
Insights into the Potential Mechanisms of JAK2V617F Somatic Mutation Contributing Distinct Phenotypes in Myeloproliferative Neoplasms


Abstract
:1. Introduction
1.1. Myeloproliferative Neoplasms
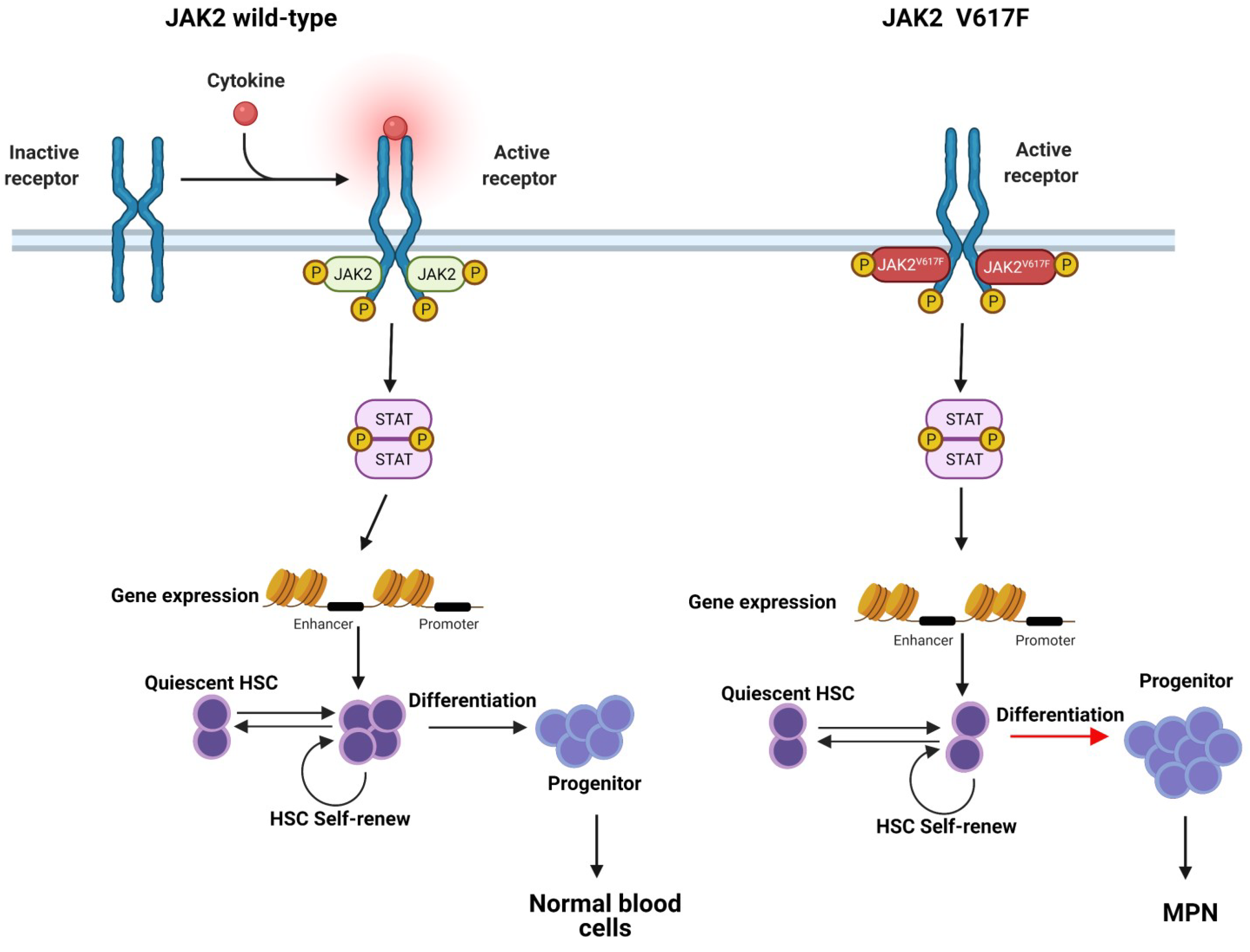
1.2. JAK-STAT Pathway
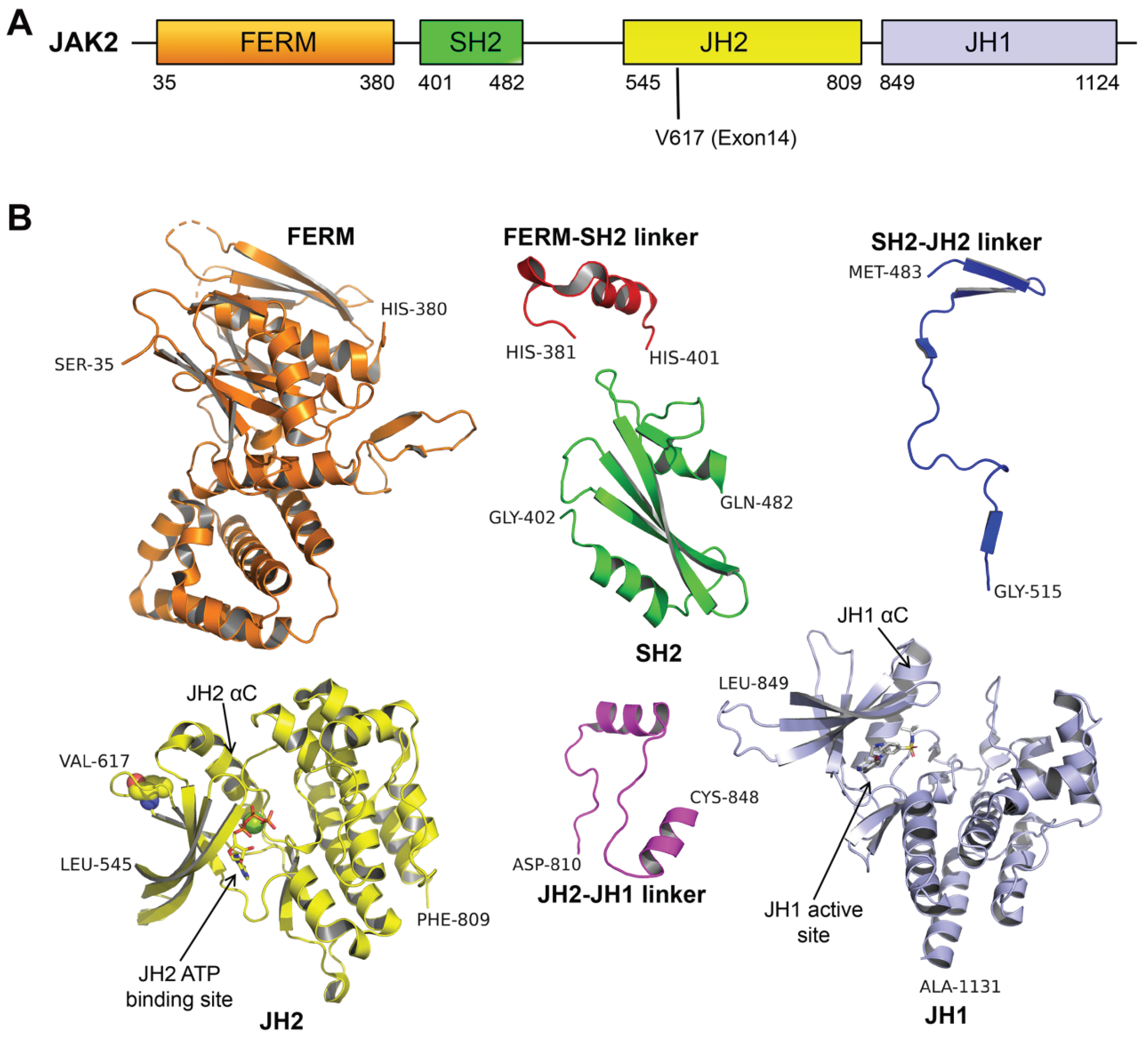
2. The Role of JAK2V617F Contributes to MPN: Basis in Structural Biology
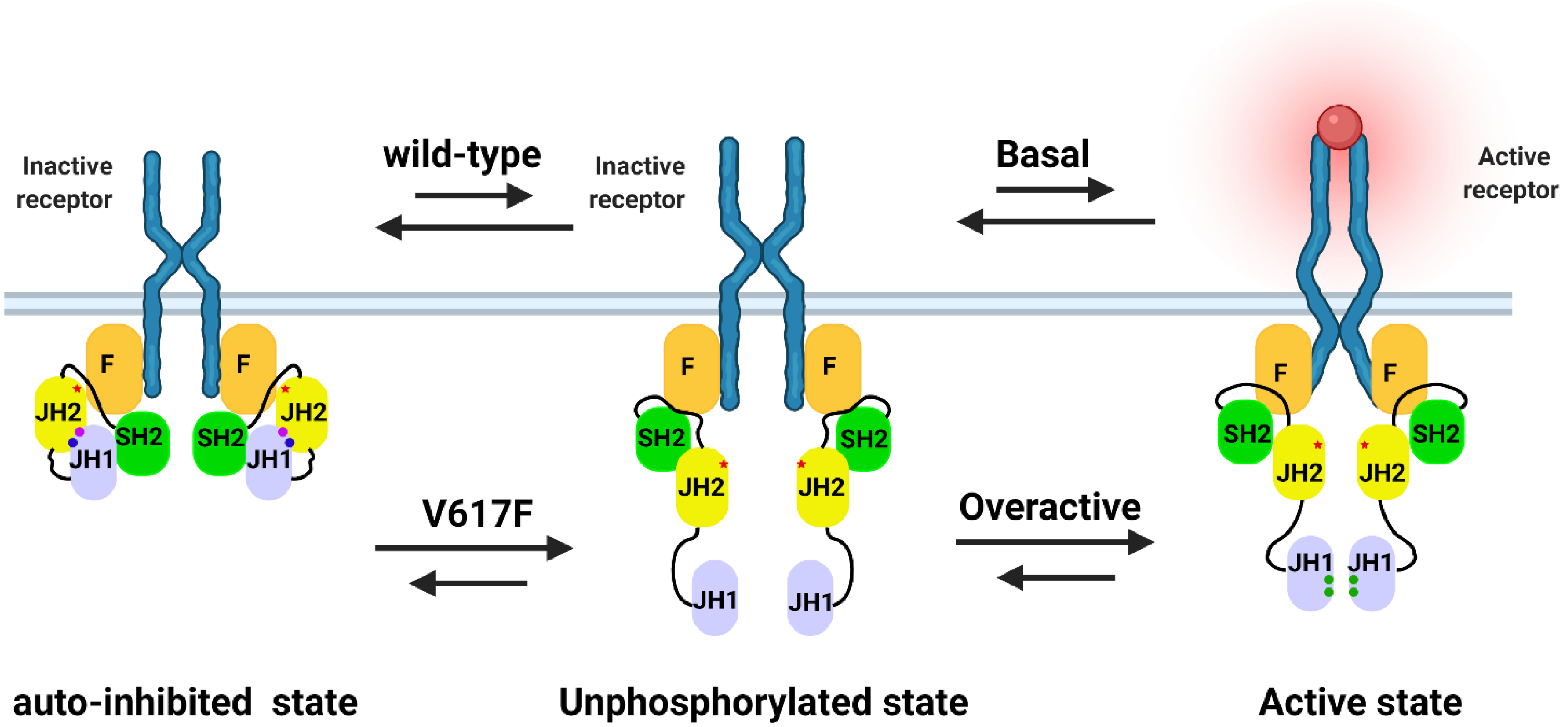
2.1. The Potential Mechanism of JAK2 Auto-Inhibition
2.2. The Potential Mechanism of JAK2V617F Hyper-Activation
3. The Role of JAK2V617F in Contributing to the MPN Phenotype

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Comments | |
|---|---|---|
| JAK2V617F allele burden |
|
|
| ET: allele burden > 50%. n = 165 [55] | Higher frequency of arterial thrombosis and splenomegaly | |
| PV: allele burden > 50%. n = 43 [56] | More severe disease status (higher HCT, RBC, HGB, and WBC) | |
| MPN: homozygous. heter = 45 homo = 13 [57] | Higher hemoglobin, increased incidence of pruritus, higher rate of fibrotic transformation | |
| Distinct signaling | RAS-ERK and phosphatidylinositol 3 kinase-AKT pathways | Dysregulated erythropoiesis in PV |
| IRS2 [58] | Increased cell viability and reduced apoptosis in JAK2-mutated cells | |
| IGF1R inhibitory [59] | Prevent the hematological disease in Jak2V617F mutation mice | |
| NT157 inhibits IRS1/2 and STAT3/5 [60] | ||
|
| |
| Epigenetic modifiers | TET2, ASXL1, IDH1, IDH2, IKZF1 and EZH2 | MPN-associated mutations |
| TET2 12%, ASXL1 5%, DNMT3a 5%, EZH2 ~3% and IDH1 ~1.5% [62] | MPN | |
|
| |
| TET2 in different phenotype of MPN | |
| Immune response | TNFa, IFNα, and IFNg pathways [66] | MPN development |
| IFNα [67] | JAK2V617F increase molecular responses to | |
| TNFα [68] | Promote expansion of JAK2V617F cells in MPN | |
| ROS and inflammatory factors [69] | influence MPN progression | |
| Effect of lifestyle | Smoking [70] | Increase the risk of MPN |
| Coffee consumption [71] | Inversely associated with the risk of PV | |
| Mediterranean dietary [72] | Decrease symptom burden in MPN | |
| Obesity [73] | Elevate overall risk for MPN especially with ET |
3.1. JAK2V617F Mutation Allele Burden
3.2. Distinct Signaling in JAK2V617F MPN Phenotype
3.3. Epigenetic Modifiers in JAK2V617F MPN Phenotype
3.4. Immune Response in JAK2V617F MPN Phenotype
3.5. Effect of Lifestyle in JAK2V617F MPN
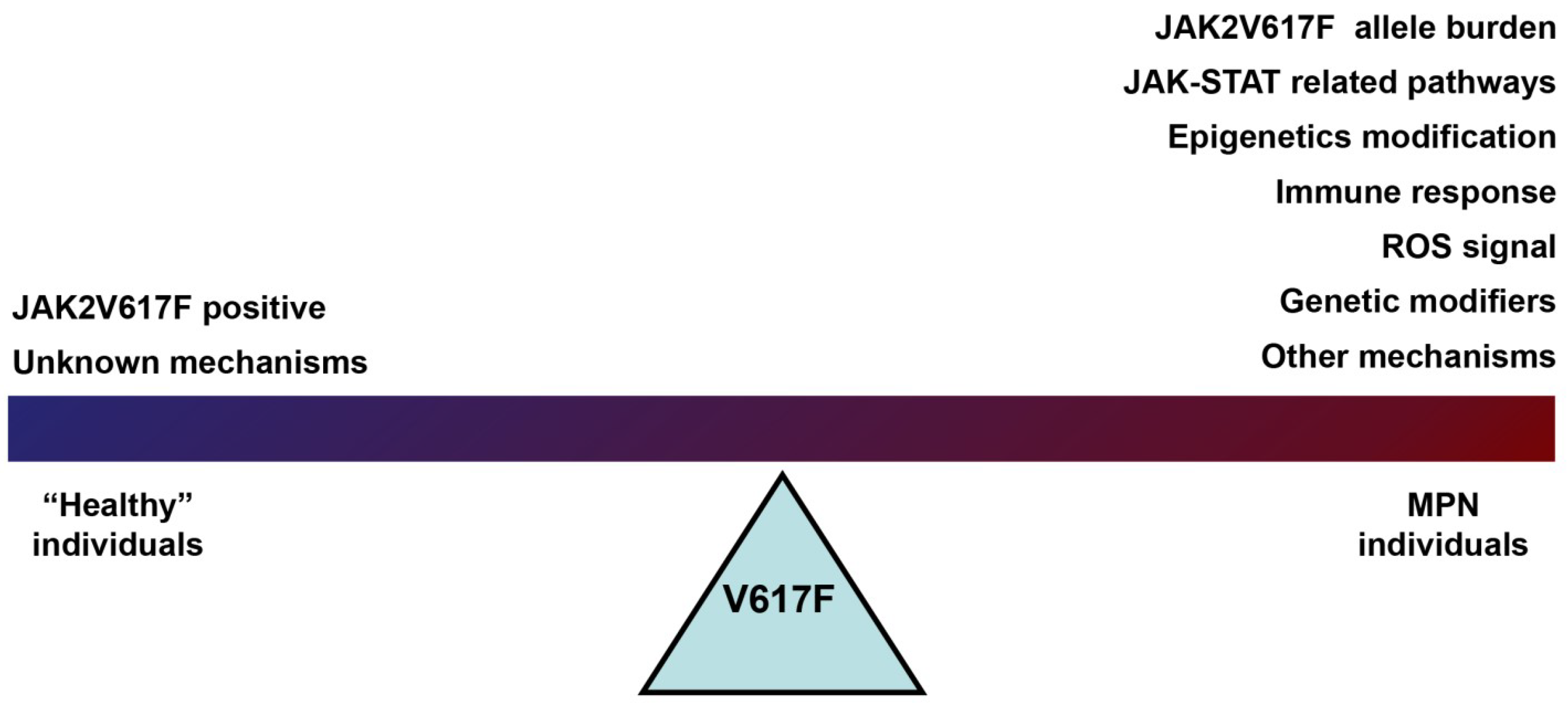
4. Summary and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dameshek, W. Some speculations on the myeloproliferative syndromes. Blood 1951, 6, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Thiele, J.; Vardiman, J.W. The 2008 World Health Organization classification system for myeloproliferative neoplasms: Order out of chaos. Cancer 2009, 115, 3842–3847. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 4, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Crisà, E.; Venturino, E.; Passera, R.; Prina, M.; Schinco, P.; Borchiellini, A.; Giai, V.; Ciocca Vasino, M.A.; Bazzan, M.; Vaccarino, A.; et al. A retrospective study on 226 polycythemia vera patients: Impact of median hematocrit value on clinical outcomes and survival improvement with anti-thrombotic prophylaxis and non-alkylating drugs. Ann. Hematol. 2010, 89, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Colafigli, G.; Scalzulli, E.; Pepe, S.; Di Prima, A.; Efficace, F.; Martelli, M.; Foà, R.; Breccia, M. The advantages and risks of ruxolitinib for the treatment of polycythemia vera. Expert Rev. Hematol. 2020, 13, 1067–1072. [Google Scholar] [CrossRef]
- Lengfelder, E.; Berger, U.; Hehlmann, R. Interferon α in the treatment of polycythemia vera. Ann. Hematol. 2000, 79, 103–109. [Google Scholar] [CrossRef]
- Cortelazzo, S. Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J. Clin. Oncol. 1990, 8, 556–562. [Google Scholar] [CrossRef]
- Fenaux, P.; Simon, M.; Caulier, M.T.; Lai, J.L.; Goudemand, J.; Bauters, F. Clinical course of essential thrombocythemia in 147 cases. Cancer 1990, 66, 549–556. [Google Scholar] [CrossRef]
- Tefferi, A.; Fonseca, R.; Pereira, D.L.; Clark Hoagland, H. A long-term retrospective study of young women with essential thrombocythemia. Mayo Clin. Proc. 2001, 76, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; Vannucchi, A.M.; Antonioli, E.; et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: An international study of 891 patients. Blood 2011, 117, 5857–5859. [Google Scholar] [CrossRef]
- Oh, S.T.; Gotlib, J. JAK2 V617F and beyond: Role of genetics and aberrant signaling in the pathogenesis of myeloproliferative neoplasms. Expert Rev. Hematol. 2010, 3, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Briére, J.B. Essential thrombocythemia. Orphanet J. Rare Dis. 2007, 2, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes, F.; Passamonti, F.; Barosi, G.; San Matteo, P. Life expectancy and prognostic factors in the classic BCR/ABL-negative myeloproliferative disorders. Leukemia 2008, 22, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International working group for myelofibrosis research and treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Barosi, G.; Mesa, R.A.; Thiele, J.; Cervantes, F.; Campbell, P.J.; Verstovsek, S.; Dupriez, B.; Levine, R.L.; Passamonti, F.; Gotlib, J.; et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A consensus statement from the international working group for myelofibrosis research and treatment. Leukemia 2008, 22, 437–438. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 1551–1560. [Google Scholar] [CrossRef] [Green Version]
- Ayalew Tefferi, W.V. Myeloproliferative neoplasms: Molecular pathophysiology, essential clinical understanding, and treatment strategies. J. Clin. Oncol. 2011, 29, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Barosi, G.; Birgegard, G.; Cervantes, F.; Finazzi, G.; Griesshammer, M.; Harrison, C.; Hasselbalch, H.C.; Hehlmann, R.; Hoffman, R.; et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European leukemiaNet. J. Clin. Oncol. 2011, 29, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 domains: Bringing specificity to JAK-receptor interactions. Front. Endocrinol. (Lausanne) 2017, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Toms, A.V.; Deshpande, A.; McNally, R.; Jeong, Y.; Rogers, J.M.; Kim, C.U.; Gruner, S.M.; Ficarro, S.B.; Marto, J.A.; Sattler, M.; et al. Structure of a pseudokinase-domain switch that controls oncogenic activation of Jak kinases. Nat. Struct. Mol. Biol. 2013, 20, 1221–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, M.D.; Kuo, F.I.; Smith, P.A. Targeting the Janus Kinase Family in Autoimmune Skin Diseases. Front. Immunol. 2019, 10, 2342. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.K.; Bamert, R.S.; Patel, O.; Wang, C.; Walden, P.M.; Wilks, A.F.; Fantino, E.; Rossjohn, J.; Lucet, I.S. Dissecting specificity in the Janus kinases: The structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J. Mol. Biol. 2009, 387, 219–232. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in Immunity, Immunodeficiency, and Cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Prchal, J.F.; Axelrad, A.A. Letter: Bone-marrow responses in polycythemia vera. N. Engl. J. Med. 1974, 290, 1382. [Google Scholar] [CrossRef]
- Argetsinger, L.S.; Kouadio, J.-L.K.; Steen, H.; Stensballe, A.; Jensen, O.N.; Carter-Su, C. Autophosphorylation of JAK2 on Tyrosines 221 and 570 Regulates Its Activity. Mol. Cell. Biol. 2004, 24, 4955–4967. [Google Scholar] [CrossRef] [Green Version]
- Feener, E.P.; Rosario, F.; Dunn, S.L.; Stancheva, Z.; Myers, M.G. Tyrosine Phosphorylation of Jak2 in the JH2 Domain Inhibits Cytokine Signaling. Mol. Cell. Biol. 2004, 24, 4968–4978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida-Takahashi, R.; Rosario, F.; Gong, Y.; Kopp, K.; Stancheva, Z.; Chen, X.; Feener, E.P.; Myers, M.G. Phosphorylation of Jak2 on Ser523 InhibitsJak2-Dependent Leptin ReceptorSignaling. Mol. Cell. Biol. 2006, 26, 4063–4073. [Google Scholar] [CrossRef] [Green Version]
- Mazurkiewicz-Munoz, A.M.; Argetsinger, L.S.; Kouadio, J.-L.K.; Stensballe, A.; Jensen, O.N.; Cline, J.M.; Carter-Su, C. Phosphorylation of JAK2 at Serine 523: A Negative Regulator of JAK2 That Is Stimulated by GrowthHormone and Epidermal Growth Factor. Mol. Cell. Biol. 2006, 26, 4052–4062. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Gnanasambandan, K.; Ungureanu, D.; Kim, E.T.; Hammarén, H.; Yamashita, K.; Silvennoinen, O.; Shaw, D.E.; Hubbard, S.R. Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat. Struct. Mol. Biol. 2014, 21, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Witthuhn, B.A.; Matsuda, T.; Kohlhuber, F.; Kerr, I.M.; Ihle, J.N. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol. Cell. Biol. 1997, 17, 2497–2501. [Google Scholar] [CrossRef] [Green Version]
- McNally, R.; Li, Q.; Li, K.; Dekker, C.; Vangrevelinghe, E.; Jones, M.; Chène, P.; MacHauer, R.; Radimerski, T.; Eck, M.J. Discovery and Structural Characterization of ATP-Site Ligands for the Wild-Type and V617F Mutant JAK2 Pseudokinase Domain. ACS Chem. Biol. 2019, 14, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Ultsch, M.; Wallweber, H.; Kohli, P.B.; Johnson, A.R.; Eigenbrot, C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 8025–8030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, S.R. Mechanistic insights into regulation of JAK2 tyrosine kinase. Front. Endocrinol. (Lausanne) 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, L.N.; Ungureanu, D.; Liau, N.P.D.; Young, S.N.; Laktyushin, A.; Hammaren, H.; Lucet, I.S.; Nicola, N.A.; Silvennoinen, O.; Babon, J.J.; et al. Mechanistic insights into activation and SOCS3-mediated inhibition of myeloproliferative neoplasm-associated JAK2 mutants from biochemical and structural analyses. Biochem. J. 2014, 458, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Ayaz, P.; Hammarén, H.M.; Raivola, J.; Sharon, D.; Hubbard, S.R.; Silvennoinen, O.; Shan, Y.; Shaw, D.E. Structural models of full-length JAK2 kinase. bioRxiv 2019, 3, 727727. [Google Scholar] [CrossRef]
- Leroy, E.; Dusa, A.; Colau, D.; Motamedi, A.; Cahu, X.; Mouton, C.; Huang, L.J.; Shiau, A.K.; Constantinescu, S.N. Uncoupling JAK2 V617F activation from cytokine-induced signalling by modulation of JH2 αC helix. Biochem. J. 2016, 473, 1579–1591. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Zhang, C.; Sarmiento, J.; Standley, D.M. Protein loop modeling with optimized backbone potential functions. J. Chem. Theory Comput. 2012, 8, 1820–1827. [Google Scholar] [CrossRef]
- Nagar, B.; Hantschel, O.; Young, M.A.; Scheffzek, K.; Veach, D.; Bornmann, W.; Clarkson, B.; Superti-Furga, G.; Kuriyan, J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 2003, 112, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Seeliger, M.A.; Eastwood, M.P.; Frank, F.; Xu, H.; Jensen, M.; Dror, R.O.; Kuriyan, J.; Shaw, D.E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrao, R.D.; Wallweber, H.J.A.; Lupardus, P.J. Receptor-mediated dimerization of JAK2 FERM domains is required for JAK2 activation. Elife 2018, 7, e38089. [Google Scholar] [CrossRef] [PubMed]
- De Vos, A.M.; Ultsch, M.; Kossiakoff, A.A. Human growth hormone and extracellular domain of its receptor: Crystal structure of the complex. Science 1992, 255, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Syed, R.S.; Reid, S.W.; Li, C.; Cheetham, J.C.; Aoki, K.H.; Liu, B.; Zhan, H.; Osslund, T.D.; Chirino, A.J.; Zhang, J.; et al. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature 1998, 395, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Dusa, A.; Mouton, C.; Pecquet, C.; Herman, M.; Constantinescu, S.N. JAK2 V617F constitutive activation requires JH2 residue F595: A pseudokinase domain target for specific inhibitors. PLoS ONE 2010, 5, e11157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Dong, H.; Zhang, C.C.; Kinch, L.; Osawa, M.; Iacovino, M.; Grishin, N.V.; Kyba, M.; Huang, L.J.S. A JAK2 interdomain linker relays epo receptor engagement signals to kinase activation. J. Biol. Chem. 2009, 284, 26988–26998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnanasambandan, K.; Magis, A.; Sayeski, P.P. The constitutive activation of Jak2-V617F is mediated by a π stacking mechanism involving Phenylalanines 595 and 617. Biochemistry 2010, 49, 9972–9984. [Google Scholar] [CrossRef] [Green Version]
- Anand, S.; Stedham, F.; Beer, P.; Gudgin, E.; Ortmann, C.A.; Bench, A.; Erber, W.; Green, A.R.; Huntly, B.J.P. Effects of the JAK2 mutation on the hematopoietic stem and progenitor compartment in human myeloproliferative neoplasms. Blood 2011, 118, 177–181. [Google Scholar] [CrossRef]
- James, C.; Mazurier, F.; Dupont, S.; Chaligne, R.; Lamrissi-Garcia, I.; Tulliez, M.; Lippert, E.; Marion, F.X.; Pasquet, J.M.; Etienne, G.; et al. The hematopoietic stem cell compartment of JAK2V617F-positive myeloproliferative disorders is a reflection of disease heterogeneity. Blood 2008, 112, 2429–2438. [Google Scholar] [CrossRef] [Green Version]
- Larsen, T.S.; Christensen, J.H.; Hasselbalch, H.C.; Pallisgaard, N. The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders. Br. J. Haematol. 2007, 136, 745–751. [Google Scholar] [CrossRef]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F Expression Causes a Lethal Myeloproliferative Neoplasm with Differential Effects on Hematopoietic Stem and Progenitor Cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Kent, D.G.; Godfrey, A.L.; Manning, H.; Nangalia, J.; Aziz, A.; Chen, E.; Saeb-Parsy, K.; Fink, J.; Sneade, R.; et al. JAK2V617F homozygosity drives a phenotypic switch in myeloproliferative neoplasms, but is insufficient to sustain disease. Blood 2014, 123, 3139–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, D.G.; Li, J.; Tanna, H.; Fink, J.; Kirschner, K.; Pask, D.C.; Silber, Y.; Hamilton, T.L.; Sneade, R.; Simons, B.D.; et al. Self-Renewal of Single Mouse Hematopoietic Stem Cells Is Reduced by JAK2V617F Without Compromising Progenitor Cell Expansion. PLoS Biol. 2013, 11, e100157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007, 110, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, E.; Guglielmelli, P.; Poli, G.; Bogani, C.; Pancrazzi, A.; Longo, G.; Ponziani, V.; Tozzi, L.; Pieri, L.; Santini, V.; et al. Influence of JAK2V617F allele burden on phenotype in essential thrombocythemia. Haematologica 2008, 93, 41–48. [Google Scholar] [CrossRef]
- Michiels, J.J.; Berneman, Z.; Schroyens, W.; De Raeve, H. Changing concepts of diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms: From Dameshek 1950 to Vainchenker 2005 and beyond. Acta Haematol. 2015, 133, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Tiedt, R.; Hao-Shen, H.; Sobas, M.A.; Looser, R.; Dirnhofer, S.; Schwaller, J.; Skoda, R.C. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008, 111, 3931–3940. [Google Scholar] [CrossRef]
- de Melo Campos, P.; Machado-Neto, J.A.; Eide, C.A.; Savage, S.L.; Scopim-Ribeiro, R.; da Silva Souza Duarte, A.; Favaro, P.; Lorand-Metze, I.; Costa, F.F.; Tognon, C.E.; et al. IRS2 silencing increases apoptosis and potentiates the effects of ruxolitinib in JAK2V617F-positive myeloproliferative neoplasms. Oncotarget 2016, 7, 6948–6959. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Soria-Valles, C.; Santiago-Fernández, O.; Bernal, T.; Mittelbrunn, M.; Colado, E.; Rodríguez, F.; Bonzon-Kulichenko, E.; Vázquez, J.; Porta-De-La-Riva, M.; et al. Loss of the proteostasis factor AIRAPL causes myeloid transformation by deregulating IGF-1 signaling. Nat. Med. 2016, 22, 91–96. [Google Scholar] [CrossRef]
- Fenerich, B.A.; Fernandes, J.C.; Rodrigues Alves, A.P.N.; Coelho-Silva, J.L.; Scopim-Ribeiro, R.; Scheucher, P.S.; Eide, C.A.; Tognon, C.E.; Druker, B.J.; Rego, E.M.; et al. NT157 has antineoplastic effects and inhibits IRS1/2 and STAT3/5 in JAK2(V617F)-positive myeloproliferative neoplasm cells. Signal Transduct. Target. Ther. 2020, 5, 5. [Google Scholar] [CrossRef]
- Oku, S.; Takenaka, K.; Kuriyama, T.; Shide, K.; Kumano, T.; Kikushige, Y.; Urata, S.; Yamauchi, T.; Iwamoto, C.; Shimoda, H.K.; et al. JAK2 V617F uses distinct signalling pathways to induce cell proliferation and neutrophil activation: Research paper. Br. J. Haematol. 2010, 150, 334–344. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Pardanani, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009, 23, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef]
- Koschmieder, S.; Chatain, N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev. 2020, 42, 100711. [Google Scholar] [CrossRef]
- Czech, J.; Cordua, S.; Weinbergerova, B.; Baumeister, J.; Crepcia, A.; Han, L.; Maié, T.; Costa, I.G.; Denecke, B.; Maurer, A.; et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia 2019, 33, 995–1010. [Google Scholar] [CrossRef]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. Smoking as a contributing factor for development of polycythemia vera and related neoplasms. Leuk. Res. 2015, 39, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Podoltsev, N.A.; Wang, X.; Wang, R.; Hofmann, J.N.; Liao, L.M.; Zeidan, A.M.; Mesa, R.; Ma, X. Lifestyle factors and risk of myeloproliferative neoplasms in the NIH-AARP diet and health study. Int. J. Cancer 2020, 147, 948–957. [Google Scholar] [CrossRef]
- Martínez-González, M.A.; García-Arellano, A.; Toledo, E.; Salas-Salvadó, J.; Buil-Cosiales, P.; Corella, D.; Covas, M.I.; Schröder, H.; Arós, F.; Gómez-Gracia, E.; et al. A 14-item Mediterranean diet assessment tool and obesity indexes among high-risk subjects: The PREDIMED trial. PLoS ONE 2012, 7, e43134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncombe, A.S.; Anderson, L.A.; James, G.; de Vocht, F.; Fritschi, L.; Mesa, R.; Clarke, M.; McMullin, M.F. Modifiable Lifestyle and Medical Risk Factors Associated With Myeloproliferative Neoplasms. HemaSphere 2020, 4, e327. [Google Scholar] [CrossRef]
- Campbell, P.J.; Scott, L.M.; Buck, G.; Wheatley, K.; East, C.L.; Marsden, J.T.; Duffy, A.; Boyd, E.M.; Bench, A.J.; Scott, M.A.; et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: A prospective study. Lancet 2005, 366, 1945–1953. [Google Scholar] [CrossRef]
- Wolanskyj, A.P.; Lasho, T.L.; Schwager, S.M.; McClure, R.F.; Wadleigh, M.; Lee, S.J.; Gilliland, D.G.; Tefferi, A. JAK2V617F mutation in essential thrombocythaemia: Clinical associations and long-term prognostic relevance. Br. J. Haematol. 2005, 131, 208–213. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Schwager, S.M.; Steensma, D.P.; Mesa, R.A.; Li, C.Y.; Wadleigh, M.; Gary Gilliland, D. The JAK2V617F tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: Lineage specificity and clinical correlates. Br. J. Haematol. 2005, 131, 320–328. [Google Scholar] [CrossRef]
- Kim, D.Y.; Tariq, H.; Brown, A.F.; Vadlamudi, K.; Wang, Y.; Ikoma, S.; Holder, K.N.; Higgins, R.A.; Fan, H. JAK2 V617F Mutation Allele Burden (MAB) and Its Correlation with Hematologic Characteristics in Myeloproliferative Neoplasms. Blood 2017, 130, 5267. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Schwager, S.M.; Strand, J.S.; Elliott, M.; Mesa, R.; Li, C.-Y.; Wadleigh, M.; Lee, S.J.; Gilliland, D.G. The clinical phenotype of wild-type, heterozygous, and homozygous JAK2V617F in polycythemia vera. Cancer 2006, 106, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Huang, L.J.S.; Lodish, H.F. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J. Biol. Chem. 2008, 283, 5258–5266. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Levine, R.; Tong, W.; Wernig, G.; Pikman, Y.; Zarnegar, S.; Gilliland, D.G.; Lodish, H. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc. Natl. Acad. Sci. USA 2005, 102, 18962–18967. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Laubach, J.P.; Fu, P.; Jiang, X.; Salter, K.H.; Potti, A.; Arcasoy, M.O. Polycythemia vera erythroid precursors exhibit increased proliferation and apoptosis resistance associated with abnormal RAS and PI3K pathway activation. Exp. Hematol. 2009, 37, 1411–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funakoshi-Tago, M.; Tago, K.; Abe, M.; Sonoda, Y.; Kasahara, T. STAT5 activation is critical for the transformation mediated by myeloproliferative disorder-associated JAK2 V617F mutant. J. Biol. Chem. 2010, 285, 5296–5307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grebien, F.; Kerenyi, M.A.; Kovacic, B.; Kolbe, T.; Becker, V.; Dolznig, H.; Pfeffer, K.; Klingmüller, U.; Müller, M.; Beug, H.; et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood 2008, 111, 4511–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachs, Z.; Been, R.A.; Decoursin, K.J.; Nguyen, H.T.; Mohd Hassan, N.A.; Noble-Orcutt, K.E.; Eckfeldt, C.E.; Pomeroy, E.J.; Diaz-Flores, E.; Geurts, J.L.; et al. Stat5 is critical for the development and maintenance of myeloproliferative neoplasm initiated by Nf1 deficiency. Haematologica 2016, 101, 1190–1199. [Google Scholar] [CrossRef]
- Chen, E.; Beer, P.A.; Godfrey, A.L.; Ortmann, C.A.; Li, J.; Costa-Pereira, A.P.; Ingle, C.E.; Dermitzakis, E.T.; Campbell, P.J.; Green, A.R. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell 2010, 18, 524–535. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Richmond, T.D.; Muntean, A.G.; Barber, D.L.; Weiss, M.J.; Crispino, J.D. STAT1 promotes megakaryopoiesis downstream of GATA-1 in mice. J. Clin. Investig. 2007, 117, 3890–3899. [Google Scholar] [CrossRef]
- Machado-Neto, J.A.; Favaro, P.; Lazarini, M.; da Silva Santos Duarte, A.; Archangelo, L.F.; Lorand-Metze, I.; Costa, F.F.; Saad, S.T.O.; Traina, F. Downregulation of IRS2 in myelodysplastic syndrome: A possible role in impaired hematopoietic cell differentiation. Leuk. Res. 2012, 36, 931–935. [Google Scholar] [CrossRef]
- Schacher, D.H.; VanHoy, R.W.; Liu, Q.; Arkins, S.; Dantzer, R.; Freund, G.G.; Kelley, K.W. Developmental Expression of Insulin Receptor Substrate-2 During Dimethylsulfoxide-Induced Differentiation of Human HL-60 Cells. J. Immunol. 2000, 164, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Lima, K.; Lopes, L.R.; Machado-Neto, J.A. Exploring redox vulnerabilities in JAK2(V617F)-positive cellular models. Hematol. Transfus. Cell Ther. 2021, 43, 430–436. [Google Scholar] [CrossRef]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [Green Version]
- Vener, C.; Novembrino, C.; Bamonti Catena, F.; Fracchiolla, N.S.; Gianelli, U.; Savi, F.; Radaelli, F.; Fermo, E.; Cortelezzi, A.; Lonati, S.; et al. Oxidative stress is increased in primary and post-polycythemia vera myelofibrosis. Exp. Hematol. 2010, 38, 1058–1065. [Google Scholar] [CrossRef]
- Hurtado-Nedelec, M.; Csillag, M.J.G.; Boussetta, T.; Belambri, S.A.; Fay, M.; Cassinat, B.; Gougerot, M.A.P.; Dang, P.M.C.; El-Benna, J. Increased reactive oxygen species production and p47phox phosphorylation in neutrophils from myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica 2013, 98, 1517–1524. [Google Scholar] [CrossRef]
- Allahverdi, N.; Yassin, M.; Ibrahim, M. Environmental Factors, Lifestyle Risk Factors, and Host Characteristics Associated With Philadelphia Negative Myeloproliferative Neoplasm: A Systematic Review. Cancer Control 2021, 28, 10732748211046802. [Google Scholar] [CrossRef]
- Chen, C.C.; You, J.Y.; Lung, J.; Huang, C.E.; Chen, Y.Y.; Leu, Y.W.; Ho, H.Y.; Li, C.P.; Lu, C.H.; Der Lee, K.; et al. Aberrant let7a/HMGA2 signaling activity with unique clinical phenotype in JAK2-mutated myeloproliferative neoplasms. Haematologica 2017, 102, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Uras, I.Z.; Maurer, B.; Nivarthi, H.; Jodl, P.; Kollmann, K.; Prchal-Murphy, M.; Milosevic Feenstra, J.D.; Zojer, M.; Lagger, S.; Grausenburger, R.; et al. CDK6 coordinates JAK2V617F mutant MPN via NF-kB and apoptotic networks. Blood 2019, 133, 1677–1690. [Google Scholar] [CrossRef] [Green Version]
- Catherwood, M.A.; McAllister, R.; McCallion, P.; McGimpsey, J.E.; Hindley, A.; Feerick, J.; Greenfield, G.; Kennedy, P.; Benson, G.; Arnold, C.; et al. A molecular diagnostic algorithm for JAK2 V617F investigations in suspected myeloproliferative neoplasms. Ir. J. Med. Sci. 2020, 189, 621–626. [Google Scholar] [CrossRef]
- McPherson, S.; McMullin, M.F.; Mills, K. Epigenetics in Myeloproliferative Neoplasms. J. Cell. Mol. Med. 2017, 21, 1660–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milosevic, J.D.; Kralovics, R. Genetic and epigenetic alterations of myeloproliferative disorders. Int. J. Hematol. 2012, 97, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of Mutation Order on Myeloproliferative Neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Schneider, R.K.; Breyfogle, L.J.; Rosen, E.A.; Poveromo, L.; Elf, S.; Ko, A.; Brumme, K.; Levine, R.; Ebert, B.L.; et al. Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood 2015, 125, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staehle, H.F.; Heinemann, J.; Gruender, A.; Omlor, A.M.; Pahl, H.L.; Jutzi, J.S. Jmjd1c is dispensable for healthy adult hematopoiesis and Jak2V617F-driven myeloproliferative disease initiation in mice. PLoS ONE 2020, 15, e228362. [Google Scholar] [CrossRef]
- McKenney, A.S.; Lau, A.N.; Hanasoge Somasundara, A.V.; Spitzer, B.; Intlekofer, A.M.; Ahn, J.; Shank, K.; Rapaport, F.T.; Patel, M.A.; Papalexi, E.; et al. JAK2/IDH-mutant–driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J. Clin. Investig. 2018, 128, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef]
- Anthony Green, P.B. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl. J. Med. 2010, 362, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Bolton-Gillespie, E.; Schemionek, M.; Klein, H.U.; Flis, S.; Hoser, G.; Lange, T.; Nieborowska-Skorska, M.; Maier, J.; Kerstiens, L.; Koptyra, M.; et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood 2013, 121, 4175–4183. [Google Scholar] [CrossRef]
- Chen, E.; Ahn, J.S.; Sykes, D.B.; Breyfogle, L.J.; Godfrey, A.L.; Nangalia, J.; Ko, A.; DeAngelo, D.J.; Green, A.R.; Mullally, A. RECQL5 Suppresses Oncogenic JAK2-Induced Replication Stress and Genomic Instability. Cell Rep. 2015, 13, 2345–2352. [Google Scholar] [CrossRef]
- Holmström, M.O.; Hjortsø, M.D.; Ahmad, S.M.; Met, Ö.; Martinenaite, E.; Riley, C.; Straten, P.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The JAK2V617F mutation is a target for specific T cells in the JAK2V617F-positive myeloproliferative neoplasms. Leukemia 2017, 31, 495–498. [Google Scholar] [CrossRef]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F.; Rumi, E.; Pietra, D.; Lazzarino, M.; Cazzola, M. JAK2 (V617F) mutation in healthy individuals. Br. J. Haematol. 2007, 136, 678–679. [Google Scholar] [CrossRef]
- Williams, N.; Lee, J.; Moore, L.; Baxter, J.E.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; Campbell, P.J.; et al. Driver mutation acquisition in utero and childhood followed by lifelong clonal evolution underlie myeloproliferative neoplasms. Blood 2020, 136, LBA-1. [Google Scholar] [CrossRef]
- Williams, N.; Lee, J.; Moore, L.; Baxter, E.J.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; Campbell, P.J.; et al. Phylogenetic reconstruction of myeloproliferative neoplasm reveals very early origins and lifelong evolution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Van Egeren, D.; Escabi, J.; Nguyen, M.; Liu, S.; Reilly, C.R.; Patel, S.; Kamaz, B.; Kalyva, M.; DeAngelo, D.J.; Galinsky, I.; et al. Reconstructing the lineage histories and differentiation trajectories of individual cancer cells in JAK2-mutant myeloproliferative neoplasms. bioRxiv 2020, 1–10. [Google Scholar] [CrossRef]
- Godfrey, A.L.; Chen, E.; Pagano, F.; Silber, Y.; Campbell, P.J.; Green, A.R. Clonal analyses reveal associations of JAK2V617F homozygosity with hematologic features, age and gender in polycythemia vera and essential thrombocythemia. Haematologica 2013, 98, 718–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, A.D.; Thompson, C.A.; Wang, A.H.; Vierkant, R.A.; Habermann, T.M.; Ross, J.A.; Mesa, R.A.; Virnig, B.A.; Cerhan, J.R. Anthropometric, medical history and lifestyle risk factors for myeloproliferative neoplasms in the Iowa Women’s Health Study cohort. Int. J. Cancer 2014, 134, 1741–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm Sørensen, A.; Hasselbalch, H.C. Smoking and philadelphia-negative chronic myeloproliferative neoplasms. Eur. J. Haematol. 2016, 97, 63–69. [Google Scholar] [CrossRef]
- Tefferi, A.; Wassie, E.A.; Lasho, T.L.; Finke, C.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.; Gangat, N.; Wolanskyj, A.P. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014, 28, 2300–2303. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Li, J.; Spensberger, D.; Ahn, J.S.; Anand, S.; Beer, P.A.; Ghevaert, C.; Chen, E.; Forrai, A.; Scott, L.M.; Ferreira, R.; et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood 2010, 116, 1528–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gou, P.; Zhang, W.; Giraudier, S. Insights into the Potential Mechanisms of JAK2V617F Somatic Mutation Contributing Distinct Phenotypes in Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2022, 23, 1013. https://doi.org/10.3390/ijms23031013
Gou P, Zhang W, Giraudier S. Insights into the Potential Mechanisms of JAK2V617F Somatic Mutation Contributing Distinct Phenotypes in Myeloproliferative Neoplasms. International Journal of Molecular Sciences. 2022; 23(3):1013. https://doi.org/10.3390/ijms23031013
Chicago/Turabian StyleGou, Panhong, Wenchao Zhang, and Stephane Giraudier. 2022. "Insights into the Potential Mechanisms of JAK2V617F Somatic Mutation Contributing Distinct Phenotypes in Myeloproliferative Neoplasms" International Journal of Molecular Sciences 23, no. 3: 1013. https://doi.org/10.3390/ijms23031013