
Analysis of Intrinsic Breast Cancer Subtypes: The Clinical Utility of Epigenetic Biomarkers and TP53 Mutation Status in Triple-Negative Cases
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
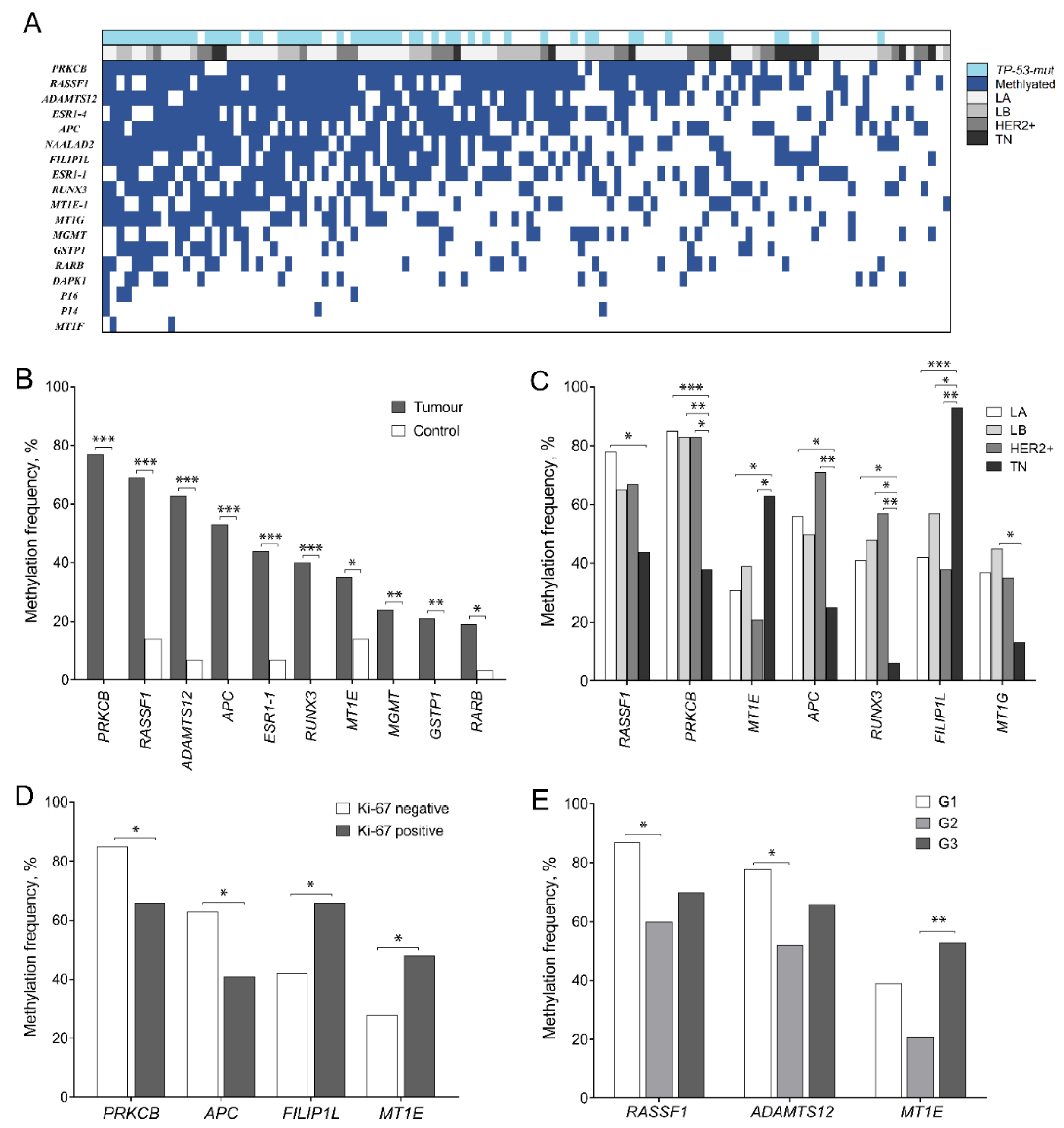
2.1. DNA Methylation Spectrum
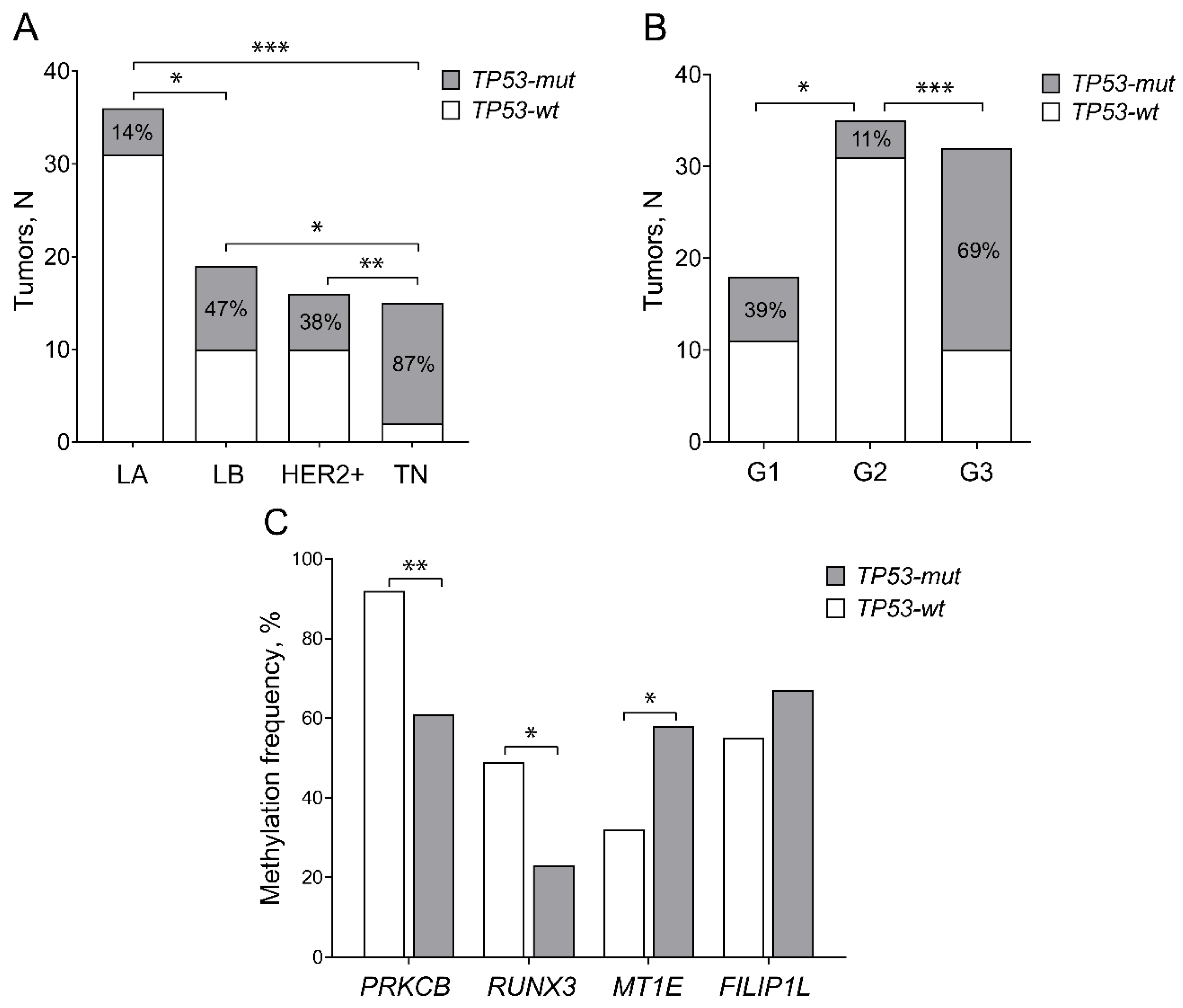
2.2. TP53 Mutation Spectrum
2.3. TP53 Mutations Predominate in TN BC Subtype
2.4. Prediction of Overall Survival
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. DNA Extraction
4.3. DNA Methylation Assay
4.4. TP53 Mutation Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 in breast cancer: Potential as a therapeutic target and biomarker. Breast Cancer Res. Treat. 2018, 170, 213–219. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor Supressive Functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Huszno, J.; Grzybowska, E. TP53 mutations and SNPs as prognostic and predictive factors in patients with breast cancer. Oncol. Lett. 2018, 16, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovic, J.; Ronneberg, J.A.; Tost, J.; Kristensen, V. The epigenetics of breast cancer. Mol. Oncol. 2010, 4, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Lo, P.K.; Sukumar, S. Epigenomics and breast cancer. Pharmacogenomics 2008, 9, 1879–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sporikova, Z.; Koudelakova, V.; Trojanec, R.; Hajduch, M. Genetic Markers in Triple-Negative Breast Cancer. Clin. Breast Cancer 2018, 18, e841–e850. [Google Scholar] [CrossRef]
- Holm, K.; Hegardt, C.; Staaf, J.; Vallon-Christersson, J.; Jonsson, G.; Olsson, H.; Borg, A.; Ringner, M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010, 12, R36. [Google Scholar] [CrossRef]
- Titus, A.J.; Way, G.P.; Johnson, K.C.; Christensen, B.C. Deconvolution of DNA methylation identifies differentially methylated gene regions on 1p36 across breast cancer subtypes. Sci. Rep. 2017, 7, 11594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunami, E.; Shinozaki, M.; Sim, M.; Nguyen, S.; Vu, A.; Giuliano, A.; Hoon, D. Estrogen receptor and HER2/neu status affect epigenetic differences of tumor-related genes in primary breast tumors. Breast Cancer Res. 2008, 10, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Turcan, S.; Rimner, A.; Kaufman, A.; Giri, D.; Morris, L.G.T.; Shen, R.; Seshan, V.; Mo, Q.; Heguy, A.; et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci. Transl. Med. 2011, 3, 75ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Temian, D.C.; Pop, L.A.; Irimie, A.J.; Berindan-Neagoe, I. The Epigenetics of Triple-Negative and Basal-Like Breast Cancer: Current Knowledge. J. Breast Cancer 2018, 21, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Pineda, B.; Diaz-Lagares, A.; Perez-Fidalgo, J.A.; Burgues, O.; Gonzalez-Barrallo, I.; Crujeiras, A.B.; Sandoval, J.; Esteller, M.; Lluch, A.; Eroles, P. A two-gene epigenetic signature for the prediction of response to neoadjuvant chemotherapy in triple-negative breast cancer patients. Clin. Epigenetics 2019, 11, 33. [Google Scholar] [CrossRef]
- Liu, X.P.; Hou, J.; Chen, C.; Guan, L.; Hu, H.K.; Li, S. A DNA Methylation-Based Panel for the Prognosis and Diagnosis of Patients with Breast Cancer and Its Mechanisms. Front. Mol. Biosci. 2020, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.J.; Stone, A.; Armstrong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Ma, Y.; Zhu, A.; Han, Y. An early biomarker and potential therapeutic target of RUNX3 hyper methylation in breast cancer, a system review and meta-analysis. Oncotarget 2017, 8, 22166–22174. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Kim, J.H.; Rybak, Y.; Luna, A.; Choi, C.H.; Chung, J.Y.; Hewitt, S.M.; Adem, A.; Tubridy, E.; Lin, J.; et al. Reduced expression of FILIP1L, a novel WNT pathway inhibitor, is associated with poor survival, progression and chemoresistance in ovarian cancer. Oncotarget 2016, 7, 77052–77070. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Jung, S.; Jung, T.; Moon, K.; Kim, I.; Jeong, Y.; Jin, S.; Pei, J.; Wen, M.; Jang, W. Role of metallothionein 1E in the migration and invasion of human glioma cell lines. Int. J. Oncol. 2012, 41, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.P.; Zhang, X.M.; Zhang, Z.; Zheng, L.H.; Jindal, S.; Liu, Y.J. Association of p53 expression with poor prognosis in patients with triple-negative breast invasive ductal carcinoma. Medicine 2019, 98, e15449. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T. p53 alterations in human cancer: More questions than answers. Oncogene 2007, 26, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Muggerud, A.A.; Vu, P.; Due, E.U.; Sorlie, T.; Borresen-Dale, A.L.; Warnberg, F.; Langerod, A. Full sequencing of TP53 identifies identical mutations within in situ and invasive components in breast cancer suggesting clonal evolution. Mol. Oncol. 2009, 3, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbandi, A.; Nguyen, H.D.; Jackson, J.G. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer 2020, 6, 98–110. [Google Scholar] [CrossRef]
- Daniunaite, K.; Bakavicius, A.; Zukauskaite, K.; Rauluseviciute, I.; Lazutka, J.R.; Ulys, A.; Jankevicius, F.; Jarmalaite, S. Promoter Methylation of PRKCB, ADAMTS12, and NAALAD2 Is Specific to Prostate Cancer and Predicts Biochemical Disease Recurrence. Int. J. Mol. Sci. 2021, 22, 6091. [Google Scholar] [CrossRef]
- Serenaite, I.; Daniunaite, K.; Jankevicius, F.; Laurinavicius, A.; Petroska, D.; Lazutka, J.R.; Jarmalaite, S. Heterogeneity of DNA methylation in multifocal prostate cancer. Virchows Arch. 2015, 466, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Maleckaite, R.; Zalimas, A.; Bakavicius, A.; Jankevicius, F.; Jarmalaite, S.; Daniunaite, K. DNA methylation of metallothionein genes is associated with the clinical features of renal cell carcinoma. Oncol. Rep. 2019, 41, 3535–3544. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.C.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Holmila, R.; Husgafvel-Pursiainen, K. Analysis of TP53 gene mutations in human lung cancer: Comparison of capillary electrophoresis single strand conformation polymorphism assay with denaturing gradient gel electrophoresis and direct sequencing. Cancer Detect. Prev. 2006, 38, 1–6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Characteristics | TP53-mut, N (%) | TP53-wt, N (%) | p-Value |
|---|---|---|---|---|
| BC patients, N | 33 | 53 | ||
| Median age at BC diagnosis | yrs. (IQR) | 55 (24) | 61 (19) | 0.377 |
| Histological type | Ductal; N (%) | 32 (38) | 46 (55) | 0.396 |
| Lobular; N (%) | 1 (1) | 5 (6) | ||
| Pathological stage | T1; N (%) | 15 (17) | 24 (28) | 1.0 |
| T2; N (%) | 18 (21) | 29 (34) | ||
| Lymph node metastasis | No (N0); N (%) | 21 (25) | 25 (29) | 0.186 |
| Yes (N1); N (%) | 12 (14) | 27 (32) | ||
| Tumor differentiation grade | G1; N (%) | 7 (8) | 11 (13) | (G1 vs. G2) 0.032 (G1 vs. G3) 0.072. (G2 vs. G3) < 0.0001 |
| G2; N (%) | 4 (5) | 31 (36) | ||
| G3; N (%) | 22 (26) | 10 (12) | ||
| Intrinsic BC subtype | LA; N (%) | 5 (6) | 31 (36) | (LA vs. LB) 0.010 (LA vs. HER2+) 0.073 (LA vs. TN) < 0.0001 (LB vs. HER2+) 0.734 (LB vs. TN) 0.030 (HER2 + vs. TN) 0.010 |
| LB; N (%) | 9 (10) | 10 (12) | ||
| Her2+; N (%) | 6 (7) | 10 (12) | ||
| TN; N (%) | 13 (15) | 2 (2) | ||
| ER status | Negative; N (%) | 15 (18) | 4 (5) | <0.0001 |
| Positive; N (%) | 18 (21) | 48 (56) | ||
| PR status | Negative; N (%) | 16 (19) | 12 (14) | 0.018 |
| Positive; N (%) | 17 (20) | 41 (48) | ||
| HER2 status | Negative; N (%) | 28 (33) | 43 (50) | 0.775 |
| Positive; N (%) | 5 (6) | 10 (12) | ||
| Ki-67 | Negative; N (%) | 8 (9) | 39 (46) | <0.0001 |
| Positive; N (%) | 24 (28) | 14 (16) | ||
| Survival | Remission; N (%) | 18 (31) | 24 (41) | 0.773 |
| Death; N (%) | 6 (10) | 10 (17) |
| Covariate | Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|---|
| HR (95% CI) | p-Value | HR (95% CI) | p-Value | ||
| Methylated: yes/no | FILIP1L | 3.33 (0.92−11.97) | 0.067 | ||
| P16 | 3.14 (0.72−13.67) | 0.128 | |||
| RUNX3 | 2.07 (0.7−5.50) | 0.147 | 4.64 (1.15−18.75) | 0.032 | |
| P14 | 2.14 (0.29−16.03) | 0.462 | |||
| MT1F | 2.02 (0.27−15.15) | 0.497 | |||
| ADAMTS12 | 1.42 (0.51−3.98) | 0.502 | |||
| DAPK1 | 1.34 (0.39−4.62) | 0.643 | |||
| MT1E | 1.20 (0.47−3.11) | 0.704 | |||
| NAALAD2 | 1.15 (0.43−3.11) | 0.779 | |||
| Clinical–pathological charact. | Age (cont.) | 1.04 (1.00−1.09) | 0.049 | 1.07 (1.02−1.12) | 0.010 |
| T (1 vs. 2) | 1.06 (0.42−2.69) | 0.897 | |||
| N (yes/no) | 2.15 (0.84−5.53) | 0.113 | |||
| G (≤2 vs. 3) | 1.55 (0.62−3.88) | 0.357 | |||
| TN subtype (yes/no) | 2.91 (1.04−8.18) | 0.044 | 13.92 (2.97−65.20) | 0.010 | |
| Molecular biomarkers (high/low): | |||||
| ER | 0.61 (0.22−1.71) | 0.349 | |||
| PR | 0.68 (0.26−1.74) | 0.422 | |||
| HER2 | 0.25 (0.03−1.86) | 0.177 | |||
| Ki-67 | 1.02 (0.40−2.58) | 0.963 | |||
| TP53-mut (yes/no) | 0.95 (0.34−2.65) | 0.919 | |||
| BC Features | BC Subtypes | |||||
|---|---|---|---|---|---|---|
| LA | LB | HER2+ * | TN | Total BC Cases | ||
| N = 55 (%) | N = 24 (%) | N = 21 (%) | N = 16 (%) | N = 116 (%) | ||
| Median age at BC diagnosis | yrs. (IQR) | 61 (19) | 58 (22) | 55 (23) | 52 (26) | |
| Histology | Ductal; N (%) | 45 (82) | 22 (92) | 18 (86) | 16 (100) | 101 (87) |
| Lobular; N (%) | 8 (15) | 2 (8) | 3 (14) | 0 (0) | 13 (11) | |
| Apocrine; N (%) | 2 (3) | 0 (0) | 0 (0) | 0 (0) | 2 (2) | |
| Tumor stage | T1; N (%) | 33 (60) | 13 (54) | 11 (52) | 6 (38) | 63 (54) |
| T2; N (%) | 22 (40) | 11 (46) | 10 (48) | 10 (63) | 53 (46) | |
| Spread to lymph nodes | N0; N (%) | 33 (54) | 13 (54) | 11 (52) | 10 (63) | 67 (58) |
| N1; N (%) | 21 (61) | 11 (46) | 10 (48) | 6 (38) | 48 (42) | |
| Grade | G1; N (%) | 16 (29) | 3 (13) | 2 (10) | 2 (13) | 23 (20) |
| G2; N (%) | 35 (64) | 7 (29) | 9 (43) | 1 (6) | 52 (45) | |
| G3; N (%) | 4 (7) | 14 (58) | 10 (48) | 13 (81) | 41 (35) | |
| Ki-67 expression | Ki-67 neg; N (%) | 53 (96) | 0 (0) | 15 (71) | 0 (0) | 68 (59) |
| Ki-67 pos; N (%) | 2 (4) | 24 (100) | 6 (29) | 16 (100) | 48 (41) | |
| Survival (N = 78) | Remission; N (%) | 25 (71) | 13 (68) | 12 (92) | 6 (55) | 56 (72) |
| Death; N (%) | 10 (29) | 5 (26) | 1 (8) | 5 (45) | 21 (27) | |
| Relapse; N (%) | 0 (0) | 1 (5) | 0 (0) | 0 (0) | 1 (1) | |
| 5-year survival (%) | 25 (71) | 16 (94) | 11 (92) | 6 (55) | 58 (74) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadzeviciene, I.; Snipaitiene, K.; Scesnaite-Jerdiakova, A.; Daniunaite, K.; Sabaliauskaite, R.; Laurinaviciene, A.; Drobniene, M.; Ostapenko, V.; Jarmalaite, S. Analysis of Intrinsic Breast Cancer Subtypes: The Clinical Utility of Epigenetic Biomarkers and TP53 Mutation Status in Triple-Negative Cases. Int. J. Mol. Sci. 2022, 23, 15429. https://doi.org/10.3390/ijms232315429
Sadzeviciene I, Snipaitiene K, Scesnaite-Jerdiakova A, Daniunaite K, Sabaliauskaite R, Laurinaviciene A, Drobniene M, Ostapenko V, Jarmalaite S. Analysis of Intrinsic Breast Cancer Subtypes: The Clinical Utility of Epigenetic Biomarkers and TP53 Mutation Status in Triple-Negative Cases. International Journal of Molecular Sciences. 2022; 23(23):15429. https://doi.org/10.3390/ijms232315429
Chicago/Turabian StyleSadzeviciene, Ieva, Kristina Snipaitiene, Asta Scesnaite-Jerdiakova, Kristina Daniunaite, Rasa Sabaliauskaite, Aida Laurinaviciene, Monika Drobniene, Valerijus Ostapenko, and Sonata Jarmalaite. 2022. "Analysis of Intrinsic Breast Cancer Subtypes: The Clinical Utility of Epigenetic Biomarkers and TP53 Mutation Status in Triple-Negative Cases" International Journal of Molecular Sciences 23, no. 23: 15429. https://doi.org/10.3390/ijms232315429