
Application and Challenge of 3rd Generation Sequencing for Clinical Bacterial Studies

 ,
,
Abstract
:1. Transformation of Genome Sequencing Landscape
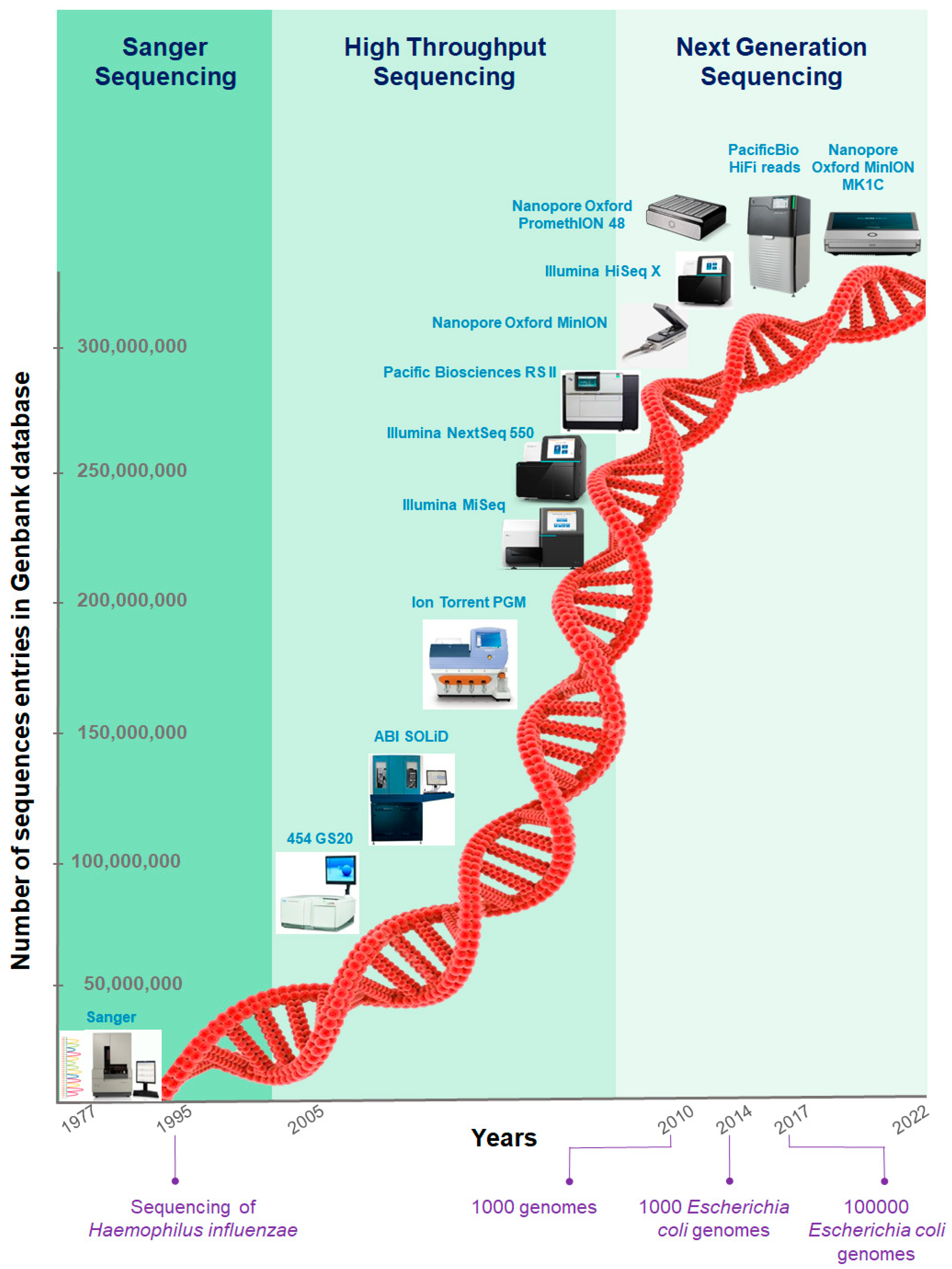
1.1. Emergence of Nucleic Acid Sequencing
1.2. Awake of Microbial Genomics
1.3. Short-Read Sequencing Limitations
1.4. Long-Read Sequencing Developments
2. Disruption of Clinical Studies on Prokaryotes
2.1. Molecular Detection and Identification of Pathogens
2.2. SNPs Genotyping
- (i)
- MLST allows the characterization of a genus (or species) that is already known to identify the species (or subspecies) thanks to the SNPs comparisons within a set of housekeeping genes [41,42,43]. It is commonly the reference technique to discriminate between different strains. The sequences of these housekeeping genes have the particularity to present a stable polymorphism in time but are divergent enough to distinguish strains between them. MLST analyses have become common because they provide good resolution while being easily reproducible and standardizable. Challagundla et al. analyzed 598 genome sequences of Staphylococcus aureus to track the evolution of Clonal Complex 5 Methicillin-Resistant, which caused several hospital-associated infections in the Western hemisphere [44]. Their analysis based on MLST comparisons was able to identify and characterize the geographical spread of S. aureus. CC5-MRSA clones over the world.
- (ii)
- The study of SNPs at the level of complete genomes is obviously more efficient and accurate than using only a set of housekeeping genes. This global approach is being developed at the same time as the WGS is being facilitated. An example is the tracking of diffusion and monitoring the evolution of M. tuberculosis Beijing lineage [41], a very virulent and potentially antibiotic-resistant strain. Using a large dataset of a single M. tuberculosis lineage, Merker et al. identified the biogeographic structure and evolutionary history of the Beijing lineage worldwide through the SNPs analysis of 4987 isolates from 99 countries [45]. They showed that this lineage originated in the Far East, from where it spread throughout the world in several waves. In addition, global SNPs genotyping was applied to Mycobacterium abscessus, a human skin bacterium. Choo et al. described the migration of the clinical isolates through different geographical locations, from India to Southeast Asia, Europe and then to the USA [46]. The outbreak of Vibrio cholerae in Haiti [47] is another example of the ability of SNPs genotyping to track strains. Talkington et al. have sequenced 122 isolates, genotyped and compared with isolates from other countries. The authors used SNP analyses to establish phylogeny and trace the origins of these outbreaks. Characterization based on genomes proves that Haiti isolates are clonally and genetically similar to isolates originating in southern Asia and Africa.
2.3. Phenotype Prediction to Track Virulence Factors and Antimicrobial Resistance
2.4. Comparative Genomics to Understand bacterial Strains Evolution
2.5. Taxogenomics
2.6. Metagenomics
2.7. Transcriptomics and MetaTranscriptomics
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sanger, F.; Brownlee, G.; Barrell, B. A two-dimensional fractionation procedure for radioactive nucleotides. J. Mol. Biol. 1965, 13, 373–398, IN1–IN4. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeoch, D.J.; Davison, A.J. Alphaherpesviruses possess a gene homologous to the protein kinase gene family of eukaryotes and retroviruses. Nucleic Acids Res. 1986, 14, 1765–1777. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M.; et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-Throughput Sequencing Technologies. Molecular Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morey, M.; Fernández-Marmiesse, A.; Castiñeiras, D.; Fraga, J.M.; Couce, M.L.; Cocho, J.A. A Glimpse into Past, Present, and Future DNA Sequencing. Mol. Genet. Metab. 2013, 110, 3–24. [Google Scholar] [CrossRef]
- Qiang-long, Z.; Shi, L.; Peng, G.; Fei-shi, L. High-Throughput Sequencing Technology and Its Application. J. Northeast. Agric. Univ. 2014, 21, 84–96. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2009, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Metzker, M.L. Emerging technologies in DNA sequencing. Genome Res. 2005, 15, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.D.; Du, J.; Lam, H.; Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. Identification of genomic indels and structural variations using split reads. BMC Genom. 2011, 12, 375. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Xu, N.; Li, Z.; Zhang, S.; Wu, J.; Kim, D.H.; Marma, M.S.; Meng, Q.; Cao, H.; Li, X.; et al. Four-color DNA sequencing with 3’-O-modified nucleotide reversible terminators and chemically cleavable fluorescent dideoxynucleotides. Proc. Natl. Acad. Sci. USA 2008, 105, 9145–9150. [Google Scholar] [CrossRef] [Green Version]
- Kingsford, C.; Schatz, M.C.; Pop, M. Assembly complexity of prokaryotic genomes using short reads. BMC Bioinform. 2010, 11, 21. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.; Gorrie, C.; Holt, K. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef]
- Mavromatis, K.; Land, M.L.; Brettin, T.S.; Quest, D.J.; Copeland, A.; Clum, A.; Goodwin, L.; Woyke, T.; Lapidus, A.; Klenk, H.P.; et al. The Fast Changing Landscape of Sequencing Technologies and Their Impact on Microbial Genome Assemblies and Annotation. PLoS ONE 2012, 7, e48837. [Google Scholar] [CrossRef]
- Pollard, M.O.; Gurdasani, D.; Mentzer, A.J.; Porter, T.; Sandhu, M.S. Long Reads: Their Purpose and Place. Hum. Mol. Genet. 2018, 27, R234–R241. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Venkatesan, B.M.; Bashir, R. Nanopore sensors for nucleic acid analysis. Nat. Nanotechnol. 2011, 6, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Yao, Y.; Bratus-Neuenschwander, A.; Patrignani, A.; Robinson, M.D.; Hussain, S. A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Begum, G.; Albanna, A.; Bankapur, A.; Nassir, N.; Tambi, R.; Berdiev, B.; Akter, H.; Karuvantevida, N.; Kellam, B.; Alhashmi, D.; et al. Long-Read Sequencing Improves the Detection of Structural Variations Impacting Complex Non-Coding Elements of the Genome. Int. J. Mol. Sci. 2021, 22, 2060. [Google Scholar] [CrossRef]
- Feng, Z.; Clemente, J.C.; Wong, B.; Schadt, E.E. Detecting and phasing minor single-nucleotide variants from long-read sequencing data. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Chang, P.-C.; Nattestad, M.; Kolesnikov, A.; Goel, S.; Baid, G.; Eizenga, J.M.; Miga, K.H.; Carnevali, P.; et al. Haplotype-Aware Variant Calling Enables High Accuracy in Nanopore Long-Reads Using Deep Neural Networks. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rhee, M.; Burns, M.A. Nanopore sequencing technology: Research trends and applications. Trends Biotechnol. 2006, 24, 580–586. [Google Scholar] [CrossRef]
- Leggett, R.M.; Clark, M.D. A world of opportunities with nanopore sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2015, 107, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.-S.; Roukos, D. From next-generation sequencing to nanopore sequencing technology: Paving the way to personalized genomic medicine. Expert Rev. Med. Devices 2013, 10, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loman, N.j.; Pallen, M.j. Twenty years of bacterial genome sequencing. Nat. Rev. Genet. 2015, 13, 787–794. [Google Scholar] [CrossRef] [PubMed]
- McGinn, S.; Gut, I.G. DNA sequencing—spanning the generations. New Biotechnol. 2013, 30, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, E.C. Pint-sized DNA sequencer impresses first users. Nature 2015, 521, 15–16. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, E.; Lärkeryd, A.; Sjödin, A.; Forsman, M.; Stenberg, P. Scaffolding of a bacterial genome using MinION nanopore sequencing. Sci. Rep. 2015, 5, 11996. [Google Scholar] [CrossRef] [Green Version]
- Judge, K.; Harris, S.R.; Reuter, S.; Parkhill, J.; Peacock, S.J. Early insights into the potential of the Oxford Nanopore MinION for the detection of antimicrobial resistance genes. J. Antimicrob. Chemother. 2015, 70, 2775–2778. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Laing, C.R.; Whiteside, M.D.; Gannon, V.P.J. Pan-genome Analyses of the Species Salmonella enterica, and Identification of Genomic Markers Predictive for Species, Subspecies, and Serovar. Front. Microbiol. 2017, 8, 1345. [Google Scholar] [CrossRef] [Green Version]
- Weingarten, R.A.; Johnson, R.; Conlan, S.; Ramsburg, A.M.; Dekker, J.P.; Lau, A.F.; Khil, P.; Odom, R.T.; Deming, C.; Park, M.; et al. Genomic Analysis of Hospital Plumbing Reveals Diverse Reservoir of Bacterial Plasmids Conferring Carbapenem Resistance. mBio 2018, 9, e02011-17. [Google Scholar] [CrossRef] [Green Version]
- Aanensen, D.M.; Feil, E.J.; Holden, M.T.G.; Dordel, J.; Yeats, C.A.; Fedosejev, A.; Goater, R.; Castillo-Ramírez, S.; Corander, J.; Colijn, C.; et al. Whole-Genome Sequencing for Routine Pathogen Surveillance in Public Health: A Population Snapshot of Invasive Staphylococcus aureus in Europe. mBio 2016, 7, e00444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Been, M.; Pinholt, M.; Top, J.; Bletz, S.; Mellmann, A.; van Schaik, W.; Brouwer, E.; Rogers, M.; Kraat, Y.; Bonten, M.; et al. Core Genome Multilocus Sequence Typing Scheme for High-Resolution Typing of Enterococcus faecium. J. Clin. Microbiol. 2015, 53, 3788–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feijao, P.; Yao, H.-T.; Fornika, D.; Gardy, J.; Hsiao, W.; Chauve, C.; Chindelevitch, L. MentaLiST—A fast MLST caller for large MLST schemes. Microb. Genom. 2018, 4, e000146. [Google Scholar] [CrossRef] [PubMed]
- Challagundla, L.; Reyes, J.; Rafiqullah, I.; Sordelli, D.O.; Echaniz-Aviles, G.; Velazquez-Meza, M.E.; Castillo-Ramírez, S.; Fittipaldi, N.; Feldgarden, M.; Chapman, S.B.; et al. Phylogenomic Classification and the Evolution of Clonal Complex 5 Methicillin-Resistant Staphylococcus aureus in the Western Hemisphere. Front. Microbiol. 2018, 9, 1901. [Google Scholar] [CrossRef] [Green Version]
- Merker, M.; Blin, C.; Mona, S.; Duforet-Frebourg, N.; Lecher, S.; Willery, E.; Blum, M.; Rüsch-Gerdes, S.; Mokrousov, I.; Aleksic, E.; et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet. 2015, 47, 242–249. [Google Scholar] [CrossRef]
- Choo, S.W.; Wee, W.Y.; Ngeow, Y.F.; Mitchell, W.; Tan, J.L.; Wong, G.J.; Zhao, Y.; Xiao, J. Genomic reconnaissance of clinical isolates of emerging human pathogen Mycobacterium abscessus reveals high evolutionary potential. Sci. Rep. 2014, 4, 4061. [Google Scholar] [CrossRef]
- Talkington, D.; Bopp, C.; Tarr, C.; Parsons, M.B.; Dahourou, G.; Freeman, M.; Joyce, K.; Turnsek, M.; Garrett, N.; Humphrys, M.; et al. Characterization of Toxigenic Vibrio cholerae from Haiti, 2010–Emerg. Infect. Dis. 2011, 17, 2122–2129. [Google Scholar] [CrossRef]
- Slobounov, S.; Cao, C.; Jaiswal, N.; Newell, K.M. Neural basis of postural instability identified by VTC and EEG. Exp. Brain Res. 2009, 199, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Armougom, F.; Bitam, I.; Croce, O.; Merhej, V.; Barassi, L.; Nguyen, T.-T.; La Scola, B.; Raoult, D. Genomic Insights into a New Citrobacter koseri Strain Revealed Gene Exchanges with the Virulence-Associated Yersinia pestis pPCP1 Plasmid. Front. Microbiol. 2016, 7, 340. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Zhong, J.; Zhang, J.; Li, C.; Yu, X.; Xiao, J.; Jia, X.; Ding, N.; Ma, G.; Wang, G.; et al. Pan-Genomic Study of Mycobacterium tuberculosis Reflecting the Primary/Secondary Genes, Generality/Individuality, and the Interconversion Through Copy Number Variations. Front. Microbiol. 2018, 9, 1886. [Google Scholar] [CrossRef] [Green Version]
- Codoñer, F.M.; Pou, C.; Thielen, A.; García, F.; Delgado, R.; Dalmau, D.; Alvarez-Tejado, M.; Ruiz, L.; Clotet, B.; Paredes, R. Added Value of Deep Sequencing Relative to Population Sequencing in Heavily Pre-Treated HIV-1-Infected Subjects. PLoS ONE 2011, 6, e194612011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falgenhauer, L.; Waezsada, S.E.; Yao, Y.; Imirzalioglu, C.; Käsbohrer, A.; Roesler, U.; Brenner Michael, G.; Schwarz, S.; Werner, G.; Kreienbrock, L.; et al. Colistin Resistance Gene Mcr-1 in Extended- Spectrum β-Lactamase- Producing Gram-Negative Bacteria in Germany. Lancet 2016, 16, 282–283. [Google Scholar] [CrossRef] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Jeukens, J.; Freschi, L.; Vincent, A.T.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Charette, S.J.; Levesque, R.C. A Pan-Genomic Approach to Understand the Basis of Host Adaptation in Achromobacter. Genome Biol. Evol. 2017, 9, 1030–1046. [Google Scholar] [CrossRef] [PubMed]
- Juraschek, K.; Borowiak, M.; Tausch, S.; Malorny, B.; Käsbohrer, A.; Otani, S.; Schwarz, S.; Meemken, D.; Deneke, C.; Hammerl, J. Outcome of Different Sequencing and Assembly Approaches on the Detection of Plasmids and Localization of Antimicrobial Resistance Genes in Commensal Escherichia coli. Microorganisms 2021, 9, 598. [Google Scholar] [CrossRef] [PubMed]
- Arredondo-Alonso, S.; Willems, R.; van Schaik, W.; Schürch, A.C. On the (im)possibility of reconstructing plasmids from whole-genome short-read sequencing data. Microb. Genom. 2017, 3, e000128. [Google Scholar] [CrossRef]
- Nguyen, M.; Brettin, T.; Long, S.W.; Musser, J.M.; Olsen, R.J.; Olson, R.; Shukla, M.; Stevens, R.L.; Xia, F.; Yoo, H.; et al. Developing an in silico minimum inhibitory concentration panel test for Klebsiella pneumoniae. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Long, S.W.; McDermott, P.F.; Olsen, R.J.; Olson, R.; Stevens, R.L.; Tyson, G.H.; Zhao, S.; Davis, J.J. Using Machine Learning to Predict Antimicrobial MICs and Associated Genomic Features for Nontyphoidal Salmonella. J. Clin. Microbiol. 2019, 57, e01260-18. [Google Scholar] [CrossRef] [Green Version]
- Eyre, D.W.; De Silva, D.; Cole, K.; Peters, J.; Cole, M.; Grad, Y.H.; Demczuk, W.; Martin, I.; Mulvey, M.R.; Crook, D.W.; et al. WGS to predict antibiotic MICs for Neisseria gonorrhoeae. J. Antimicrob. Chemother. 2017, 72, 1937–1947. [Google Scholar] [CrossRef] [Green Version]
- Decano, A.G.; Ludden, C.; Feltwell, T.; Judge, K.; Parkhill, J.; Downing, T. Complete Assembly of Escherichia coli Sequence Type 131 Genomes Using Long Reads Demonstrates Antibiotic Resistance Gene Variation within Diverse Plasmid and Chromosomal Contexts. mSphere 2019, 4, e00130–19. [Google Scholar] [CrossRef] [Green Version]
- Golparian, D.; Donà, V.; Sánchez-Busó, L.; Förster, S.; Harris, S.; Endimiani, A.; Low, N.; Unemo, M. Antimicrobial resistance prediction and phylogenetic analysis of Neisseria gonorrhoeae isolates using the Oxford Nanopore MinION sequencer. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wu, Y.; Kudinha, T.; Jia, P.; Wang, L.; Xu, Y.; Yang, Q. Comprehensive Pathogen Identification, Antibiotic Resistance, and Virulence Genes Prediction Directly from Simulated Blood Samples and Positive Blood Cultures by Nanopore Metagenomic Sequencing. Front. Genet. 2021, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Freddolino, P.L.; Goodarzi, H.; Tavazoie, S. Revealing the Genetic Basis of Natural Bacterial Phenotypic Divergence. J. Bacteriol. 2014, 196, 825–839. [Google Scholar] [CrossRef] [Green Version]
- Brbic, M.; Piškorec, M.; Vidulin, V.; Kriško, A.; Šmuc, T.; Supek, F. The landscape of microbial phenotypic traits and associated genes. Nucleic Acids Res. 2016, 44, 10074–10090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, J.A.; Tien Mai, T.; Galardini, M.; Wheeler, N.E.; Horsfield, S.T.; Parkhill, J.; Corander, J.; Lees, C.J.; Jacques Ravel, E.; Wilson, D. Improved Prediction of Bacterial Genotype-Phenotype Associations Using Interpretable Pangenome-Spanning Regressions. ASM J. 2020, 4, e01344-20. [Google Scholar] [CrossRef]
- Goberna, M.; Verdú, M. Predicting microbial traits with phylogenies. ISME J. 2015, 10, 959–967. [Google Scholar] [CrossRef]
- Weimann, A.; Mooren, K.; Frank, J.; Pope, P.B.; Bremges, A.; McHardy, A.C. From Genomes to Phenotypes: Traitar, the Microbial Trait Analyzer. mSystems 2016, 1, e00101-16. [Google Scholar] [CrossRef] [Green Version]
- Fraser, C.M.; Gocayne, J.D.; White, O.; Adams, M.D.; Clayton, R.A.; Fleischmann, R.D.; Bult, C.J.; Kerlavage, A.R.; Sutton, G.; Kelley, J.M.; et al. The Minimal Gene Complement of Mycoplasma genitalium. Science 1995, 270, 397–404. [Google Scholar] [CrossRef]
- Schmid, M.; Frei, D.; Patrignani, A.; Schlapbach, R.; Frey, J.E.; Remus-Emsermann, M.; Ahrens, C.H. Pushing the limits of de novo genome assembly for complex prokaryotic genomes harboring very long, near identical repeats. Nucleic Acids Res. 2018, 46, 8953–8965. [Google Scholar] [CrossRef]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.; Chaisson, M.J.; Steinberg, K.M.; Warren, W.; Hoekzema, K.; Gordon, D.; Graves-Lindsay, T.A.; Munson, K.; Kronenberg, Z.; Vives, L.; et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2016, 27, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tattini, L.; D’Aurizio, R.; Magi, A.; Tattini, L.; D’Aurizio, R.; Magi, A. Detection of Genomic Structural Variants from Next-Generation Sequencing Data. Front. Bioeng. Biotechnol. 2015, 3, 92. [Google Scholar] [CrossRef] [Green Version]
- Noll, N.; Urich, E.; Wüthrich, D.; Hinic, V.; Egli, A.; Neher, R.A. Resolving Structural Diversity of Carbapenemase-Producing Gram-Negative Bacteria Using Single Molecule Sequencing. bioRxiv 2018. [Google Scholar] [CrossRef]
- Periwal, V.; Scaria, V. Insights into structural variations and genome rearrangements in prokaryotic genomes. Bioinformatics 2014, 31, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.S.; Urban, A.E.; Mills, R.E. Structural variation in the sequencing era. Nat. Rev. Genet. 2019, 21, 171–189. [Google Scholar] [CrossRef]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675.e19. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Risse, J.; Thomson, M.; Patrick, S.; Blakely, G.; Koutsovoulos, G.; Blaxter, M.; Watson, M. A single chromosome assembly of Bacteroides fragilis strain BE1 from Illumina and MinION nanopore sequencing data. GigaScience 2015, 4, 60. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.; Beka, L.; Graf, J.; Klassen, J.L. Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genom. 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, F.; Aigrain, L.; Quail, M.A.; Coupland, P.; Bonfield, J.K.; Davies, R.M.; Tischler, G.; Jackson, D.K.; Keane, T.M.; Li, J.; et al. De novo yeast genome assemblies from MinION, PacBio and MiSeq platforms. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Krogerus, K.; Magalhães, F.; Castillo, S.; Peddinti, G.; Vidgren, V.; de Chiara, M.; Yue, J.-X.; Liti, G.; Gibson, B. Lager Yeast Design through Meiotic Segregation of a Fertile Saccharomyces Cerevisiae x Saccharomyces Eubayanus Hybrid. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yue, J.-X.; Liti, G. Long-read sequencing data analysis for yeasts. Nat. Protoc. 2018, 13, 1213–1231. [Google Scholar] [CrossRef]
- Yue, J.-X.; Li, J.; Aigrain, L.; Hallin, J.; Persson, K.; Oliver, K.; Bergström, A.; Coupland, P.; Warringer, J.; Lagomarsino, M.C.; et al. Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat. Genet. 2017, 49, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Peter, J.; de Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome Evolution across 1,011 Saccharomyces Cerevisiae Isolates Species-Wide Genetic and Phenotypic Diversity. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Broad Institute of MIT and Harvard. Assembly Polishing with Pilon—De. Available online: https://github.com/broadinstitute/pilon (accessed on 20 November 2021).
- Error Correction Using Pilon|Long-Read, Long Reach Bioinformatics Tutorials.
- Institut de Génomique. NaS. Available online: https://github.com/institut-de-genomique/NaS (accessed on 20 November 2021).
- James, G. Nanocorr: Error Correction for Oxford Nanopore Data. Available online: https://github.com/jgurtowski/nanocorr (accessed on 20 November 2021).
- La, S.; Haghshenas, E.; Chauve, C. LRCstats, a tool for evaluating long reads correction methods. Bioinformatics 2017, 33, 3652–3654. [Google Scholar] [CrossRef] [Green Version]
- Ben Khedher, M.; Nindo, F.; Chevalier, A.; Bonacorsi, S.; Dubourg, G.; Fenollar, F.; Casagrande, C.; Lotte, R.; Boyer, L.; Gallet, A.; et al. Complete Circular Genome Sequences of ThreeBacillus CereusGroup Strains Isolated from Positive Blood Cultures FromPreterm and Immunocompromised Infants Hospitalized InFrance. Clin. Microbiol. Rev. 2010, 23, 382–398. [Google Scholar]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, 1–29. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Cerdeira, L.T.; Hawkey, J.; Méric, G.; Vezina, B.; Wyres, K.L.; Holt, K.E. Trycycler: Consensus long-read assemblies for bacterial genomes. Genome Biol. 2021, 22, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stackebrandt, E.; Frederiksen, W.; Garrity, G.M.; Grimont, P.A.D.; Kampfer, P.; Maiden, M.C.J.; Nesme, X.; Rosselló-Mora, R.; Swings, J.; Trüper, H.G.; et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 2002, 52, 1043–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiden, M.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Fournier, P.-E.; Drancourt, M. New Microbes New Infections promotes modern prokaryotic taxonomy: A new section “TaxonoGenomics: New genomes of microorganisms in humans”. New Microbes New Infect. 2015, 7, 48–49. [Google Scholar] [CrossRef] [Green Version]
- Patil, P.P.; Kumar, S.; Midha, S.; Gautam, V.; Patil, P. Taxonogenomics reveal multiple novel genomospecies associated with clinical isolates of Stenotrophomonas maltophilia. Microb. Genom. 2018, 4, e000207. [Google Scholar] [CrossRef]
- Saati-Santamaría, Z.; Peral-Aranega, E.; Velázquez, E.; Rivas, R.; García-Fraile, P. Phylogenomic Analyses of the Genus Pseudomonas Lead to the Rearrangement of Several Species and the Definition of New Genera. Biology 2021, 10, 782. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Handelsman, J. Metagenomics for studying unculturable microorganisms: Cutting the Gordian knot. Genome Biol. 2005, 6, 229. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.; Handelsman, J. Biotechnological prospects from metagenomics. Curr. Opin. Biotechnol. 2003, 14, 303–310. [Google Scholar] [CrossRef]
- Hilton, S.K.; Castro-Nallar, E.; Perez-Losada, M.; Toma, I.; McCaffrey, T.A.; Hoffman, E.; Siegel, M.O.; Simon, G.L.; Johnson, W.; Crandall, K.A. Metataxonomic and Metagenomic Approaches vs. Culture-Based Techniques for Clinical Pathology. Front. Microbiol. 2016, 7, 484. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; A Beutler, N.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Quick, J.; Claro, I.; Thézé, J.; De Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; Da Costa, A.C.; et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef]
- Salipante, S.J.; Sengupta, D.J.; Rosenthal, C.; Costa, G.; Spangler, J.; Sims, E.H.; Jacobs, M.A.; Miller, S.I.; Hoogestraat, D.R.; Cookson, B.T.; et al. Rapid 16S rRNA Next-Generation Sequencing of Polymicrobial Clinical Samples for Diagnosis of Complex Bacterial Infections. PLoS ONE 2013, 8, e65226. [Google Scholar] [CrossRef] [Green Version]
- Langelier, C.; Zinter, M.S.; Kalantar, K.; Yanik, G.A.; Christenson, S.; O’Donovan, B.; White, C.; Wilson, M.; Sapru, A.; Dvorak, C.C.; et al. Metagenomic Sequencing Detects Respiratory Pathogens in Hematopoietic Cellular Transplant Patients. Am. J. Respir. Crit. Care Med. 2018, 197, 524–528. [Google Scholar] [CrossRef]
- Zhou, Y.; Wylie, K.M.; El Feghaly, R.E.; Mihindukulasuriya, K.; Elward, A.; Haslam, D.B.; Storch, G.A.; Weinstock, G.M. Metagenomic Approach for Identification of the Pathogens Associated with Diarrhea in Stool Specimens. J. Clin. Microbiol. 2016, 54, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Blauwkamp, T.A.; Thair, S.; Rosen, M.J.; Blair, L.; Lindner, M.S.; Vilfan, I.D.; Kawli, T.; Christians, F.C.; Venkatasubrahmanyam, S.; Wall, G.D.; et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat. Microbiol. 2019, 4, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Langelier, C.; Kalantar, K.L.; Moazed, F.; Wilson, M.R.; Crawford, E.D.; Deiss, T.; Belzer, A.; Bolourchi, S.; Caldera, S.; Fung, M.; et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc. Natl. Acad. Sci. USA 2018, 115, E12353–E12362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputo, A.; Dubourg, G.; Croce, O.; Gupta, S.; Robert, C.; Papazian, L.; Rolain, J.M.; Raoult, D. Whole-genome assembly of Akkermansia muciniphila sequenced directly from human stoo. Biol. Direct 2015, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somerville, V.; Lutz, S.; Schmid, M.; Frei, D.; Moser, A.; Irmler, S.; Frey, J.E.; Ahrens, C.H. Long-read based de novo assembly of low-complexity metagenome samples results in finished genomes and reveals insights into strain diversity and an active phage system. BMC Microbiol. 2019, 19, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearman, W.S.; Freed, N.E.; Silander, O.K. Testing the advantages and disadvantages of short- and long- read eukaryotic metagenomics using simulated reads. BMC Bioinform. 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Yang, C.; Sun, Y.; Igarashi, Y.; Jin, T.; Luo, F. PacBio Long Reads Improve Metagenomic Assemblies, Gene Catalogs, and Genome Binning. Front. Genet. 2020, 11, 516269. [Google Scholar] [CrossRef]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Wang, A.H.-J.; Jennings, M.P. Discovery of virulence factors of pathogenic bacteria. Curr. Opin. Chem. Biol. 2008, 12, 93–101. [Google Scholar] [CrossRef]
- Suzuki, S.; Horinouchi, T.; Furusawa, C. Prediction of antibiotic resistance by gene expression profiles. Nat. Commun. 2014, 5, 5792. [Google Scholar] [CrossRef] [Green Version]
- Westermann, A.; Gorski, S.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Genet. 2012, 10, 618–630. [Google Scholar] [CrossRef]
- Durmus, S.; Çakir, T.; Özgür, A.; Guthke, R. A Review on Computational Systems Biology of Pathogen-Host Interactions. Front. Microbiol. 2015, 6, 235. [Google Scholar] [CrossRef]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef]
- Byron, S.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Van, T.T.H.; Lacey, J.A.; Vezina, B.; Phung, C.; Anwar, A.; Scott, P.C.; Moore, R.J. Survival Mechanisms of Campylobacter hepaticus Identified by Genomic Analysis and Comparative Transcriptomic Analysis of in vivo and in vitro Derived Bacteria. Front. Microbiol. 2019, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Au, K.F.; Sebastiano, V.; Afshar, P.T.; Durruthy, J.D.; Lee, L.; Williams, B.A.; van Bakel, H.; Schadt, E.E.; Reijo-Pera, R.A.; Underwood, J.G.; et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, E4821–E4830. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T.; DuBois, R.M.; Forsberg, E.C.; Akeson, M.; Vollmers, C. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017, 8, 16027. [Google Scholar] [CrossRef] [Green Version]
- Kuosmanen, A.; Norri, T.; Mäkinen, V. Evaluating approaches to find exon chains based on long reads. Briefings Bioinform. 2017, 19, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Tilgner, H.; Grubert, F.; Sharon, D.; Snyder, M.P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. USA 2014, 111, 9869–9874. [Google Scholar] [CrossRef] [Green Version]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, H.-J.; Buck, D.; Au, K.F. Comprehensive Comparison of Pacific Biosciences and Oxford Nanopore Technologies and Their Applications to Transcriptome Analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef]
- Howorka, S.; Siwy, Z.S. Reading amino acids in a nanopore. Nat. Biotechnol. 2020, 38, 159–160. [Google Scholar] [CrossRef]
- Tang, L. Next-generation peptide sequencing. Nat. Chem. Biol. 2018, 15, 997. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Sloggett, C.; Goonasekera, N.; Makunin, I.; Benson, D.; Crowe, M.; Gladman, S.; Kowsar, Y.; Pheasant, M.; Horst, R.; et al. Genomics Virtual Laboratory: A Practical Bioinformatics Workbench for the Cloud. PLoS ONE 2015, 10, e0140829. [Google Scholar] [CrossRef] [Green Version]
- Blankenberg, D.; Taylor, J.; Schenck, I.; He, J.; Zhang, Y.; Ghent, M.; Veeraraghavan, N.; Albert, I.; Miller, W.; Makova, K.D.; et al. A framework for collaborative analysis of ENCODE data: Making large-scale analyses biologist-friendly. Genome Res. 2007, 17, 960–964. [Google Scholar] [CrossRef] [Green Version]
- Fisch, K.M.; Meißner, T.; Gioia, L.; Ducom, J.-C.; Carland, T.M.; Loguercio, S.; Su, A.I. Omics Pipe: A community-based framework for reproducible multi-omics data analysis. Bioinformatics 2015, 31, 1724–1728. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, V.; Ceol, A.; Ogier, A.G.E.; de Pretis, S.; Galeota, E.; Kishore, K.; Bora, P.; Croci, O.; Campaner, S.; Amati, B.; et al. Integrated Systems for NGS Data Management and Analysis: Open Issues and Available Solutions. Front. Genet. 2016, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Reisinger, E.; Genthner, L.; Kerssemakers, J.; Kensche, P.; Borufka, S.; Jugold, A.; Kling, A.; Prinz, M.; Scholz, I.; Zipprich, G.; et al. OTP: An automatized system for managing and processing NGS data. J. Biotechnol. 2017, 261, 53–62. [Google Scholar] [CrossRef]
- Kallio, M.A.; Tuimala, J.T.; Hupponen, T.; Klemelä, P.; Gentile, M.; Scheinin, I.; Koski, M.; Käki, J.; Korpelainen, E.I. Chipster: User-friendly analysis software for microarray and other high-throughput data. BMC Genom. 2011, 12, 507. [Google Scholar] [CrossRef] [Green Version]
- McLellan, A.S.; Dubin, R.A.; Jing, Q.; Broin, P.; Moskowitz, D.; Suzuki, M.; Calder, R.B.; Hargitai, J.; Golden, A.; Greally, J.M. The Wasp System: An open source environment for managing and analyzing genomic data. Genomics 2012, 100, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Wagle, P.; Nikolić, M.; Frommolt, P. QuickNGS elevates Next-Generation Sequencing data analysis to a new level of automation. BMC Genom. 2015, 16, 487. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Short-Read Technologies | Long-Read Technologies |
|---|---|
| Fixed run time: - Increased time to results and inability to identify workflow errors before completed sequencing - Additional practical complexities associated with handling and storing large volumes of sequence data | Real-time data acquisition: - Achieve rapid turnaround with immediate access to results - Enrich single targets during sequencing, with no additional sample prep using adaptive sampling - Identify microbiome composition and resistance in real-time |
| Limited flexibility: - Sample batching often required for optimal efficiency - Potentially leads to long turnaround times - Benchtop devices confine sequencing to centralized locations | Scalable and flexible: - Scale to suit the throughput needs - Decentralize sequencing - No sample batching needed |
| Read length typically 50–300 bp | Unrestricted read length (>4 Mb achieved) |
| Limited genomic characterization: - Short reads do not span entire structural variants or important classes of genomic aberrations (repeat expansions and repeat-rich regions) - fragmented genome assemblies and ambiguous isoforms identification - Short sequencing reads may not span complex genomic regions such as genes duplications, transposons and prophage sequences - Potentially missing important genomic information | Comprehensive genomic characterization: - Identify mutations in complex and repetitive genomic regions - Accurately phase single nucleotide variants, structural variants, and base modifications - Can fully assemble genomes more easily - Simplify de novo assembly and correct microbial reference genomes - Possibility to completely assemble genomes and plasmids from metagenomic samples - Resolving complex genomic regions and similar species |
| Amplification required: - Amplification can introduce bias reducing uniformity of coverage and removes base modifications - Necessitating additional sample prep and sequencing runs | Amplification-free protocols: - Detect and phase base modifications as standard - No additional preparation required |
| Constrained to the lab: - Traditional sequencing technologies are typically expensive and require substantial site infrastructure - Usually limited its usage to well-resourced environments - Delay in transmitting the results | Sequence anywhere: - Sequence in your lab or in the field - Sequence at sample source and eliminate sample shipping delays - Scale-up with high-throughput |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Khedher, M.; Ghedira, K.; Rolain, J.-M.; Ruimy, R.; Croce, O. Application and Challenge of 3rd Generation Sequencing for Clinical Bacterial Studies. Int. J. Mol. Sci. 2022, 23, 1395. https://doi.org/10.3390/ijms23031395
Ben Khedher M, Ghedira K, Rolain J-M, Ruimy R, Croce O. Application and Challenge of 3rd Generation Sequencing for Clinical Bacterial Studies. International Journal of Molecular Sciences. 2022; 23(3):1395. https://doi.org/10.3390/ijms23031395
Chicago/Turabian StyleBen Khedher, Mariem, Kais Ghedira, Jean-Marc Rolain, Raymond Ruimy, and Olivier Croce. 2022. "Application and Challenge of 3rd Generation Sequencing for Clinical Bacterial Studies" International Journal of Molecular Sciences 23, no. 3: 1395. https://doi.org/10.3390/ijms23031395