
Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma

 , , and
, , and
Abstract
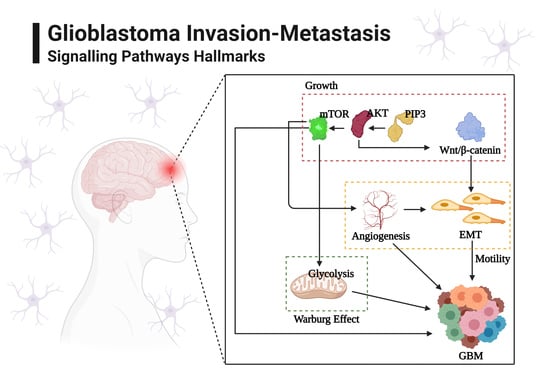
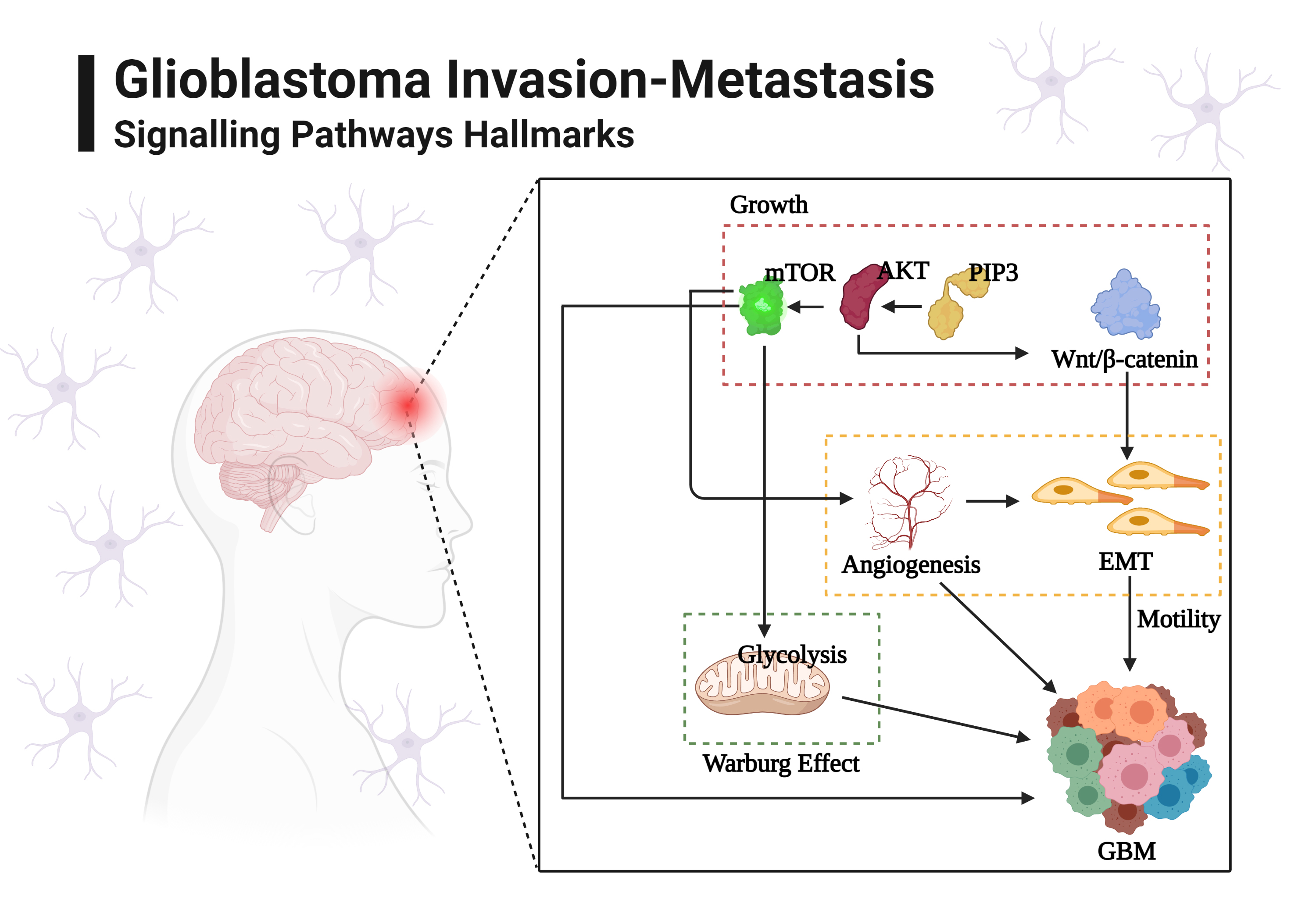
:
1. Introduction
2. Glioblastoma Invasion: Programmed Signaling Pathways
2.1. Wnt Pathway
2.1.1. Wnt Signaling in Glioblastoma
2.1.2. Involvement of Wnt Signaling in Glioblastoma Invasion
2.1.3. Emerging Links between Wnt Signaling and Autophagy in Glioma
3. Different Forms of Epithelial-Mesenchymal Transition (EMT)
A Wnt Perspective of EMT in Glioblastoma Invasion
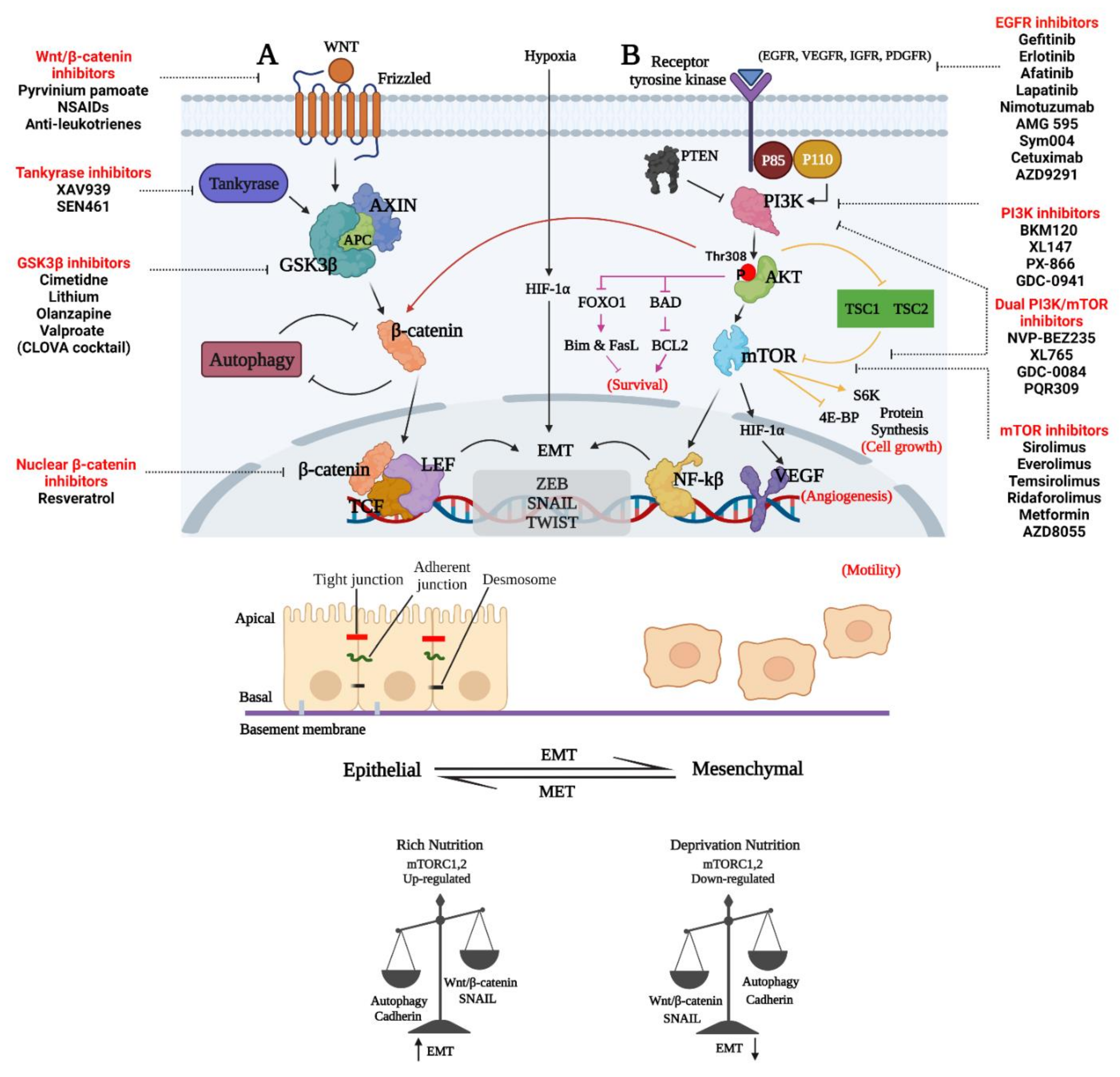
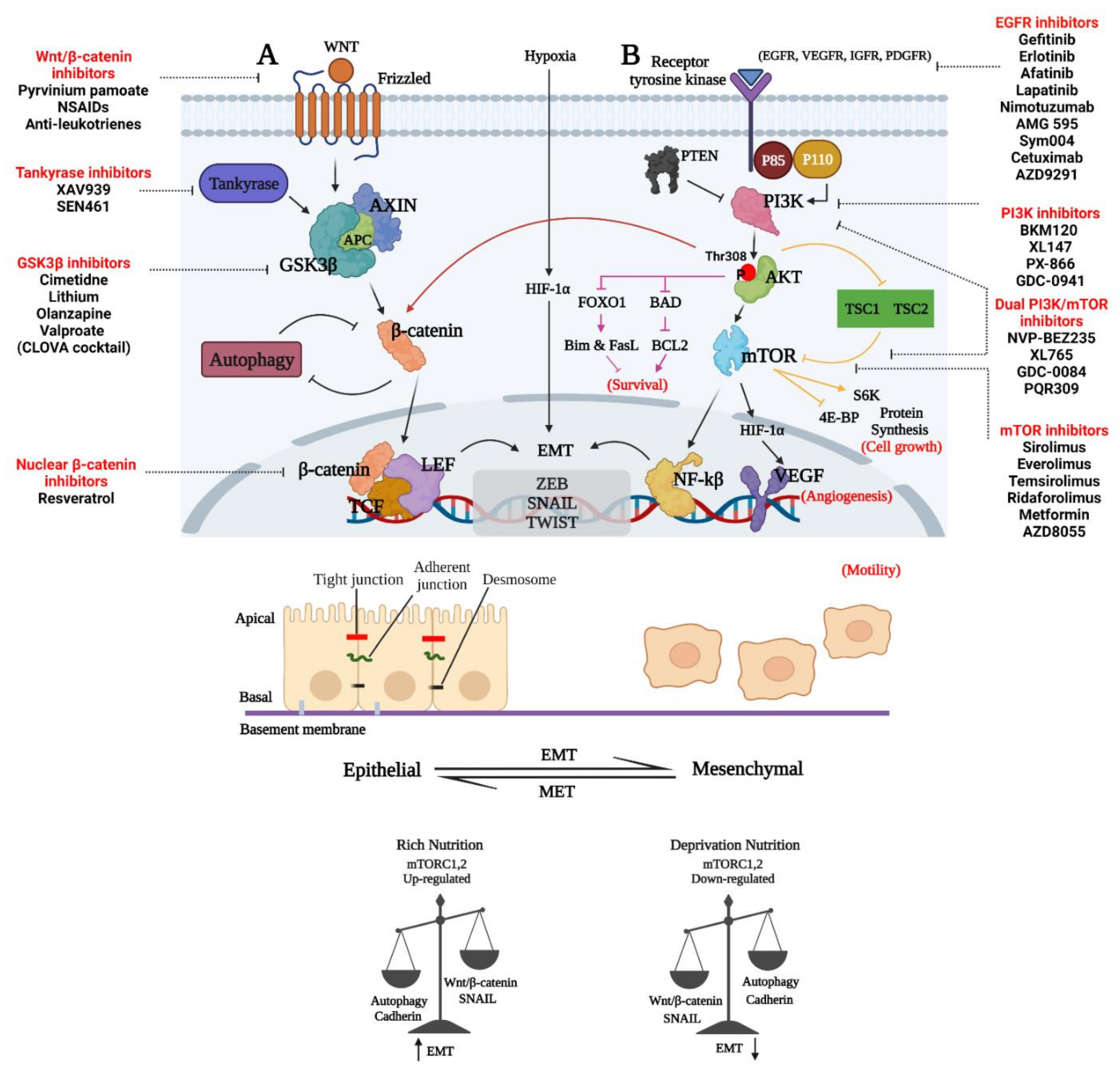
4. Therapeutic Approaches to Decrease the Invasion and Survival of GBM Cells by Inhibiting Wnt/β-Catenin and Emt
5. PI3K/Akt/mTOR (PAM) Signaling Cascade in Glioblastoma Invasion
5.1. PAM in Gliomagenesis
5.1.1. Role of PAM in Glioma Growth and Invasion
5.1.2. PAM in Angiogenesis of Brain Tumors
6. Therapeutic Approaches Targeting PAM in GBM
7. GBM Cell Metabolic Functions as a Target of Wnt and PAM Therapeutics
7.1. Glucose and Lactate Are Main Fuel Sources for GBM Cells
7.2. Role of Fatty Acids in GBM Metabolism
7.3. Amino Acid Metabolism in GBM
7.4. Research Progress on the Changes in Glycolysis and HIF in GBM Invasion
8. Therapeutic Targeting Wnt and PAM Metabolic Functions to Decrease Invasion and Survival of GBM
9. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Alizadeh, J.; Thliveris, J.; Koleini, N.; Kardami, E.; Hatch, G.M.; Xu, F.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Statins: A new approach to combat temozolomide chemoresistance in glioblastoma. J. Investig. Med. 2018, 66, 1083–1087. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, I.; Vosoughi, A.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Cascio, C.L. Developmentally regulated signaling pathways in glioma invasion. Cell. Mol. Life Sci. 2018, 75, 385–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, J.; Shojaei, S.; Sepanjnia, A.; Hashemi, M.; Eftekharpour, E.; Ghavami, S. Simultaneous Detection of Autophagy and Epithelial to Mesenchymal Transition in the Non-small Cell Lung Cancer Cells. Methods Mol. Biol. 2019, 1854, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Virshup, D.M. Targeting Wnts at the Source—New Mechanisms, New Biomarkers, New Drugs. Mol. Cancer Ther. 2015, 14, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, U.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef]
- Foltz, G.; Yoon, J.-G.; Lee, H.; Ma, L.; Tian, Q.; Hood, L.; Madan, A. Epigenetic Regulation of Wnt Pathway Antagonists in Human Glioblastoma Multiforme. Genes Cancer 2010, 1, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, J.M.; Rank, K.B.; Zeng, Y.; Capen, A.; Yadav, V.; Manro, J.R.; Engler, T.A.; Chedid, M. Activating the Wnt/β-catenin pathway for the treatment of melanoma–application of LY2090314, a novel selective inhibitor of glycogen synthase kinase. PLoS ONE 2015, 10, e0125028. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Tu, Y.; Sun, X.; Jiang, J.; Jin, X.; Bo, X.; Li, Z.; Bian, A.; Wang, X.; Liu, D.; et al. Wnt/beta-Catenin pathway in human glioma: Expression pattern and clinical/prognostic correlations. Clin. Exp. Med. 2010, 11, 105–112. [Google Scholar] [CrossRef]
- Zhang, Y.; Ng, P.K.-S.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; De Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.e3. [Google Scholar] [CrossRef] [Green Version]
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro-Oncology 2010, 12, 882–889. [Google Scholar] [CrossRef]
- Zhong, C.; Li, X.; Tao, B.; Peng, L.; Peng, T.; Yang, X.; Xia, X.; Chen, L. LIM and SH3 protein 1 induces glioma growth and invasion through PI3K/AKT signaling and epithelial-mesenchymal transition. Biomed. Pharmacother. 2019, 116, 109013. [Google Scholar] [CrossRef]
- Batsios, G.; Viswanath, P.; Subramani, E.; Najac, C.; Gillespie, A.M.; Santos, R.D.; Molloy, A.R.; Pieper, R.O.; Ronen, S.M. PI3K/mTOR inhibition of IDH1 mutant glioma leads to reduced 2HG production that is associated with increased survival. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Buzzai, M.; Bauer, E.D.; Jones, R.G.; DeBerardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid β-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef] [Green Version]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, B.C. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.K.; Fan, Q.-W.; Weiss, W.A. PI3K Signaling in Glioma-Animal Models and Therapeutic Challenges. Brain Pathol. 2009, 19, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, R.; Zhang, X.; Guo, M. Glioblastoma stem cells and Wnt signaling pathway: Molecular mechanisms and therapeutic targets. Chin. Neurosurg. J. 2020, 6, 1–6. [Google Scholar]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/β-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118926. [Google Scholar] [CrossRef]
- Morris, L.G.T.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.; Turcan, S.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Nager, M.; Bhardwaj, D.; Cantí, C.; Medina, L.; Nogués, P.; Herreros, J. β-Catenin Signalling in Glioblastoma Multiforme and Glioma-Initiating Cells. Chemother. Res. Pract. 2012, 2012, 192362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Guo, J.; Chen, H.; Yao, C.-J.; Zhuang, D.-X.; Wang, Y.; Tang, W.-J.; Ren, G.; Yao, Y.; Wu, J.-S.; et al. Gene mutation profiling of primary glioblastoma through multiple tumor biopsy guided by 1H-magnetic resonance spectroscopy. Int. J. Clin. Exp. Pathol. 2015, 8, 5327–5335. [Google Scholar]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2010, 8, 97–106. [Google Scholar] [CrossRef]
- Petherick, K.J.; Williams, A.C.; Lane, J.D.; Ordóñez-Morán, P.; Huelsken, J.; Collard, T.J.; Smartt, H.J.M.; Batson, J.; Malik, K.; Paraskeva, C.; et al. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013, 32, 1903–1916. [Google Scholar] [CrossRef]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.; Ghavami, S. Alzheimer’s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef]
- Colella, B.; Faienza, F.; Carinci, M.; D’Alessandro, G.; Catalano, M.; Santoro, A.; Cecconi, F.; Limatola, C.; Di Bartolomeo, S. Autophagy induction impairs Wnt/β-catenin signalling through β-catenin relocalisation in glioblastoma cells. Cell Signal. 2019, 53, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Lorzadeh, S.; Ghavami, S. Autophagy and cancer metastasis: A Trojan horse. J. Investig. Med. 2021, 69, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wang, J.; Nika, H.; Hawke, D.; Keezer, S.; Ge, Q.; Fang, B.; Fang, X.; Litchfield, D.W.; Aldape, K.; et al. EGF-induced ERK activation promotes CK2-mediated disassociation of α-catenin from β-catenin and transactivation of β-catenin. Mol. Cell 2009, 36, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, U.; Nikkhah, G.; Maciaczyk, J. Epithelial-to-mesenchymal(-like) transition as a relevant molecular event in malignant gliomas. Cancer Lett. 2013, 331, 131–138. [Google Scholar] [CrossRef]
- Koen, E.J.; Collier, A.B.; Maharaj, S.K. Particle-in-cell simulations of beam-driven electrostatic waves in a plasma. Phys. Plasmas 2012, 19, 042101. [Google Scholar] [CrossRef]
- Imran, S.A.M.; Yazid, M.D.; Idrus, R.B.H.; Maarof, M.; Nordin, A.; Razali, R.A.; Lokanathan, Y. Is There an Interconnection between Epithelial-Mesenchymal Transition (EMT) and Telomere Shortening in Aging? Int. J. Mol. Sci. 2021, 22, 3888. [Google Scholar] [CrossRef]
- Tan, W.; Wang, Y.; Chen, Y.; Chen, C. Cell tracing reveals the transdifferentiation fate of mouse lung epithelial cells during pulmonary fibrosis in vivo. Exp. Ther. Med. 2021, 22, 1–9. [Google Scholar] [CrossRef]
- Abolhassani, A.; Riazi, G.H.; Azizi, E.; Amanpour, S.; Muhammadnejad, S.; Haddadi, M.; Zekri, A.; Shirkoohi, R. FGF10: Type III Epithelial Mesenchymal Transition and Invasion in Breast Cancer Cell Lines. J. Cancer 2014, 5, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Bierie, B.; Moses, H.L. TGFβ: The molecular Jekyll and Hyde of cancer. Nat. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Castanon, I.; Baylies, M.K. A Twist in fate: Evolutionary comparison of Twist structure and function. Gene 2002, 287, 11–22. [Google Scholar] [CrossRef]
- Elias, M.C.; Tozer, K.R.; Silber, J.R.; Mikheeva, S.; Deng, M.; Morrison, R.S.; Manning, T.C.; Silbergeld, D.L.; Glackin, C.A.; Reh, T.A.; et al. TWIST is Expressed in Human Gliomas, Promotes Invasion. Neoplasia 2005, 7, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.-P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014, 16, 671–685. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.-Y.; Kandel, J.J.; Yamashiro, D.J.; Canoll, P.; Anastassiou, D. A Multi-Cancer Mesenchymal Transition Gene Expression Signature Is Associated with Prolonged Time to Recurrence in Glioblastoma. PLoS ONE 2012, 7, e34705. [Google Scholar] [CrossRef]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers 2019, 11, 312. [Google Scholar] [CrossRef] [Green Version]
- Pang, H.; Zheng, Y.; Zhao, Y.; Xiu, X.; Wang, J. miR-590-3p suppresses cancer cell migration, invasion and epithelial-mesenchymal transition in glioblas-toma multiforme by targeting ZEB1 and ZEB. Biochem. Biophys. Res. Commun. 2015, 468, 739–745. [Google Scholar] [CrossRef]
- Qi, S.; Song, Y.; Peng, Y.; Wang, H.; Long, H.; Yu, X.; Li, Z.; Fang, L.; Wu, A.; Luo, W.; et al. ZEB2 Mediates Multiple Pathways Regulating Cell Proliferation, Migration, Invasion, and Apoptosis in Glioma. PLoS ONE 2012, 7, e38842. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Tilló, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef]
- Wang, Q.; Li, X.; Zhu, Y.; Yang, P. MicroRNA-16 suppresses epithelial-mesenchymal transition-related gene expression in human glioma. Mol. Med. Rep. 2014, 10, 3310–3314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Chen, Z.; Shen, L.; Tang, T.; Yang, M.; Zheng, X. Long Noncoding RNA LINC-PINT Suppresses Cell Proliferation, Invasion, and EMT by Blocking Wnt/β-Catenin Signaling in Glioblastoma. Front. Pharmacol. 2021, 11, 11. [Google Scholar] [CrossRef]
- Li, L.; Shao, M.-Y.; Zou, S.-C.; Xiao, Z.-F.; Chen, Z.-C. MiR-101-3p inhibits EMT to attenuate proliferation and metastasis in glioblastoma by targeting TRIM. J. Neuro-Oncol. 2019, 141, 19–30. [Google Scholar] [CrossRef]
- Chen, W.; Kong, K.; Xu, X.; Chen, C.; Li, H.; Wang, F.; Peng, X.; Zhang, Z.; Li, P.; Li, J.; et al. Downregulation of miR-205 is associated with glioblastoma cell migration, invasion, and the epithelial-mesenchymal transition, by targeting ZEB1 via the Akt/mTOR signaling pathway. Int. J. Oncol. 2018, 52, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Yang, Y.; Xue, J.; Xi, Z.; Hu, L.; Pan, S.-J.; Sun, Q. Long noncoding RNA SNHG7 promotes the progression and growth of glioblastoma via inhibition of miR-5095. Biochem. Biophys. Res. Commun. 2018, 496, 712–718. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, G.; Zhang, Y.; Jiang, H.; Wang, W.; Zhao, D.; Hong, J.; Yu, H.; Qi, L. Mesenchymal stem cell-derived exosomal microRNA-133b suppresses glioma progression via Wnt/β-catenin signaling pathway by targeting EZH2. Stem Cell Res. Ther. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Guo, R.; Guan, F.; Ma, S.; Li, M.; Wu, J.; Liu, X.; Li, H.; Yang, B. MicroRNA-128-3p Enhances the Chemosensitivity of Temozolomide in Glioblastoma by Targeting c-Met and EMT. Sci. Rep. 2020, 10, 9471. [Google Scholar] [CrossRef]
- Osuka, S.; Zhu, D.; Zhang, Z.; Li, C.; Stackhouse, C.T.; Sampetrean, O.; Olson, J.J.; Gillespie, G.Y.; Saya, H.; Willey, C.D.; et al. IGF1/N-cadherin/b-catenin/Clusterin signaling axis can mediates adaptive radioresistance in glioblastoma. In Proceedings of the AACR Annual Meeting 2021, Philadelphia, PA, USA, 10–15 April 2021. [Google Scholar]
- Suwala, A.K.; Koch, K.; Rios, D.H.; Aretz, P.; Uhlmann, C.; Ogorek, I.; Felsberg, J.; Reifenberger, G.; Köhrer, K.; Deenen, R.; et al. Inhibition of Wnt/beta-catenin signaling downregulates expression of aldehyde dehydrogenase isoform 3A1 (ALDH3A1) to reduce resistance against temozolomide in glioblastoma in vitro. Oncotarget 2018, 9, 22703–22716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, E.-J.; Kim, S.; Hsieh, J.-T.; Baek, S.T. Wnt/β-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, Y.; Biswas, A.; Kumar, U.; Banerjee, I.; Das, S.; Maji, S.; Das, S.K.; Emdad, L.; Cavenee, W.K.; Mandal, M.; et al. Lumefantrine, an antimalarial drug, reverses radiation and temozolomide resistance in glioblastoma. Proc. Natl. Acad. Sci. USA 2020, 117, 12324–12331. [Google Scholar] [CrossRef] [PubMed]
- Deprez, J.; Vertommen, D.; Alessi, D.; Hue, L.; Rider, M.H. Phosphorylation and Activation of Heart 6-Phosphofructo-2-kinase by Protein Kinase B and Other Protein Kinases of the Insulin Signaling Cascades. J. Biol. Chem. 1997, 272, 17269–17275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. Erratum: The somatic genomic landscape of glioblastoma (Cell (2013) 155 (462-477)). Cell 2014, 157, 753. [Google Scholar] [CrossRef] [Green Version]
- Vadlakonda, L.; Pasupuleti, M.; Reddanna, P. Role of PI3K-AKT-mTOR and Wnt signaling pathways in transition of G1-S phase of cell cycle in cancer cells. Front. Oncol. 2013, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Von Achenbach, C.; Weller, M.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Fabbro, D.; Reifenberger, G.; Szabó, E. Synergistic growth inhibition mediated by dual PI3K/mTOR pathway targeting and genetic or direct pharmacological AKT inhibition in human glioblastoma models. J. Neurochem. 2020, 153, 510–524. [Google Scholar] [CrossRef]
- Langhans, J.; Schneele, L.; Trenkler, N.; Von Bandemer, H.; Nonnenmacher, L.; Karpel-Massler, G.; Siegelin, M.D.; Zhou, S.; Halatsch, M.-E.; Debatin, K.-M.; et al. The effects of PI3K-mediated signalling on glioblastoma cell behaviour. Oncogenesis 2017, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Holand, K.; Salm, F.; Arcaro, A. The phosphoinositide 3-kinase signaling pathway as a therapeutic target in grade IV brain tumors. Curr. Cancer Drug Targets 2011, 11, 894–918. [Google Scholar] [CrossRef]
- Hartmann, C.; Bartels, G.; Gehlhaar, C.; Holtkamp, N.; von Deimling, A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol. 2005, 109, 639–642. [Google Scholar] [CrossRef]
- Guerreiro, A.S.; Fattet, S.; Fischer, B.; Shalaby, T.; Jackson, S.P.; Schoenwaelder, S.M.; Grotzer, M.A.; Delattre, O.; Arcaro, A. Targeting the PI3K p110α isoform inhibits medulloblastoma proliferation, chemoresistance, and migration. Clin. Cancer Res. 2008, 14, 6761–6769. [Google Scholar] [CrossRef] [Green Version]
- Salm, F.; Dimitrova, V.; Von Bueren, A.O.; Ćwiek, P.; Rehrauer, H.; Djonov, V.; Anderle, P.; Arcaro, A. The Phosphoinositide 3-Kinase p110α Isoform Regulates Leukemia Inhibitory Factor Receptor Expression via c-Myc and miR-125b to Promote Cell Proliferation in Medulloblastoma. PLoS ONE 2015, 10, e0123958. [Google Scholar] [CrossRef] [Green Version]
- Koul, D.; Shen, R.; Bergh, S.; Sheng, X.; Shishodia, S.; LaFortune, T.A.; Lu, Y.; De Groot, J.F.; Mills, G.B.; Yung, W.A. Inhibition of Akt survival pathway by a small-molecule inhibitor in human glioblastoma. Mol. Cancer Ther. 2006, 5, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, S.K.; Piekorz, R.P.; Nürnberg, B.; To, S.-S.T. The catalytic phosphoinositol 3-kinase isoform p110δ is required for glioma cell migration and invasion. Eur. J. Cancer 2012, 48, 149–157. [Google Scholar] [CrossRef]
- Mueller, W.; Mizoguchi, M.; Silen, E.; D’Amore, K.; Nutt, C.L.; Louis, D.N. Mutations of the PIK3CA gene are rare in human glioblastoma. Acta Neuropathol. 2005, 109, 654–655. [Google Scholar] [CrossRef]
- Gallia, G.L.; Tyler, B.M.; Hann, C.L.; Siu, I.-M.; Giranda, V.L.; Vescovi, A.L.; Brem, H.; Riggins, G.J. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol. Cancer Ther. 2009, 8, 386–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, W.; Digon-Söntgerath, B.; Koch, A.; Waha, A.; Endl, E.; Dani, I.; Denkhaus, D.; Goodyer, C.G.; Sörensen, N.; Wiestler, O.D.; et al. Phosphatidylinositol 3′-Kinase/AKT Signaling Is Activated in Medulloblastoma Cell Proliferation and Is Associated with Reduced Expression ofPTEN. Clin. Cancer Res. 2006, 12, 3019–3027. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Fillmore, H.L.; Kadian, R.; Broaddus, W.C.; Tye, G.W.; van Meter, T.E. The alkylphospholipid perifosine induces apoptosis and p21-mediated cell cycle arrest in medulloblastoma. Mol. Cancer Res. 2009, 7, 1813–1821. [Google Scholar] [CrossRef] [Green Version]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Ali-Hemmati, A.; Rahmati, M.; Charoudeh, H.N. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef]
- Millis, S.Z.; Jardim, D.L.; Albacker, L.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Kurzrock, R. Phosphatidylinositol 3-kinase pathway genomic alterations in 60,991 diverse solid tumors informs targeted therapy opportunities. Cancer 2019, 125, 1185–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Pore, N.; Cerniglia, G.J.; Mick, R.; Georgescu, M.-M.; Bernhard, E.J.; Hahn, S.M.; Gupta, A.K.; Maity, A. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res. 2007, 67, 4467–4473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.-C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Knobbe, C.B.; Reifenberger, G. Genetic Alterations and Aberrant Expression of Genes Related to the Phosphatidyl-lnositol-3′-Kinase/Protein Kinase B (Akt) Signal Transduction Pathway in Glioblastomas. Brain Pathol. 2006, 13, 507–518. [Google Scholar] [CrossRef]
- Dey, N.; Crosswell, H.E.; De, P.; Parsons, R.; Peng, Q.; Su, J.D.; Durden, D.L. The Protein Phosphatase Activity of PTEN Regulates Src Family Kinases and Controls Glioma Migration. Cancer Res. 2008, 68, 1862–1871. [Google Scholar] [CrossRef] [Green Version]
- Mayo, L.D.; Dixon, J.E.; Durden, D.L.; Tonks, N.K.; Donner, D.B. PTEN Protects p53 from Mdm2 and Sensitizes Cancer Cells to Chemotherapy. J. Biol. Chem. 2002, 277, 5484–5489. [Google Scholar] [CrossRef] [Green Version]
- Vitucci, M.; Karpinich, N.O.; Bash, R.E.; Werneke, A.M.; Schmid, R.S.; White, K.K.; McNeill, R.S.; Huff, B.; Wang, S.; Van Dyke, T.; et al. Cooperativity between MAPK and PI3K signaling activation is required for glioblastoma pathogenesis. Neuro-Oncology 2013, 15, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Daniele, S.; Costa, B.; Zappelli, E.; Da Pozzo, E.; Sestito, S.; Nesi, G.; Campiglia, P.; Marinelli, L.; Novellino, E.; Rapposelli, S.; et al. Combined inhibition of AKT/mTOR and MDM2 enhances Glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Sci. Rep. 2015, 5, 9956. [Google Scholar] [CrossRef]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of mTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway that Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Duzgun, Z.; Eroglu, Z.; Avci, C.B. Role of mTOR in glioblastoma. Gene 2016, 575, 187–190. [Google Scholar] [CrossRef]
- Wu, S.-H.; Bi, J.-F.; Cloughesy, T.; Cavenee, W.K.; Mischel, P.S. Emerging function of mTORC2 as a core regulator in glioblastoma: Metabolic reprogramming and drug resistance. Cancer Biol. Med. 2014, 11, 255–263. [Google Scholar] [CrossRef]
- Gini, B.; Zanca, C.; Guo, D.; Matsutani, T.; Masui, K.; Ikegami, S.; Yang, H.; Nathanson, D.; Villa, G.R.; Shackelford, D.; et al. The mTOR kinase inhibitors, CC214-1 and CC214-2, preferentially block the growth of EGFRvIII-activated glioblastomas. Clin. Cancer Res. 2013, 19, 5722–5732. [Google Scholar] [CrossRef] [Green Version]
- Rich, J.N.; Hans, C.; Jones, B.; Iversen, E.S.; McLendon, R.E.; Rasheed, B.K.A.; Dobra, A.; Dressman, H.K.; Bigner, D.D.; Nevins, J.R.; et al. Gene Expression Profiling and Genetic Markers in Glioblastoma Survival. Cancer Res. 2005, 65, 4051–4058. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Han, Q.; Wang, X.; Yang, M.; Zhang, Z.; Li, P.; Chen, A.; Hu, C.; Li, S. IBP-mediated suppression of autophagy promotes growth and metastasis of breast cancer cells via activating mTORC2/Akt/FOXO3a signaling pathway. Cell Death Dis. 2013, 4, e842. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Altman, J.K.; Szilard, A.; Goussetis, D.J.; Sassano, A.; Colamonici, M.; Gounaris, E.; Frankfurt, O.; Giles, F.J.; Eklund, E.A.; Beauchamp, E.; et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin. Cancer Res. 2014, 20, 2400–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, X.; Wang, J.; Wang, C.; Sommer, E.M.; Kozasa, T.; Srinivasula, S.M.; Alessi, D.; Offermanns, S.; Simon, M.I.; Wu, D. PRR5L degradation promotes mTORC2-mediated PKC-δ phosphorylation and cell migration downstream of Gα 12. Nat. Cell Biol. 2012, 14, 686–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hütt-Cabezas, M.; Karajannis, M.A.; Zagzag, D.; Shah, S.; Horkayne-Szakaly, I.; Rushing, E.J.; Cameron, J.D.; Jain, D.; Eberhart, C.G.; Raabe, E.H.; et al. Activation of mTORC1/mTORC2 signaling in pediatric low-grade glioma and pilocytic astrocytoma reveals mTOR as a therapeutic target. Neuro-Oncology 2013, 15, 1604–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Gao, T. mTORC 2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep. 2014, 15, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Ke, Y.; Sun, X.; Yu, L.; Yang, Z.; Zhang, Y.; Du, M.; Wang, J.; Liu, X.; Huang, S. Mammalian target of rapamycin signaling is involved in the vasculogenic mimicry of glioma via hypoxia-inducible factor-1α. Oncol. Rep. 2014, 32, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Jhanwar-Uniyal, M.; Gulati, N.; Karsy, M.; Albert, L.; Murali, R. Involvement of mTORC1 and mTORC2 in regulation of glioblastoma multiforme growth and motility. Int. J. Oncol. 2009, 35, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.A.; Dvorak, H.F. Heterogeneity of the tumor vasculature: The need for new tumor blood vessel type-specific targets. Clin. Exp. Metastasis 2012, 29, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [Green Version]
- Gacche, R.N.; Meshram, R.J. Angiogenic factors as potential drug target: Efficacy and limitations of anti-angiogenic therapy. Biochim. Biophys. Acta 2014, 1846, 161–179. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Reardon, D.A.; De Groot, J.F.; Wick, W.; Weller, M. Antiangiogenic Therapy for Glioblastoma: Current Status and Future Prospects. Clin. Cancer Res. 2014, 20, 5612–5619. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Franklin, R.A.; Montalto, G.; Cervello, M.; Libra, M.; Candido, S.; Malaponte, G.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: How mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012, 3, 1068–1111. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, M.K.; Lukas, R.V.; Jafri, N.F.; Faoro, L.; Salgia, R. Epidermal Growth Factor Receptor–Mediated Signal Transduction in the Development and Therapy of Gliomas. Clin. Cancer Res. 2006, 12, 7261–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, P.K.; Hart, J. PI3K and STAT3: A New Alliance. Cancer Discov. 2011, 1, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Liu, X.; Zhao, P.; Zhao, H.; Gao, W.; Wang, L. Celastrol Suppresses Glioma Vasculogenic Mimicry Formation and Angiogenesis by Blocking the PI3K/Akt/mTOR Signaling Pathway. Front. Pharmacol. 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J.; Cho, B.J.; Lee, D.J.; Hwang, Y.H.; Chun, S.H.; Kim, H.H.; Kim, I.A. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: Targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer 2014, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofano, A.; Pandolfi, P.P. The multiple roles of PTEN in tumor suppression. Cell 2000, 100, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Kessler, T.; Sahm, F.; Blaes, J.; Osswald, M.; Rübmann, P.; Milford, D.; Urban, S.; Jestaedt, L.; Heiland, S.; Bendszus, M.; et al. Glioma cell VEGFR-2 confers resistance to chemotherapeutic and antiangiogenic treatments in PTEN-deficient glioblastoma. Oncotarget 2015, 6, 31050–31068. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.K.; Sharma, P.; Harbor, P.C.; Rahaman, S.O.; Haque, S.J. PI3K-AKT pathway negatively controls EGFR-dependent DNA-binding activity of Stat3 in glioblastoma multiforme cells. Oncogene 2005, 24, 7290–7300. [Google Scholar] [CrossRef] [Green Version]
- Colardo, M.; Segatto, M.; Di Bartolomeo, S. Targeting RTK-PI3K-mTOR Axis in Gliomas: An Update. Int. J. Mol. Sci. 2021, 22, 4899. [Google Scholar] [CrossRef] [PubMed]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Di Iorio, P.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Robert, J. Textbook of Cell Signalling in Cancer; Springer: Singapore, 2015. [Google Scholar]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Yamada, D.; Hoshii, T.; Tanaka, S.; Hegazy, A.M.; Kobayashi, M.; Tadokoro, Y.; Ohta, K.; Ueno, M.; Ali, M.A.; Hirao, A. Loss of Tsc1 accelerates malignant gliomagenesis when combined with oncogenic signals. J. Biochem. 2013, 155, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Byron, S.A.; Tran, N.L.; Halperin, R.F.; Phillips, J.J.; Kuhn, J.G.; De Groot, J.F.; Colman, H.; Ligon, K.L.; Wen, P.Y.; Cloughesy, T.F.; et al. Prospective Feasibility Trial for Genomics-Informed Treatment in Recurrent and Progressive Glioblastoma. Clin. Cancer Res. 2018, 24, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Zhao, G.; Xie, G.; Zhao, L.; Chen, Y.; Yu, H.; Zhang, Z.; Li, C.; Li, Y. Metformin and temozolomide act synergistically to inhibit growth of glioma cells and glioma stem cells in vitro and in vivo. Oncotarget 2015, 6, 32930–32943. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.; Dhruv, H.D.; Peng, S.; Inge, L.J.; Tuncali, S.; Pineda, M.; Millard, N.; Mayo, Z.; Eschbacher, J.M.; Loftus, J.C.; et al. EGFRvIII–Stat5 Signaling Enhances Glioblastoma Cell Migration and Survival. Mol. Cancer Res. 2018, 16, 1185–1195. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.Y.; Kim, C.G.; Kim, S.H.; Kim, Y.S.; Lim, Y.; Lee, Y.H. Chlorpromazine activates p21 Waf1/Cip1 gene transcription via early growth response-1 (Egr-1) in C6 glioma cells. Exp. Mol. Med. 2010, 42, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Liu, L.; Xue, Y.; Zheng, J.; Liu, X.; Ma, J.; Li, Z.; Liu, Y. Combination of endothelial-monocyte-activating polypeptide-II with temozolomide suppress malignant biological behaviors of human glioblastoma stem cells via miR-590-3p/MACC1 Inhibiting PI3K/AKT/mTOR signal path-way. Front. Mol. Neurosci. 2017, 10, 68. [Google Scholar] [CrossRef]
- Burger, M.T.; Pecchi, S.; Wagman, A.; Ni, Z.-J.; Knapp, M.; Hendrickson, T.; Atallah, G.; Pfister, K.; Zhang, Y.; Bartulis, S.; et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med. Chem. Lett. 2011, 2, 774–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koul, D.; Fu, J.; Shen, R.; LaFortune, T.A.; Wang, S.; Tiao, N.; Kim, Y.-W.; Liu, J.-L.; Ramnarian, D.; Yuan, Y.; et al. Antitumor Activity of NVP-BKM120—A Selective Pan Class I PI3 Kinase Inhibitor Showed Differential Forms of Cell Death Based on p53 Status of Glioma Cells. Clin. Cancer Res. 2012, 18, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: An open-label, multicenter, multi-arm, phase II trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Gwak, H.S.; Shingu, T.; Chumbalkar, V.; Hwang, Y.-H.; DeJournett, R.; Latha, K.; Koul, D.; Yung, W.K.A.; Powis, G.; Farrell, N.P.; et al. Combined action of the dinuclear platinum compound BBR3610 with the PI3-K inhibitor PX-866 in glioblastoma. Int. J. Cancer 2011, 128, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Koul, D.; Shen, R.; Kim, Y.-W.; Kondo, Y.; Lu, Y.; Bankson, J.; Ronen, S.M.; Kirkpatrick, D.L.; Powis, G.; Yung, W.K.A. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro-Oncology 2010, 12, 559–569. [Google Scholar] [CrossRef]
- Pitz, M.W.; Eisenhauer, E.A.; MacNeil, M.V.; Thiessen, B.; Easaw, J.C.; Macdonald, D.R.; Eisenstat, D.D.; Kakumanu, A.S.; Salim, M.; Chalchal, H.; et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro-Oncology 2015, 17, 1270–1274. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Hon, G.C.; Villa, G.R.; Turner, K.M.; Ikegami, S.; Yang, H.; Ye, Z.; Li, B.; Kuan, S.; Lee, A.Y.; et al. EGFR Mutation Promotes Glioblastoma through Epigenome and Transcription Factor Network Remodeling. Mol. Cell 2015, 60, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; de Jesus, J.; et al. Oncogenic EGFR signaling activates an mTORC2–NF-κB pathway that promotes chemotherapy resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Y.; Kuan, Y.-H.; Ou, Y.-C.; Li, J.-R.; Wu, C.-C.; Pan, P.-H.; Chen, W.-Y.; Huang, H.-Y.; Chen, C.-J. Autophagy contributes to gefitinib-induced glioma cell growth inhibition. Exp. Cell Res. 2014, 327, 102–112. [Google Scholar] [CrossRef]
- Pédeboscq, S.; L’Azou, B.; Passagne, I.; De Giorgi, F.; Ichas, F.; Pometan, J.-P.; Cambar, J. Cytotoxic and apoptotic effects of bortezomib and gefitinib compared to alkylating agents on human glioblastoma cells. J. Exp. Ther. Oncol. 2008, 7, 99–111. [Google Scholar]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; Mckeage, M.; Squire, C.J. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef]
- Zhao, H.-F.; Wang, J.; Shao, W.; Wu, C.-P.; Chen, Z.-P.; To, S.S.T.; Li, W.-P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, H.-E.; Yang, L.; Chen, K.; Wang, Y.; Liu, Y.-R.; Pan, S.-L.; Gaur, S.; Hu, S.; Yen, Y. The pan-PI3K inhibitor GDC-0941 activates canonical WNT signaling to confer resistance in TNBC cells: Resistance reversal with WNT inhibitor. Oncotarget 2015, 6, 11061–11073. [Google Scholar] [CrossRef]
- Raynaud, F.I.; Eccles, S.A.; Patel, S.; Alix, S.; Box, G.; Chuckowree, I.; Folkes, A.; Gowan, S.; Brandon, A.D.H.; Di Stefano, F.; et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: From PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol. Cancer Ther. 2009, 8, 1725–1738. [Google Scholar] [CrossRef] [Green Version]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The Identification of 2-(1H-Indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef]
- Ndubaku, C.O.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2-{3-[2-(1-Isopropyl-3-methyl-1 H-1, 2–4-triazol-5-yl)-5, 6-dihydrobenzo [f] imidazo [1, 2-d][1, 4] oxazepin-9-yl]-1 H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): A β-sparing phosphoinositide 3-kinase inhib-itor with high unbound exposure and robust in vivo antitumor activity. J. Med. Chem. 2013, 56, 4597–4610. [Google Scholar]
- Maira, S.-M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and Characterization of NVP-BKM120, an Orally Available Pan-Class I PI3-Kinase Inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Foster, P.; Yamaguchi, K.; Hsu, P.P.; Qian, F.; Du, X.; Wu, J.; Won, K.-A.; Yu, P.; Jaeger, C.T.; Zhang, W.; et al. The Selective PI3K Inhibitor XL147 (SAR245408) Inhibits Tumor Growth and Survival and Potentiates the Activity of Chemotherapeutic Agents in Preclinical Tumor Models. Mol. Cancer Ther. 2015, 14, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Rowley, B.R.; Bull, C.O.; Schneider, C.; Haegebarth, A.; Schatz, C.A.; Fracasso, P.R.; Wilkie, D.P.; Hentemann, M.; Wilhelm, S.M.; et al. BAY 80-6946 Is a Highly Selective Intravenous PI3K Inhibitor with Potent p110α and p110δ Activities in Tumor Cell Lines and Xenograft Models. Mol. Cancer Ther. 2013, 12, 2319–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, W.J.; Hentemann, M.F.; Rowley, R.B.; Bull, C.O.; Jenkins, S.; Bullion, A.M.; Johnson, J.; Redman, A.; Robbins, A.H.; Esler, W.; et al. Discovery and SAR of novel 2, 3-dihydroimidazo [1, 2-c] quinazoline PI3K inhibitors: Identification of copanlisib (BAY 80-6946). ChemMedChem 2016, 11, 1517–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihle, N.T.; Paine-Murrieta, G.; Berggren, M.I.; Baker, A.; Tate, W.R.; Wipf, P.; Abraham, R.T.; Kirkpatrick, D.L.; Powis, G. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non–small cell lung cancer xenografts. Mol. Cancer Ther. 2005, 4, 1349–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Gaut, D.; Hu, K.; Yan, H.; Yin, D.; Koeffler, H.P. Dual targeting of glioblastoma multiforme with a proteasome inhibitor (Velcade) and a phosphatidylinositol 3-kinase inhibitor (ZSTK474). Int. J. Oncol. 2013, 44, 557–562. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Yamori, T. ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci. 2007, 98, 1638–1642. [Google Scholar] [CrossRef]
- Norman, M.H.; Andrews, K.L.; Bo, Y.Y.; Booker, S.K.; Caenepeel, S.; Cee, V.J.; D’angelo, N.D.; Freeman, D.J.; Herberich, B.J.; Hong, F.-T.; et al. Selective Class I Phosphoinositide 3-Kinase Inhibitors: Optimization of a Series of Pyridyltriazines Leading to the Identification of a Clinical Candidate, AMG 511. J. Med. Chem. 2012, 55, 7796–7816. [Google Scholar] [CrossRef]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef]
- Lannutti, B.J.; Meadows, S.A.; Herman, S.E.M.; Kashishian, A.; Steiner, B.; Johnson, A.J.; Byrd, J.C.; Tyner, J.W.; Loriaux, M.M.; Deininger, M.; et al. CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011, 117, 591–594. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.-F.; Wang, J.; Jiang, H.-R.; Chen, Z.-P.; To, S.S.T. PI3K p110β isoform synergizes with JNK in the regulation of glioblastoma cell proliferation and migra-tion through Akt and FAK inhibition. J. Exp. Clin. Cancer Res. 2016, 35, 78. [Google Scholar] [CrossRef] [Green Version]
- Cushing, T.D.; Hao, X.; Shin, Y.; Andrews, K.; Brown, M.; Cardozo, M.; Chen, Y.; Duquette, J.; Fisher, B.; de Turiso, F.G.-L.; et al. Discovery and in Vivo Evaluation of (S)-N-(1-(7-Fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (AMG319) and Related PI3Kδ Inhibitors for Inflammation and Autoimmune Disease. J. Med. Chem. 2015, 58, 480–511. [Google Scholar] [CrossRef]
- Nylander, S.; Kull, B.; Björkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skärby, T.; Inghardt, T.; Fjellström, O.; et al. Human target validation of phosphoinositide 3-kinase (PI3K) β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef]
- Ohwada, J.; Ebiike, H.; Kawada, H.; Tsukazaki, M.; Nakamura, M.; Miyazaki, T.; Morikami, K.; Yoshinari, K.; Yoshida, M.; Kondoh, O.; et al. Discovery and biological activity of a novel class I PI3K inhibitor, CH5132799. Bioorg. Med. Chem. Lett. 2011, 21, 1767–1772. [Google Scholar] [CrossRef]
- Camps, M.; Rückle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Françon, B.; et al. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef]
- So, L.; Yea, S.S.; Oak, J.S.; Lu, M.; Manmadhan, A.; Ke, Q.H.; Janes, M.R.; Kessler, L.V.; Kucharski, J.M.; Li, L.-S.; et al. Selective Inhibition of Phosphoinositide 3-Kinase p110α Preserves Lymphocyte Function. J. Biol. Chem. 2013, 288, 5718–5731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maira, S.-M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; de Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Xie, G.; Zhou, G.; Cheng, Y.; Zhang, G.; Yao, G.; Chen, Y.; Li, Y.; Zhao, G. NVP-BEZ235, a novel dual PI3K–mTOR inhibitor displays anti-glioma activity and reduces chemoresistance to temozolomide in human glioma cells. Cancer Lett. 2015, 367, 58–68. [Google Scholar] [CrossRef]
- Knight, S.D.; Adams, N.D.; Burgess, J.L.; Chaudhari, A.M.; Darcy, M.G.; Donatelli, C.A.; Luengo, J.I.; Newlander, K.A.; Parrish, C.A.; Ridgers, L.H.; et al. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med. Chem. Lett. 2010, 1, 39–43. [Google Scholar] [CrossRef]
- Yu, P.; Laird, A.D.; Du, X.; Wu, J.; Won, K.-A.; Yamaguchi, K.; Hsu, P.P.; Qian, F.; Jaeger, C.T.; Zhang, W.; et al. Characterization of the Activity of the PI3K/mTOR Inhibitor XL765 (SAR245409) in Tumor Models with Diverse Genetic Alterations Affecting the PI3K Pathway. Mol. Cancer Ther. 2014, 13, 1078–1091. [Google Scholar] [CrossRef] [Green Version]
- Sutherlin, D.P.; Bao, L.; Berry, M.; Castanedo, G.; Chuckowree, I.; Dotson, J.; Folks, A.; Friedman, L.; Goldsmith, R.; Gunzner, J.; et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J. Med. Chem. 2011, 54, 7579–7587. [Google Scholar] [CrossRef]
- Heffron, T.P.; Ndubaku, C.O.; Salphati, L.; Alicke, B.; Cheong, J.; Drobnick, J.; Edgar, K.; Gould, S.E.; Lee, L.B.; Lesnick, J.D.; et al. Discovery of Clinical Development Candidate GDC-0084, a Brain Penetrant Inhibitor of PI3K and mTOR. ACS Med. Chem. Lett. 2016, 7, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Hart, S.; Novotny-Diermayr, V.; Goh, K.C.; Williams, M.; Tan, Y.C.; Ong, L.C.; Cheong, A.; Ng, B.K.; Amalini, C.; Madan, B.; et al. VS-5584, a Novel and Highly Selective PI3K/mTOR Kinase Inhibitor for the Treatment of Cancer. Mol. Cancer Ther. 2013, 12, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Mehta, P.P.; Yin, M.-J.; Sun, S.; Zou, A.; Chen, J.; Rafidi, K.; Feng, Z.; Nickel, J.; Engebretsen, J.; et al. PF-04691502, a Potent and Selective Oral Inhibitor of PI3K and mTOR Kinases with Antitumor Activity. Mol. Cancer Ther. 2011, 10, 2189–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesan, A.M.; Dehnhardt, C.M.; Santos, E.D.; Chen, Z.; Santos, O.D.; Ayral-Kaloustian, S.; Khafizova, G.; Brooijmans, N.; Mallon, R.; Hollander, I.; et al. Bis (morpholino-1, 3, 5-triazine) derivatives: Potent adenosine 5′-triphosphate competitive phos-phatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: Discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J. Med. Chem. 2010, 53, 2636–2645. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.A.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination with Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [Green Version]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Takano, S.; Ishikawa, E.; Nakai, K.; Masumoto, T.; Yamamoto, T.; Matsumura, A.; Matsuda, M. Bevacizumab in Japanese patients with malignant glioma: From basic research to clinical trial. OncoTargets Ther. 2014, 7, 1551–1562. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Tanaka, T.; Miyake, K.; Tabei, Y.; Ohara, K.; Sampetrean, O.; Kono, M.; Mizutani, K.; Yamamoto, Y.; Murayama, Y.; et al. Histopathological investigation of glioblastomas resected under bevacizumab treatment. Oncotarget 2016, 7, 52423–52435. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Gil-Gil, M.J.; Mesia, C.; Rey, M.; Bruna, J. Bevacizumab for the Treatment of Glioblastoma. Clin. Med. Insights Oncol. 2013, 7, 123–135. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Miyake, K.; Yoshida, K.; Sasaki, H. Bevacizumab for malignant gliomas: Current indications, mechanisms of action and resistance, and markers of response. Brain Tumor Pathol. 2017, 34, 62–77. [Google Scholar] [CrossRef]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition through a MET/VEGFR2 Complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Labussière, M.; Cheneau, C.; Prahst, C.; Pérez-Larraya, J.G.; Farina, P.; Lombardi, G.; Mokhtari, K.; Rahimian, A.; Delattre, J.; Eichmann, A.; et al. Angiopoietin-2 may be involved in the resistance to bevacizumab in recurrent glioblastoma. Cancer Investig. 2016, 34, 39–44. [Google Scholar] [CrossRef]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; de Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Becket, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef]
- Tabouret, E.; Denicolaï, E.; Delfino, C.; Graillon, T.; Boucard, C.; Nanni, I.; Padovani, L.; Figarella-Branger, D.; Chinot, O. Changes in PlGF and MET-HGF expressions in paired initial and recurrent glioblastoma. J. Neuro-Oncol. 2016, 130, 431–437. [Google Scholar] [CrossRef]
- Lassen, U.; Chinot, O.L.; McBain, C.; Mau-Sørensen, M.; Larsen, V.A.; Barrie, M.; Roth, P.; Krieter, O.; Wang, K.; Habben, K.; et al. Phase 1 dose-escalation study of the antiplacental growth factor monoclonal antibody RO5323441 combined with bevacizumab in patients with recurrent glioblastoma. Neuro-Oncology 2015, 17, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Puduvalli, V.K.; Chamberlain, M.C.; van den Bent, M.J.; Carpentier, A.F.; Cher, L.M.; Mason, W.; Weller, M.; Hong, S.; Musib, L.; et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 2010, 28, 1168–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, V.A.; Chan, J.; Datta, M.; Yee, J.L.; Jain, R.K. Effect of angiotensin system inhibitors on survival in newly diagnosed glioma patients and recurrent glioblastoma patients receiving chemotherapy and/or bevacizumab. J. Neuro-Oncol. 2017, 134, 325–330. [Google Scholar] [CrossRef]
- Carpentier, A.F.; Ferrari, D.; Bailon, O.; Ursu, R.; Banissi, C.; Dubessy, A.-L.; Belin, C.; Levy, C. Steroid-sparing effects of angiotensin-II inhibitors in glioblastoma patients. Eur. J. Neurol. 2012, 19, 1337–1342. [Google Scholar] [CrossRef]
- Januel, E.; Ursu, R.; Alkhafaji, A.; Marantidou, A.; Doridam, J.; Belin, C.; Levy-Piedbois, C.; Carpentier, A.F. Impact of renin-angiotensin system blockade on clinical outcome in glioblastoma. Eur. J. Neurol. 2015, 22, 1304–1309. [Google Scholar] [CrossRef]
- Arrieta, O.; Guevara, P.; Escobar, E.; García-Navarrete, R.; Pineda, B.; Sotelo, J. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br. J. Cancer 2005, 92, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Woolf, E.C.; Curley, K.L.; Liu, Q.; Turner, G.H.; Charlton, J.A.; Preul, M.C.; Scheck, A.C. The ketogenic diet alters the hypoxic response and affects expression of proteins associated with angio-genesis, invasive potential and vascular permeability in a mouse glioma model. PLoS ONE 2015, 10, e0130357. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Pinto, A.; Martherus, R.; de Jesus, J.P.S.; Polet, F.; Feron, O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Minami, N.; Tanaka, K.; Sasayama, T.; Kohmura, E.; Saya, H.; Sampetrean, O. Lactate Reprograms Energy and Lipid Metabolism in Glucose-Deprived Oxidative Glioma Stem Cells. Metabolites 2021, 11, 325. [Google Scholar] [CrossRef]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology 2017, 19, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13C] glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [Green Version]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.M.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dy-namic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Tao, B.-B.; He, H.; Shi, X.-H.; Wang, C.-L.; Li, W.-Q.; Li, B.; Dong, Y.; Hu, G.-H.; Hou, L.-J.; Luo, C.; et al. Up-regulation of USP2a and FASN in gliomas correlates strongly with glioma grade. J. Clin. Neurosci. 2013, 20, 717–720. [Google Scholar] [CrossRef]
- Michalak, K.P.; Maćkowska-Kędziora, A.; Sobolewski, B.; Woźniak, P. Key Roles of Glutamine Pathways in Reprogramming the Cancer Metabolism. Oxidative Med. Cell. Longev. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oizel, K.; Chauvin-Fleurence, C.; Oliver, L.; Gratas, C.; Geraldo, F.; Jarry, U.; Scotet, E.; Rabe, M.; Alves-Guerra, M.-C.; Teusan, R.; et al. Efficient Mitochondrial Glutamine Targeting Prevails Over Glioblastoma Metabolic Plasticity. Clin. Cancer Res. 2017, 23, 6292–6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieger, J.; Bähr, O.; Maurer, G.D.; Hattingen, E.; Franz, K.; Brucker, D.; Walenta, S.; Kämmerer, U.; Coy, J.F.; Weller, M.; et al. ERGO: A pilot study of ketogenic diet in recurrent glioblastoma. Int. J. Oncol. 2014, 44, 1843–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartoš, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Kuang, R.; Jahangiri, A.; Mascharak, S.; Nguyen, A.; Chandra, A.; Flanigan, P.M.; Yagnik, G.; Wagner, J.R.; de Lay, M.; Carrera, D.; et al. GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight 2017, 2, e88815. [Google Scholar] [CrossRef]
- Ruiz-Ontañon, P.; Orgaz, J.L.; Aldaz, B.; Elosegui-Artola, A.; Martino, J.; Berciano, M.T.; Montero, J.A.; Grande, L.; Nogueira, L.; Diaz-Moralli, S.; et al. Cellular Plasticity Confers Migratory and Invasive Advantages to a Population of Glioblastoma-Initiating Cells that Infiltrate Peritumoral Tissue. Stem Cells 2013, 31, 1075–1085. [Google Scholar] [CrossRef]
- Abbadi, S.; Rodarte, J.J.; Abutaleb, A.; Lavell, E.; Smith, C.L.; Ruff, W.; Schiller, J.; Olivi, A.; Levchenko, A.; Guerrero-Cazares, H.; et al. Glucose-6–phosphatase Is a Key Metabolic Regulator of Glioblastoma Invasion. Mol. Cancer Res. 2014, 12, 1547–1559. [Google Scholar] [CrossRef] [Green Version]
- Libby, C.; Tran, A.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim. Biophys. Acta 2018, 1869, 175–188. [Google Scholar] [CrossRef]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase: The energy charge hypothesis revisited. BioEssays 2001, 23, 1112–1119. [Google Scholar] [CrossRef]
- Choo, A.Y.; Roux, P.P.; Blenis, J. Mind the GAP: Wnt Steps onto the mTORC1 Train. Cell 2006, 126, 834–836. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boado, R.J.; Black, K.L.; Pardridge, W. Gene expression of GLUT3 and GLUT1 glucose transporters in human brain tumors. Mol. Brain Res. 1994, 27, 51–57. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine Stimulation Promotes Glucose Uptake via Phosphatidylinositol-3 Kinase/Akt Regulation of Glut1 Activity and Trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Birner, P.; Piribauer, M.; Fischer, I.; Gatterbauer, B.; Marosi, C.; Ambros, P.F.; Ambros, I.M.; Bredel, M.; Oberhuber, G.; Rössler, K.; et al. Vascular patterns in glioblastoma influence clinical outcome and associate with variable expression of angiogenic proteins: Evidence for distinct angiogenic subtypes. Brain Pathol. 2003, 13, 133–143. [Google Scholar] [CrossRef]
- Flynn, J.R.; Wang, L.; Gillespie, D.; Mph, G.J.S.; Reid, J.K.; Owens, J.; Bs, G.B.E.; Salzman, K.L.; Rn, A.Y.K.; Jensen, R.L. Hypoxia-regulated protein expression, patient characteristics, and preoperative imaging as predictors of survival in adults with glioblastoma multiforme. Cancer 2008, 113, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, R.; Takano, S.; Ishikawa, E.; Yamamoto, T.; Nakai, K.; Matsumura, A. Hypoxia-inducible factor 1α expression is a prognostic biomarker in patients with astrocytic tumors associated with necrosis on MR image. J. Neuro-Oncol. 2011, 102, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ju, T.; Yang, D.-I. Induction of hypoxia inducible factor-1 attenuates metabolic insults induced by 3-nitropropionic acid in rat C6 glioma cells. J. Neurochem. 2005, 93, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Yan, S.-F.; Xue, H.; Zhang, P.; Sun, J.-T.; Li, G. Cucurbitacin I Induces Protective Autophagy in Glioblastoma in Vitro and in Vivo. J. Biol. Chem. 2014, 289, 10607–10619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Reitano, R.; Magro, G.; Cavallaro, S.; Salomone, S.; D’Agata, V. PACAP and VIP inhibit the invasiveness of glioblastoma cells exposed to hypoxia through the regula-tion of HIFs and EGFR expression. Front. Pharmacol. 2016, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Gliomas: Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma. Front. Physiol. 2017, 8, 352. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.; Cayanan, G.; Simonds, E.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Kahn, J.; Hayman, T.J.; Jamal, M.; Rath, B.H.; Kramp, T.; Camphausen, K.; Tofilon, P.J. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro-Oncology 2014, 16, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.; Smith, J.M.; Miyamoto, S. Hexokinase-II Positively Regulates Glucose Starvation-Induced Autophagy through TORC1 Inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegazy, A.M.; Yamada, D.; Kobayashi, M.; Kohno, S.; Ueno, M.; Ali, M.A.E.; Ohta, K.; Tadokoro, Y.; Ino, Y.; Todo, T.; et al. Therapeutic Strategy for Targeting Aggressive Malignant Gliomas by Disrupting Their Energy Balance. J. Biol. Chem. 2016, 291, 21496–21509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanduja, S.; Feng, Y.; Mathis, R.A.; Sokol, E.S.; Reinhardt, F.; Halaban, R.; Gupta, P.B. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene 2016, 35, 5295–5303. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Lee, J.-H.; Oh, Y.; Koh, I.; Shim, J.-K.; Park, J.; Choi, J.; Yun, M.; Jeon, J.Y.; Huh, Y.M.; et al. Inhibition of glioblastoma tumorspheres by combined treatment with 2-deoxyglucose and metformin. Neuro-Oncology 2016, 19, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol. Metab. 2014, 25, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Sun, C.; Li, Y.; Li, X.; Sun, T.; Liu, C.; Zhou, Y.; Du, Z. MicroRNA-199a-3p suppresses glioma cell proliferation by regulating the AKT/mTOR signaling pathway. Tumor Biol. 2015, 36, 6929–6938. [Google Scholar] [CrossRef] [Green Version]
- Rathod, S.S.; Rani, S.B.; Khan, M.; Muzumdar, D.; Shiras, A. Tumor suppressive miRNA-34a suppresses cell proliferation and tumor growth of glioma stem cells by targeting Akt and Wnt signaling pathways. FEBS Open Bio 2014, 4, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Shi, Z.; Wei, W.; Lu, C.; Wei, Y.; Yan, W.; Li, R.; Zhang, J.; You, Y.; Wang, X. MiR-181b suppress glioblastoma multiforme growth through inhibition of SP1-mediated glucose metabolism. Cancer Cell Int. 2020, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Basso, J.; Miranda, A.; Sousa, J.; Pais, A.; Vitorino, C. Repurposing drugs for glioblastoma: From bench to bedside. Cancer Lett. 2018, 428, 173–183. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Quinoline-based antimalarial drugs: A novel class of autophagy inhibitors. Neurosurg. Focus 2015, 38, E12. [Google Scholar] [CrossRef]
- Roy, L.-O.; Poirier, M.-B.; Fortin, D. Chloroquine inhibits the malignant phenotype of glioblastoma partially by suppressing TGF-beta. Investig. New Drugs 2015, 33, 1020–1031. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Z.-G.; Xia, Q.; Lin, J.-P.; Yan, Z.-Q.; Yao, Z.-J.; Dong, J. Mefloquine inhibits chondrocytic proliferation by arresting cell cycle in G2/M phase. Int. J. Clin. Exp. Pathol. 2015, 8, 12583–12588. [Google Scholar]
- Erkoc, P.; Cingoz, A.; Onder, T.B.; Kizilel, S. Quinacrine Mediated Sensitization of Glioblastoma (GBM) Cells to TRAIL through MMP-Sensitive PEG Hydrogel Carriers. Macromol. Biosci. 2016, 17, 1600267. [Google Scholar] [CrossRef]
- Venugopal, C.; Hallett, R.M.; Vora, P.; Manoranjan, B.; Mahendram, S.; Qazi, M.A.; McFarlane, N.; Subapanditha, M.K.; Nolte, S.M.; Singh, M.; et al. Pyrvinium Targets CD133 in Human Glioblastoma Brain Tumor–Initiating Cells. Clin. Cancer Res. 2015, 21, 5324–5337. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef] [Green Version]
- Naujokat, C.; Steinhart, R. Salinomycin as a Drug for Targeting Human Cancer Stem Cells. J. Biomed. Biotechnol. 2012, 2012, 1–17. [Google Scholar] [CrossRef]
- Liu, W.-T.; Lin, C.-H.; Hsiao, M.; Gean, P.-W. Minocycline inhibits the growth of glioma by inducing autophagy. Autophagy 2011, 7, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Oliva, C.R.; Zhang, W.; Langford, C.; Suto, M.J.; Griguer, C.E. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget 2017, 8, 37568–37583. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, N.; Li, H.; Liu, S.; Chen, X.; Yu, S.; Wu, N.; Bian, X.W.; Shen, H.Y.; Li, C.; et al. Promoting oligodendroglial-oriented differentiation of glioma stem cell: A repurposing of quetiapine for the treatment of malignant glioma. Oncotarget 2017, 8, 37511–37524. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Meng, L.; Jiang, Y.; Cheng, W.; Tie, X.; Xia, J.; Wu, A. Lithium enhances the antitumour effect of temozolomide against TP53 wild-type glioblastoma cells via NFAT1/FasL signalling. Br. J. Cancer 2017, 116, 1302–1311. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.O.; Dmitrieva, N.; Stein, A.M.; Cutter, J.L.; Godlewski, J.; Saeki, Y.; Nita, M.; Berens, M.E.; Sander, L.M.; Newton, H.B.; et al. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro-Oncology 2008, 10, 690–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Michiue, H.; Yamada, H.; Takata, K.; Nakayama, H.; Wei, F.; Fujimura, A.; Tazawa, H.; Asai, A.; Ogo, N.; et al. Fluvoxamine, an anti-depressant, inhibits human glioblastoma invasion by disrupting actin polymerization. Sci. Rep. 2016, 6, 23372. [Google Scholar] [CrossRef]
- Bielecka-Wajdman, A.M.; Lesiak, M.; Ludyga, T.; Sieron, A.L.; Obuchowicz, E. Reversing glioma malignancy: A new look at the role of antidepressant drugs as adjuvant therapy for glioblastoma multiforme. Cancer Chemother. Pharmacol. 2017, 79, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Kim, S.H.; Kim, Y.; Kim, Y.S.; Lim, Y.; Lee, Y.H.; Shin, S.Y. The tricyclic antidepressant imipramine induces autophagic cell death in U-87MG glioma cells. Biochem. Biophys. Res. Commun. 2011, 413, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Shchors, K.; Massaras, A.; Hanahan, D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 2015, 28, 456–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, L.; Cruickshanks, N.; Tavallai, S.; Roberts, J.L.; Peery, M.; Poklepovic, A.; Dent, P. Regulation of dimethyl-fumarate toxicity by proteasome inhibitors. Cancer Biol. Ther. 2014, 15, 1646–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Mukthavaram, R.; Chao, Y.; Nomura, N.; Bharati, I.S.; Fogal, V.; Pastorino, S.; Teng, D.; Cong, X.; Pingle, S.C.; et al. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br. J. Cancer 2014, 111, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Jiang, H.; Lu, D.; Xiong, Y.; Qu, C.; Zhou, D.; Mahmood, A.; Chopp, M. Effect of Simvastatin on Glioma Cell Proliferation, Migration, and Apoptosis. Neurosurgery 2009, 65, 1087–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanae, M.; Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yamazoe, Y.; Nishida, S. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranyl-geranyl-pyrophosphate biosynthesis in glioblastoma. J. Exp. Clin. Cancer Res. 2011, 30, 74. [Google Scholar] [CrossRef] [Green Version]
- Keir, S.T.; Friedman, H.S.; Reardon, D.A.; Bigner, D.D.; Gray, L.A. Mibefradil, a novel therapy for glioblastoma multiforme: Cell cycle synchronization and interlaced therapy in a murine model. J. Neuro-Oncol. 2013, 111, 97–102. [Google Scholar] [CrossRef]
- Zhang, Y.; Cruickshanks, N.; Yuan, F.; Wang, B.; Pahuski, M.; Wulfkuhle, J.; Gallagher, I.; Koeppel, A.F.; Hatef, S.; Papanicolas, C.; et al. Targetable T-type Calcium Channels Drive Glioblastoma. Cancer Res. 2017, 77, 3479–3490. [Google Scholar] [CrossRef] [Green Version]
- Rivera, E.; Arrieta, O.; Guevara, P.; Duarte-Rojo, A.; Sotelo, J. AT1 receptor is present in glioma cells; its blockage reduces the growth of rat glioma. Br. J. Cancer 2001, 85, 1396–1399. [Google Scholar] [CrossRef]
- Gritti, M.; Würth, R.; Angelini, M.; Barbieri, F.; Peretti, M.; Pizzi, E.; Pattarozzi, A.; Carra, E.; Sirito, R.; Daga, A.; et al. Metformin repositioning as antitumoral agent: Selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014, 5, 11252–11268. [Google Scholar] [CrossRef] [Green Version]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.-T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Moyal, E.C.-J.; et al. Metformin Inhibits Growth of Human Glioblastoma Cells and Enhances Therapeutic Response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef] [Green Version]
- Cilibrasi, C.; Butta, V.; Riva, G.; Bentivegna, A. Pioglitazone Effect on Glioma Stem Cell Lines: Really a Promising Drug Therapy for Glioblastoma? PPAR Res. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Shi, W.; Shao, B.; Shi, J.; Shen, A.; Ma, Y.; Chen, J.; Lan, Q. Peroxisome proliferator-activated receptor γ agonist pioglitazone inhibits β-catenin-mediated glioma cell growth and invasion. Mol. Cell. Biochem. 2011, 349, 1–10. [Google Scholar] [CrossRef]
- Kast, R.E.; Ramiro, S.; Lladó, S.; Toro, S.; Coveñas, R.; Muñoz, M. Antitumor action of temozolomide, ritonavir and aprepitant against human glioma cells. J. Neuro-Oncol. 2015, 126, 425–431. [Google Scholar] [CrossRef]
- Furuta, T.; Sabit, H.; Dong, Y.; Miyashita, K.; Kinoshita, M.; Uchiyama, N.; Hayashi, Y.; Hayashi, Y.; Minamoto, T.; Nakada, M. Biological basis and clinical study of glycogen synthase kinase- 3β-targeted therapy by drug repositioning for glioblastoma. Oncotarget 2017, 8, 22811–22824. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, F.; James, S.; Camby, I.; Gaussin, J.-F.; Darro, F.; Brotchi, J.; Gabius, H.-J.; Kiss, R. Combined cimetidine and temozolomide, compared with temozolomide alone: Significant increases in survival in nude mice bearing U373 human glioblastoma multiforme orthotopic xenografts. J. Neurosurg. 2005, 102, 706–714. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, S.; Sun, Q.; Liu, B. Anthelmintic drug ivermectin inhibits angiogenesis, growth and survival of glioblastoma through inducing mitochondrial dysfunction and oxidative stress. Biochem. Biophys. Res. Commun. 2016, 480, 415–421. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [Green Version]
- Mccord, M.; Mukouyama, Y.-S.; Gilbert, M.R.; Jackson, S. Targeting WNT Signaling for Multifaceted Glioblastoma Therapy. Front. Cell. Neurosci. 2017, 11, 318. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Drug Name | IC50 (nM) | References | |||||
|---|---|---|---|---|---|---|---|---|
| p110α | p110β | p110δ | p110γ | mTORC1/2 | ||||
| Pan-PI3K inhibitors | Pictilisib | 3 | 33 | 3 | 75 | 580 | Inhibits tumor proliferation | [148,149] |
| Taselisib | 0.29 | 9.1 | 0.12 | 0.97 | 1200 | Inhibits tumor proliferation | [150] | |
| Buparlisib | 52 | 166 | 116 | H | 4600 | Apoptotic cell death and p53 deleted cells | [134,151] | |
| Pilaralisib | 39 | 383 | 36 | 23 | >15,000 | Inhibits tumor proliferation | [152] | |
| Copanlisib | 0.5 | 3.7 | 0.7 | 6.4 | 45 | Induces apoptosis | [153,154] | |
| Sonolisib | 0.1 | >300 | 2.9 | N/A | N/A | Complete tumor growth control in the early stages of treatment | [155] | |
| ZSTK474 | 16 | 44 | 4.6 | 49 | >10,000 | Cell apoptosis | [156,157] | |
| AMG511 | 4 | 6 | 2 | 1 | N/A | Inhibits tumor growth | [158] | |
| Isoform-selective inhibitors | Alpelisib | 5 | 1200 | 290 | 250 | >9100 | Inhibits tumor growth | [159] |
| Idelalisib | 820 | 565 | 2.5 | 89 | >1000 | Suppresses GBM cell proliferation and migration | [160,161] | |
| AMG319 | 3300 | 2700 | 18 | 850 | N/A | [162] | ||
| AZD6482 | 870 | 10 | 80 | 1090 | N/A | PI3K inhibition as anti-platelet therapy; mild generalized anti-platelet effect | [163] | |
| CH5132799 | 14 | 120 | 500 | 36 | 1600 | Potent antiproliferative activity | [164] | |
| AS-605240 | 60 | 270 | 300 | 8 | N/A | Leukocyte activation and migration | [165] | |
| MLN1117 | 15 | 4500 | 1900 | 13,390 | 1670 | p110α inhibition; attenuated B cell receptor (BCR)-dependent AKT activation, proliferation, and survival when supported by cytokines BAFF and IL-4 | [166] | |
| Dual PI3K/mTOR inhibitors | Dactolisib | 4 | 75 | 7 | 5 | 6 | Effective and specific blocking of dysfunctional PI3K activation in human tumor cell models; G1 arrest | [167,168] |
| NVP-BGT226 | 4 | 63 | N/A | 38 | N/A | p110α inhibition; attenuated B cell receptor (BCR)-dependent AKT activation, proliferation, and survival when supported by cytokines BAFF and IL-4 | [166] | |
| Omipalisib | 0.019 | 0.13 | 0.024 | 0.06 | 0.18/0.3 | mTOR inhibitor augmenting anti-proliferative efficacy of PI3K/AKT pathway inhibition | [169] | |
| Voxtalisib | 39 | 113 | 41 | 9 | 160/910 | Anti-proliferative, anti-angiogenic, pro-apoptotic | [170] | |
| Apitolisib | 5 | 27 | 7 | 14 | 17 | mTOR inhibition | [171] | |
| GDC-0084 | 2 | 46 | 3 | 10 | 70 | PI3K inhibition | [172] | |
| VS-5584 | 16 | 68 | 42 | 25 | 37 | anti-proliferative | [173] | |
| PF-04691502 | 1.8 | 2.1 | 1.6 | 1.9 | 16 | Inhibits cell proliferation | [174] | |
| Gedatolisib | 0.4 | N/A | N/A | 5.4 | 1.6 | Inhibits PI3K/Akt signaling; attenuates proliferation, survival, protein synthesis, and glucose metabolism | [175] | |
| Drug | Suggested Mechanism of Action | IC50 | Outcome | References |
|---|---|---|---|---|
| Chloroquine | Autophagy induction, inhibition of MMP-2 activity, TGF-β secretion and signaling | 30 μΜ (U251, LN229, U87MG) 40 μΜ (U251-TMZR, LN229-TMZR, U87MG-TMZR) | Primary cultures of GBM cell lines and specimens showed a 50% reduction in proliferation. | [242,243] |
| Hydroxy-chloroquine | Autophagy induction | - | Hydroxy-chloroquine eliminated TMZ-resistant glioma cells. | [242] |
| Mefloquine | Autophagy induction | 10 μΜ (U251, LN229, U87) 15 μΜ (U251-TMZR, LN229-TMZR, U87-TMZR) | Mefloquine was capable to killing U251 and U251-TMZ resistant cells at far lower doses than chloroquine. | [242,244] |
| Quinacrine | Autophagy induction | 5 μΜ (U251, LN229, U87) 8 μΜ (U251-TMZR, LN229-TMZR, U87-TMZR) | 50 mg/kg of quinacrine substantially slowed the growth of tumors in a subcutaneous human xenograft U87MG model. | [242,245] |
| Pyrvinium pamoate | Inhibition of Wnt/β-catenin signaling | 239.8 nmol/L (BT241), 122.5 nmol/L (BT486) | For 48 h, 200 nmol/L of pyrvinium decreased CD133POS cell fractions in primary (BT428) and recurrent (BT 566) GBM cells. | [246] |
| Itraconazole | Inhibition of cell proliferation | - | Itraconazole inhibited the proliferation of GBM cells in vitro (2–80 μM, U87MG and rat C6 glioma cells) and in vivo (75 mg/kg, nude mice with U87MG subcutaneous xenografts). | [247] |
| Salinomycin | OxPhos inhibition in mitochondria | - | Salinomycin decreased the cell viability of GL261 neurospheres and GL261 adherent cells. Salinomycin depleted neurosphere-forming GL261 stem cells from tumorspheres. | [248] |
| Minocycline | Growth inhibition, autophagy induction, caspase-3 mediated apoptosis | 30 μM (C6) | 50 μM of minocycline decreased the cell viability of U87MG, U251 and C6 glioma cells; 20 or 100 mg/kg of minocycline (IP) showed slower tumor growth compared controls in Mice injected with C6 cells. | [249] |
| Chlorpromazine | Inhibition of PAM signaling, autophagy induced cell death | 18.8–27.7 μM (C6) 15 μM (SH-SY5Y) | Cell viability was significantly lowered in cells treated with chlorpromazine (≥20 μM) for 24 h. Overall survival greatly improved for U251-TMZR orthotopic mouse xenograft models. | [250] |
| Quetiapine | Inhibition of Wnt/β-catenin signaling | - | Relatively high doses of quetiapine (>25 μM) may inhibit cell proliferation by retarding cell cycle in the G2-M phase; 20 mg/kg of quetiapine (IP) alone or combined with TMZ slowed tumor development in orthotopic xenograft mouse model. | [251] |
| Lithium | Inhibition of GSK-3 activation | - | 20 mM lithium for 48 h reduced in viability of 20% of U87MG cells. Through GSK-3 inhibition, lithium concentrations above 5 mM can affect the proliferation, apoptosis and migration of glioma cells. Combination of 1.2 mM lithium and TMZ increased cell death in TP53wt glioma cells and prevented tumor growth in vivo with increased median survival times of mice. | [252,253] |
| Fluvoxamine | Inhibition of FAK and Akt/mTOR | 30 μM | 20–30 μM of fluvoxamine inhibited lamellipodia formation, migration and invasion of U87MG and U251 cells in vitro; 50 mg/kg of fluvoxamine inhibited GBM cell invasion and prolonged survival in mice bearing glioma tumors. | [254] |
| Imipramine | Autophagy induction; inhibition of PI3K/Akt/mTOR | - | 60 μM of imipramine was cytotoxic and strongly reduced colony formation of U87MG and C6 cells, but not primary cultured rat astrocytes; 10 μM of imipramine inhibited mitochondrial activity relative to oxygen content in the atmosphere (from 6% in hypoxia, 11% in mild hypoxia, to 19% in medium re-oxygenated at 26% oxygen). | [255,256,257] |
| Dimethyl fumarate | Autophagy induction | - | - | [258] |
| Simvastatin | Inhibition of cell growth and migration; inhibition of Ras/ERK and Ras/Akt pathways to induce caspase-3 mediated apoptosis; downregulation of PI3K/Akt | - | 10 μM of simvastatin was cytotoxic to U251 and U87MG by inducing aopotosis. | [259,260,261] |
| Mevastatin, fluvastatin | Inhibition of cell growth; inhibition of Ras/ERK and Ras/Akt; induction of apoptosis | 0.922 μM (A 172) | 5 and 10 μM of mevastatin and fluvastatin are cytotoxic. | [259,261] |
| Mibefradil | Inhibition of tumor growth; cell cycle inhibition; activation of pro-apoptotic survivin and BAX pathways; inhibition of Akt/mTOR | - | 2.5–5 μmol/L greatly inhibited cell growth and enhanced the inhibition of GSC growth by TMZ; 24 mg/kg (bodyweight) of mibefradil (oral gavage) significantly inhibited growth of tumor. | [262,263] |
| Losartan | Reducing tumor and cell growth; reduced number of capillary blood vessels; decreased levels of VEGF, PDGF and FGF; apoptosis induction | [193,264] | ||
| Metformin | Autophagy and induction of apoptosis; activated AMPK and down-regulation of Akt/mTOR pathway; inhibition of CLIC1 activity with G1 cell arrest | - | 10 mM significantly decreased GBM cell proliferation (U87MG, U251, LN18 and SF767). | [265,266] |
| Pioglitazone | Inhibition of β-catenin expression; reduced cell viability; apoptosis induction | 85 μM (U87MG) | 100–200 μM significantly reduced the viability of glioma cells (U251, T98G, and U87MG) in a concentration- and time-dependent manner; 100 μM pioglitazone via reducing MMP-2 expression can inhibit U251 cell migration; 50 μM significantly reduced the metabolic activity of G144 cells; 10 μM promoted a minor decrease in metabolic activity in GliNS2 cells. | [267,268] |
| Aprepitant | Enhanced blockage in Akt phosphorylation due to NK-1 signaling | 32 µM (GAMG) | Maximum inhibition at 70 μM after 48 h, with no surviving cells (GAMG glioma cell line). | [269] |
| Cimetidine | GSK-3β inhibition; reduction in endogenous receptors required for cell adhesion and migration | - | Decreased growth rates of U373 GBM and 9L gliosarcoma cells at concentrations equal to or higher than 100 mM; 100 and 1000 mM cimetidine significantly decreased migration of both cell lines; doses <100 mM did not affect cell cycle kinetics or apoptosis. | [270,271] |
| Ivermectin | Deactivation of the Akt/mTOR pathways | - | 1, 5, and 10 μM inhibited proliferation of U87MG and T98G cells in a dose-dependent manner with ED50 of ∼5 μM. | [272] |
| Rapamycin | Interfering with the AMPK/mTORC1 axis | [108] | ||
| Gefitinib and Erlotinib | ATP-competitive and reversible EGFR inhibitors | [141] | ||
| Sonolisib | Reduced invasion and angiogenesis in GBM cell lines in vitro; improved survival in orthotopic xenograft models | [136,137] | ||
| Buparlisib | Well-tolerated, BBB permeable PI3K inhibitor (most often utilized PI3K inhibitor in clinical studies for GBM therapy) | [135] | ||
| Pimozide | Inhibition of migration and survival of GBM cells in a STAT5- dependent manner | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. https://doi.org/10.3390/ijms23031353
Barzegar Behrooz A, Talaie Z, Jusheghani F, Łos MJ, Klonisch T, Ghavami S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. International Journal of Molecular Sciences. 2022; 23(3):1353. https://doi.org/10.3390/ijms23031353
Chicago/Turabian StyleBarzegar Behrooz, Amir, Zahra Talaie, Fatemeh Jusheghani, Marek J. Łos, Thomas Klonisch, and Saeid Ghavami. 2022. "Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma" International Journal of Molecular Sciences 23, no. 3: 1353. https://doi.org/10.3390/ijms23031353