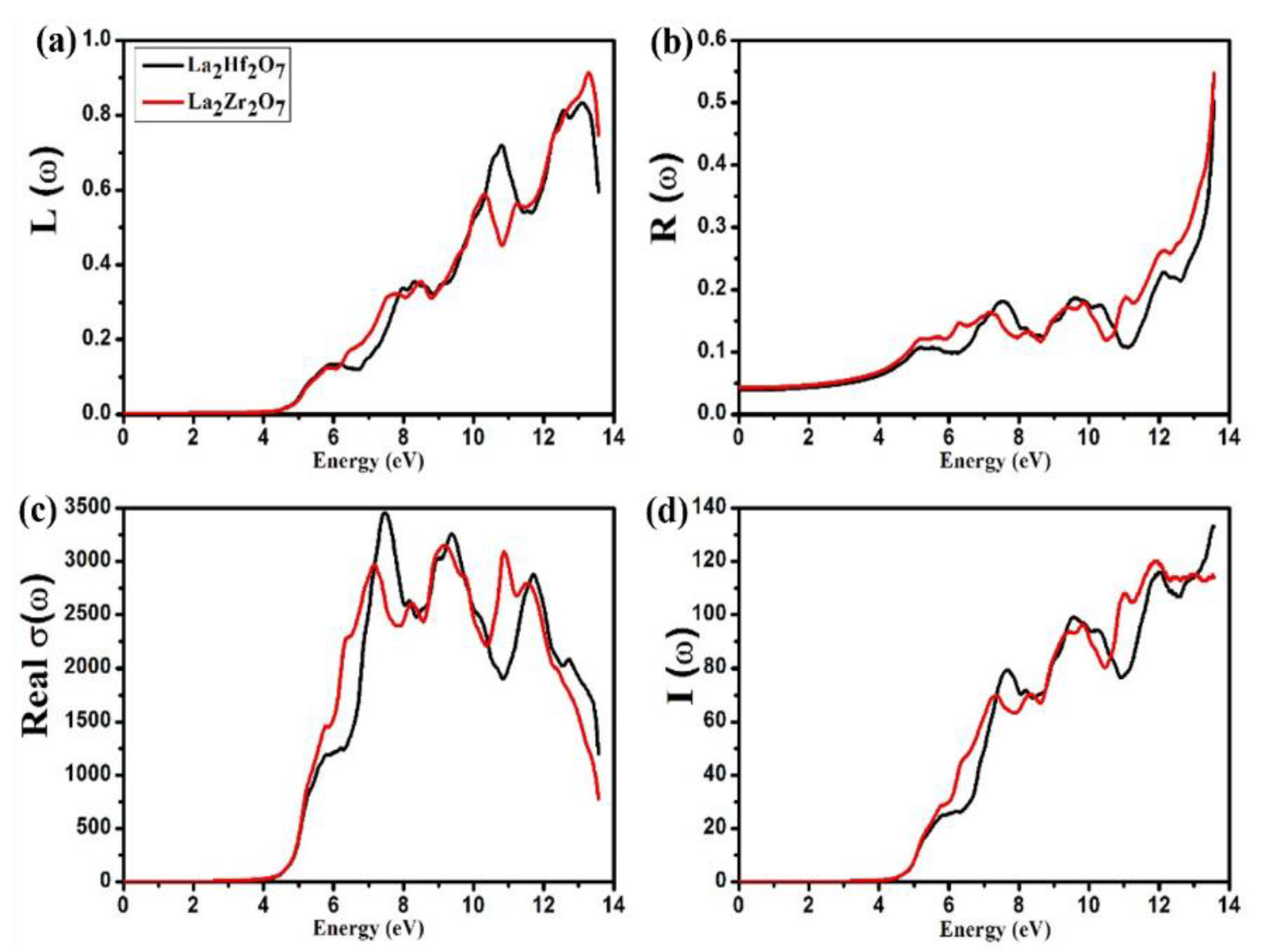
2.3. Optical Properties
2.4. Thermoelectric Properties
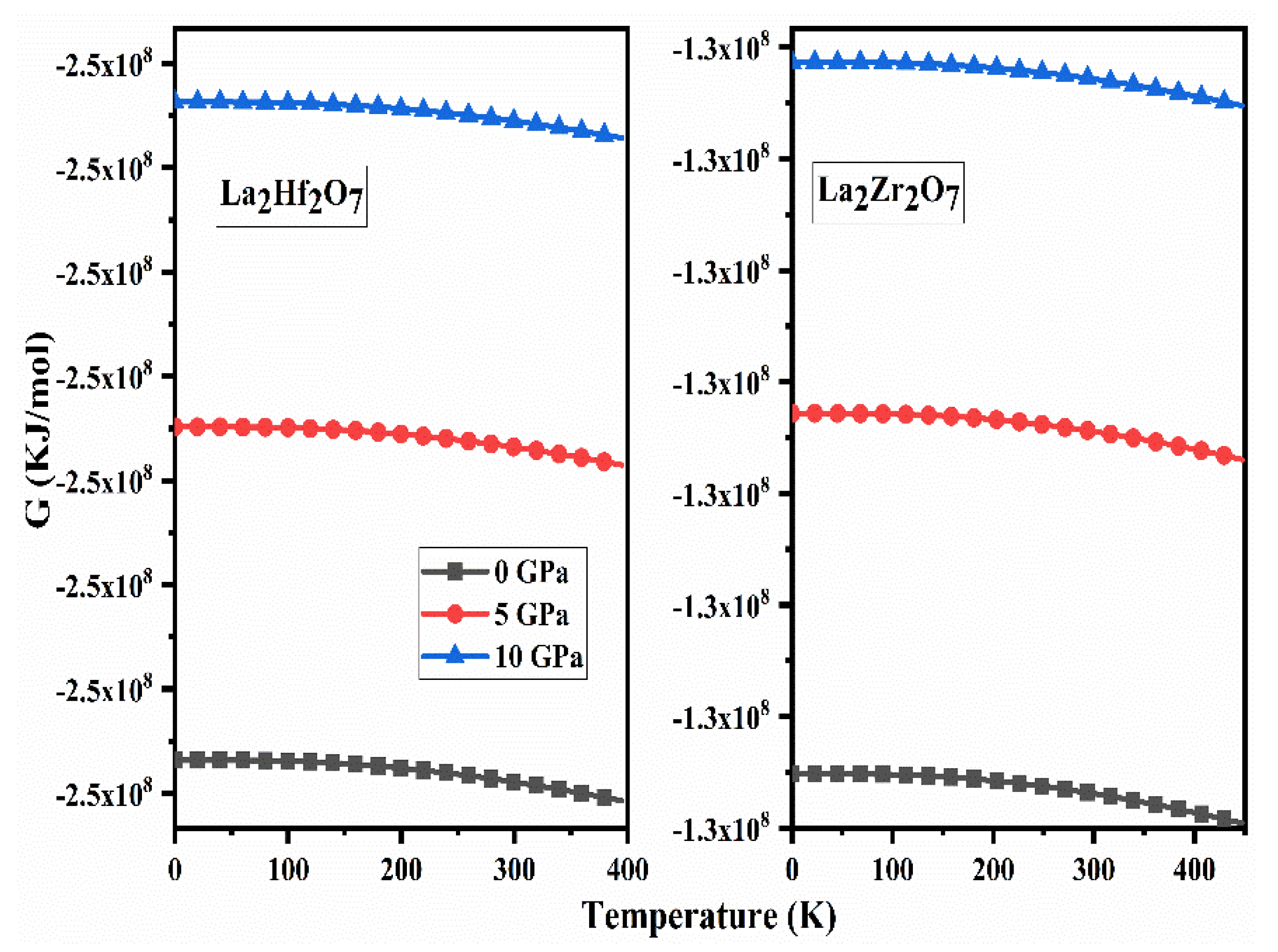
2.5. Thermodynamic Properties
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McQueen, T.; West, D.; Muegge, B.; Huang, Q.; Noble, K.; Zandbergen, H.; Cava, R. Frustrated ferroelectricity in niobate pyrochlores. J. Phys. Condens. Matter 2008, 20, 235210. [Google Scholar] [CrossRef]
- Atiq, S.; Mustafa, G.M.; Naseem, S.; Abbas, S.K.; Ramay, S.M.; Mahmood, A. Magneto-dielectric and ferroelectric tunability of multifunctional Ce-substituted neodymium zirconate pyrochlores. Mater. Lett. 2019, 243, 21–25. [Google Scholar] [CrossRef]
- Saitzek, S.; Shao, Z.; Bayart, A.; Ferri, A.; Huvé, M.; Roussel, P.; Desfeux, R. Ferroelectricity in La2Zr2O7 thin films with a frustrated pyrochlore-type structure. J. Mater. Chem. C 2014, 2, 4037–4043. [Google Scholar] [CrossRef]
- Fatima, K.; Abbas, Z.; Naz, A.; Alshahrani, T.; Chaib, Y.; Jaffery, S.H.A.; Muhammad, S.; Hussain, S.; Jung, J.; Algarni, H. Shedding light on the structural, optoelectronic, and thermoelectric properties of pyrochlore oxides (La2Q2O7 (Q = Ge, Sn)) for energy applications: A first-principles investigation. J. Solid State Chem. 2022, 313, 123305. [Google Scholar] [CrossRef]
- Kaliyaperumal, C.; Sankarakumar, A.; Palanisamy, J.; Paramasivam, T. Fluorite to pyrochlore phase transformation in nanocrystalline Nd2Zr2O7. Mater. Lett. 2018, 228, 493–496. [Google Scholar] [CrossRef]
- Mustafa, G.M.; Atiq, S.; Abbas, S.K.; Riaz, S.; Naseem, S. Tunable structural and electrical impedance properties of pyrochlores based Nd doped lanthanum zirconate nanoparticles for capacitive applications. Ceram. Int. 2018, 44, 2170–2177. [Google Scholar] [CrossRef]
- Antonova, E.; Tropin, E.; Khodimchuk, A. Alkaline earth–doped La2Zr2O7 oxides with a pyrochlore structure: Phase equilibria and electrical properties. Ionics 2022, 28, 5181–5188. [Google Scholar] [CrossRef]
- Kumar, A.; Manam, J. Observation of up conversion/down conversion luminescence and structural analysis of La2Zr2O7: Pr3+ nano phosphors. Mater. Sci. Semicond. Process. 2022, 148, 106828. [Google Scholar] [CrossRef]
- Lingjie, Z.; Tao, S.; Qianhong, S.; Ji, Z.; Lawson, C.; Xianping, F.; Hui, Y. Anti-arc erosion properties of Ag-La2Sn2O7/SnO2 contacts. Rare Met. Mater. Eng. 2016, 45, 1664–1668. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Haule, K.; Vanderbilt, D. Metal-insulator transition and topological properties of pyrochlore iridates. Phys. Rev. Lett. 2017, 118, 026404. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, N.; Xu, C.; Li, Y.; Zhu, H.; Zhu, P.; Wang, X.; Yang, W. Abnormal pressure-induced photoluminescence enhancement and phase decomposition in pyrochlore La2Sn2O7. Adv. Mater. 2017, 29, 1701513. [Google Scholar] [CrossRef] [PubMed]
- Rekhila, G.; Brahimi, R.; Bessekhouad, Y.; Trari, M. Physical and photoelectrochemical characterizations of the pyrochlore La1. 9Ba0. 1Sn2O7: Application to chromate reduction under solar light. J. Photochem. Photobiol. A Chem. 2017, 332, 345–350. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, J.; Fuentes, A.F.; Zhang, F.; Lang, M.; Lu, F.; Ewing, R.C. Enhanced radiation resistance of nanocrystalline pyrochlore Gd2(Ti0.65Zr0.35)2O7. Appl. Phys. Lett. 2009, 94, 243110. [Google Scholar] [CrossRef]
- Tong, Y.; Qian, X.; Zhao, W.; Lu, L. Synthesis and Catalytic Peoperties of TiO2/Nd2Zr2O7 Nanocomposites. J. Chin. Ceram. Soc. 2013, 41, 34–37. [Google Scholar]
- Chen, S.; Pan, B.; Zeng, L.; Luo, S.; Wang, X.; Su, W. La2Sn2O7 enhanced photocatalytic CO2 reduction with H2O by deposition of Au co-catalyst. RSC Adv. 2017, 7, 14186–14191. [Google Scholar] [CrossRef] [Green Version]
- Denisova, L.; Kargin, Y.F.; Denisov, V. Heat capacity of rare-earth stannates in the range 350–1000 K. Inorg. Mater. 2017, 53, 956–961. [Google Scholar] [CrossRef]
- Child, M.; Koskinen, O.; Linnanen, L.; Breyer, C. Sustainability guardrails for energy scenarios of the global energy transition. Renew. Sustain. Energy Rev. 2018, 91, 321–334. [Google Scholar] [CrossRef]
- Jacob, K.T.; Lwin, K.T.; Waseda, Y. System La-Pd-O: Phase diagram and thermodynamic properties of ternary oxides. Solid State Sci. 2002, 4, 205–215. [Google Scholar] [CrossRef]
- Duran, R.R.; Falsetti, P.E.; Muhr, L.; Privat, R.; Barth, D. Phase equilibrium study of the ternary system CO2+ H2O+ ethanol at elevated pressure: Thermodynamic model selection. Application to supercritical extraction of polar compounds. J. Supercrit. Fluids 2018, 138, 17–28. [Google Scholar] [CrossRef]
- Shibasaki, S.; Terasaki, I. Thermoelectric Properties of Layered Pd Oxide R2PdO4 (R = La, Nd, Sm, and Gd). J. Phys. Soc. Jpn. 2006, 75, 024705. [Google Scholar] [CrossRef] [Green Version]
- Attfield, J.P.; Férey, G. Structural correlations within the lanthanum palladium oxide family. J. Solid State Chem. 1989, 80, 286–298. [Google Scholar] [CrossRef]
- Abbas, Z.; Fatima, K.; Gorczyca, I.; Irfan, M.; Alotaibi, N.; Alshahrani, T.; Raza, H.H.; Muhammad, S. Proposition of new stable rare-earth ternary semiconductor sulfides of type LaTlS2 (La= Er, Eu, Tb): Ab-initio study and prospects for optoelectronic, spintronic and thermoelectric applications. Mater. Sci. Semicond. Process. 2022, 146, 106662. [Google Scholar] [CrossRef]
- Abbas, Z.; Fatima, K.; Abubakr, M.; Gorczyca, I.; Alshahrani, T.; Muhammad, S.; Al-Sehemi, A.G. A DFT+ U study of the effect of transition metal replacements on optoelectronic and elastic properties of TmCu3S4 (Tm = V, Ta, Nb). Optik 2022, 250, 168289. [Google Scholar] [CrossRef]
- Abbas, Z.; Jabeen, N.; Hussain, A.; Kabir, F.; Alshahrani, T.; Raza, H.H.; Muhammad, S.; Azam, S.; Gorczyca, I. Effect of Nb, Ta and V replacements on electronic, optical and elastic properties of NbCu3Se4: A GGA+ U study. J. Solid State Chem. 2021, 301, 122338. [Google Scholar] [CrossRef]
- Abubakr, M.; Fatima, K.; Abbas, Z.; Gorczyca, I.; Irfan, M.; Muhammad, S.; Khan, M.A.; Alarfaji, S.S. Study of structural, optoelectronic and magnetic properties of Half-Heusler compounds QEuPa (Q = Ba, be, Mg, Sr) using first-principles method. J. Solid State Chem. 2021, 304, 122612. [Google Scholar] [CrossRef]
- Azam, S.; Abbas, Z.; Gul, B.; Khan, M.S.; Irfan, M.; Sohail, M.; Khan, S.A.; Naseer, F.; Irfan, A.; Khan, G. First-principles calculations of optoelectronic properties of CaO: Eu+ 2 (SrO: Eu+ 2) for energy applications. Int. J. Mod. Phys. B 2018, 32, 1850333. [Google Scholar] [CrossRef]
- Abbas, Z.; Fatima, K.; Jaffery, S.H.A.; Ali, A.; Raza, H.H.; Muhammad, S.; Algarni, H.; Hussain, S.; Jung, J. Ab-initio study of Nb-based complex materials: A new class of materials for optoelectronic applications. J. Comput. Sci. 2022, 63, 101791. [Google Scholar] [CrossRef]
- Irfan, M.; Abbas, Z.; Khan, S.; Sohail, M.; Rani, M.; Azam, S.; Kityk, I. Effects of compressed strain on thermoelectric properties of Cu3SbSe4. J. Alloy. Compd. 2018, 750, 804–810. [Google Scholar] [CrossRef]
- Khan, A.A.; Reshak, A.H.; Zada, Z.; Saqib, M.; Abbas, Z.; Ismail, M.; Zada, S.; Murtaza, G.; Ali, S.; Laref, A. Thermoelectric, structural, electronic, magnetic, and thermodynamic properties of CaZn2Ge2 compound. Eur. Phys. J. Plus 2022, 137, 1–12. [Google Scholar] [CrossRef]
- Abbas, Z.; Fatima, K.; Gorczyca, I.; Jaffery, S.H.A.; Ali, A.; Irfan, M.; Raza, H.H.; Algarni, H.; Muhammad, S.; Teisseyre, H. First-principles calculations to investigate electronic, optical, and thermoelectric properties of Na2GeX3 (X = S, Se, Te) for energy applications. Mater. Sci. Semicond. Process. 2023, 154, 107206. [Google Scholar] [CrossRef]
- Petit, A.; Dulong, P. Study on the measurement of specific heat of solids. Ann. Chim. Phys. 1819, 10, 395. [Google Scholar]
- Abubakr, M.; Fatima, K.; Abbas, Z.; Hussain, A.; Jabeen, N.; Raza, H.H.; Chaib, Y.; Muhammad, S.; Siddeeg, S.M.; Gorczyca, I. Effect of S, Se and Te replacement on structural, optoelectronic and transport properties of SrXO4 (X = S, Se, Te) for energy applications: A first principles study. J. Solid State Chem. 2022, 305, 122689. [Google Scholar] [CrossRef]
- Azam, S.; Abbas, Z.; Bilal, Q.; Irfan, M.; Khan, M.A.; Naqib, S.; Khenata, R.; Muhammad, S.; Algarni, H.; Al-Sehemi, A.G. Effect of Fe doping on optoelectronic properties of CdS nanostructure: Insights from DFT calculations. Phys. B Condens. Matter 2020, 583, 412056. [Google Scholar] [CrossRef]
- Abbas, Z.; Munaf, S.; Azam, S.; Abubakar, M.; Irfan, M. First-principles Calculations of Optoelectronic Properties of Sn1-xInxA (A = S and Se) for Solar Cell Applications; EasyChair: Oxford, UK, 2020; pp. 2314–2516. [Google Scholar]
- Lin, G.; Coillet, A.; Chembo, Y.K. Nonlinear photonics with high-Q whispering-gallery-mode resonators. Adv. Opt. Photonics 2017, 9, 828–890. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a (Å) | (Å)3 | B (GPa) | B′ (Gpa) | ||
|---|---|---|---|---|---|
| La2Hf2O7 | 6.83 | 2145.42 | 173.39 | 4.38 | −190,874.76 |
| La2Zr2O7 | 6.85 | 2167.93 | 167.77 | 4.39 | −98,885.84 |
| Element | X | Y | Z |
|---|---|---|---|
| O1 | 0.625 | 0.625 | 0.625 |
| O2 | 0.375 | 0.375 | 0.375 |
| O3 | 0.332 | 0.918 | 0.918 |
| O4 | 0.082 | 0.668 | 0.668 |
| O5 | 0.918 | 0.332 | 0.918 |
| O6 | 0.918 | 0.918 | 0.332 |
| O7 | 0.332 | 0.332 | 0.918 |
| O8 | 0.332 | 0.918 | 0.332 |
| O9 | 0.918 | 0.332 | 0.332 |
| O10 | 0.668 | 0.668 | 0.082 |
| O11 | 0.082 | 0.082 | 0.668 |
| O12 | 0.668 | 0.082 | 0.082 |
| O13 | 0.668 | 0.082 | 0.668 |
| O14 | 0.082 | 0.668 | 0.082 |
| Hf1/Zr1 | 0.000 | 0.500 | 0.000 |
| Hf2/Zr2 | 0.500 | 0.000 | 0.000 |
| Hf3/Zr3 | 0.000 | 0.000 | 0.500 |
| Hf4/Zr4 | 0.000 | 0.000 | 0.000 |
| La1 | 0.500 | 0.000 | 0.500 |
| La2 | 0.000 | 0.500 | 0.500 |
| La3 | 0.500 | 0.500 | 0.000 |
| La4 | 0.500 | 0.500 | 0.500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbas, Z.; Hussain, S.; Muhammad, S.; Siddeeg, S.M.; Jung, J. A First-Principles Investigation on the Structural, Optoelectronic, and Thermoelectric Properties of Pyrochlore Oxides (La2Tm2O7 (Tm = Hf, Zr)) for Energy Applications. Int. J. Mol. Sci. 2022, 23, 15266. https://doi.org/10.3390/ijms232315266
Abbas Z, Hussain S, Muhammad S, Siddeeg SM, Jung J. A First-Principles Investigation on the Structural, Optoelectronic, and Thermoelectric Properties of Pyrochlore Oxides (La2Tm2O7 (Tm = Hf, Zr)) for Energy Applications. International Journal of Molecular Sciences. 2022; 23(23):15266. https://doi.org/10.3390/ijms232315266
Chicago/Turabian StyleAbbas, Zeesham, Sajjad Hussain, Shabbir Muhammad, Saifeldin M. Siddeeg, and Jongwan Jung. 2022. "A First-Principles Investigation on the Structural, Optoelectronic, and Thermoelectric Properties of Pyrochlore Oxides (La2Tm2O7 (Tm = Hf, Zr)) for Energy Applications" International Journal of Molecular Sciences 23, no. 23: 15266. https://doi.org/10.3390/ijms232315266