
An Interleukin-4 and Interleukin-13 Induced Atopic Dermatitis Human Skin Equivalent Model by a Skin-On-A-Chip



Abstract
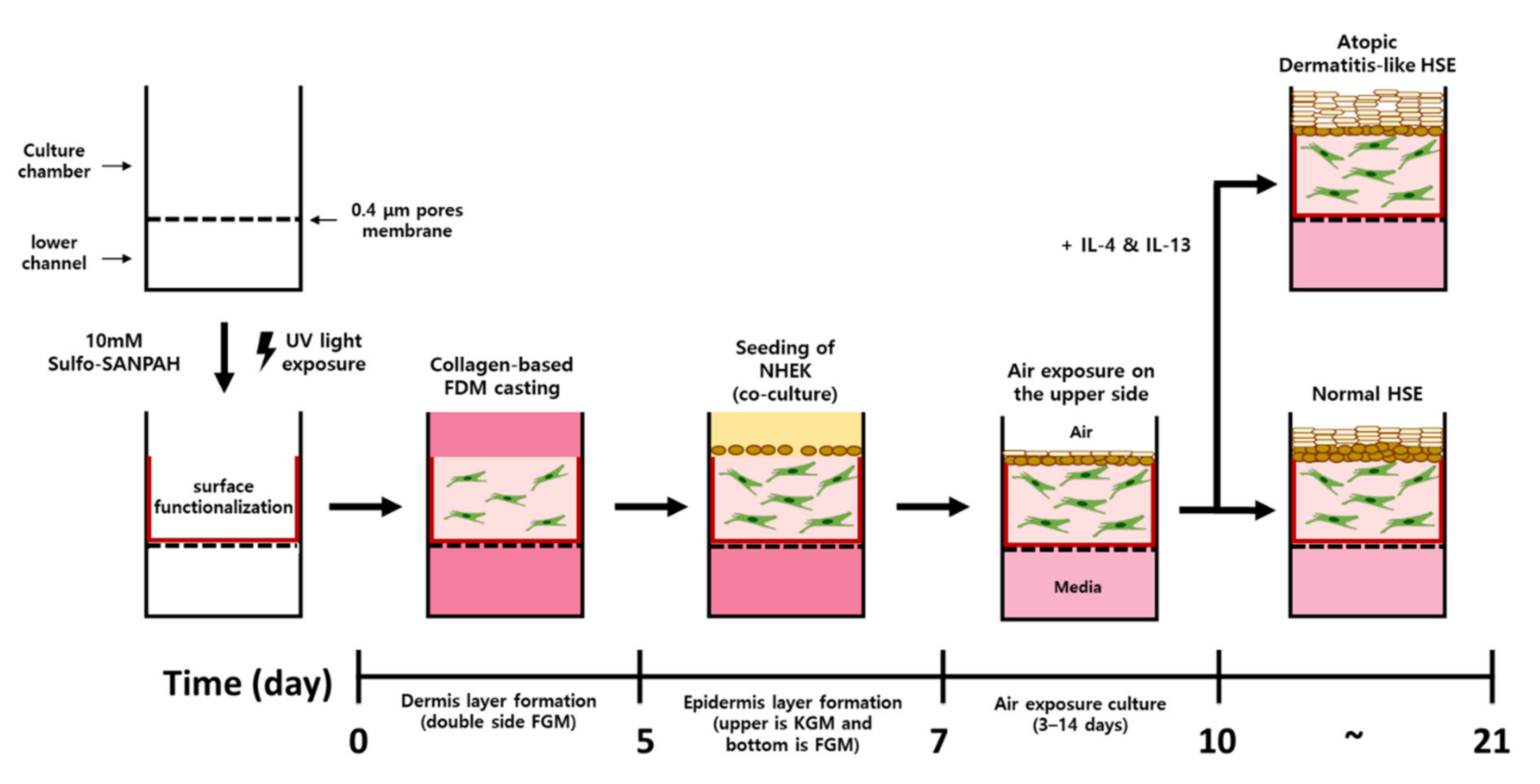
:1. Introduction
2. Results
2.1. Alteration of Tissue Morphology of the AD-like HSE Model
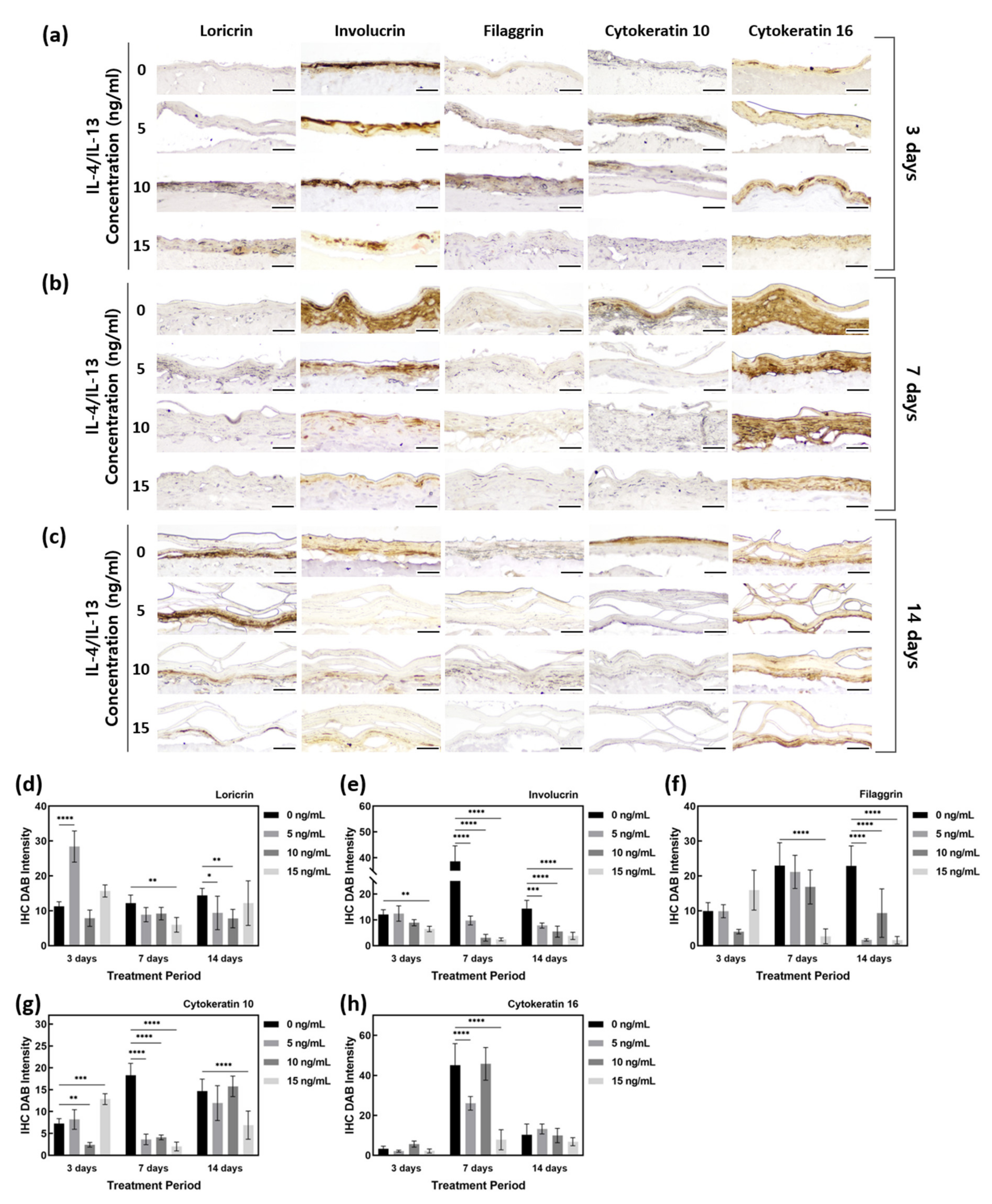
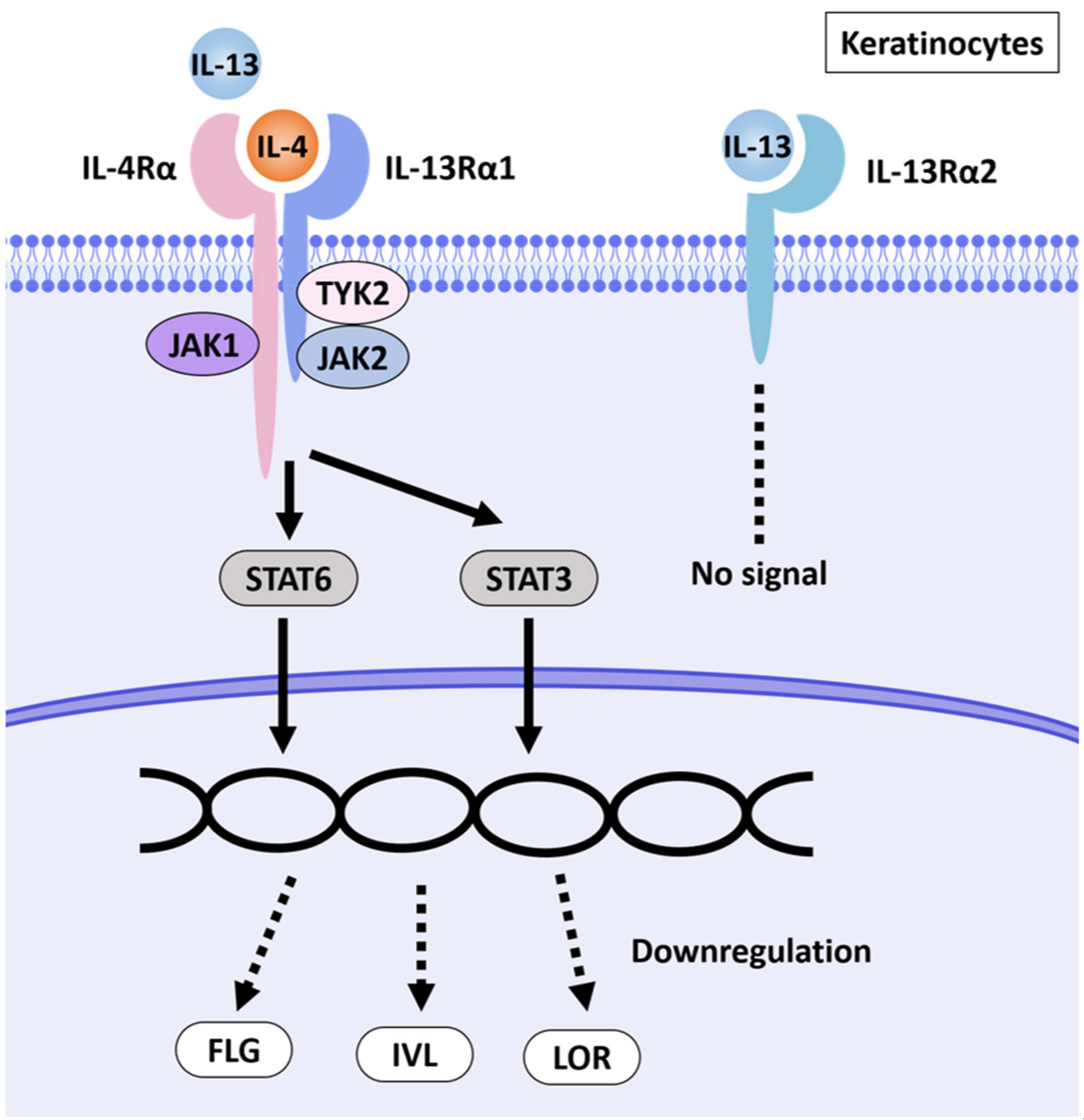
2.2. Downregulation of Barrier Function-Related Proteins in AD-HSE Stimulated with IL-4 and IL-13
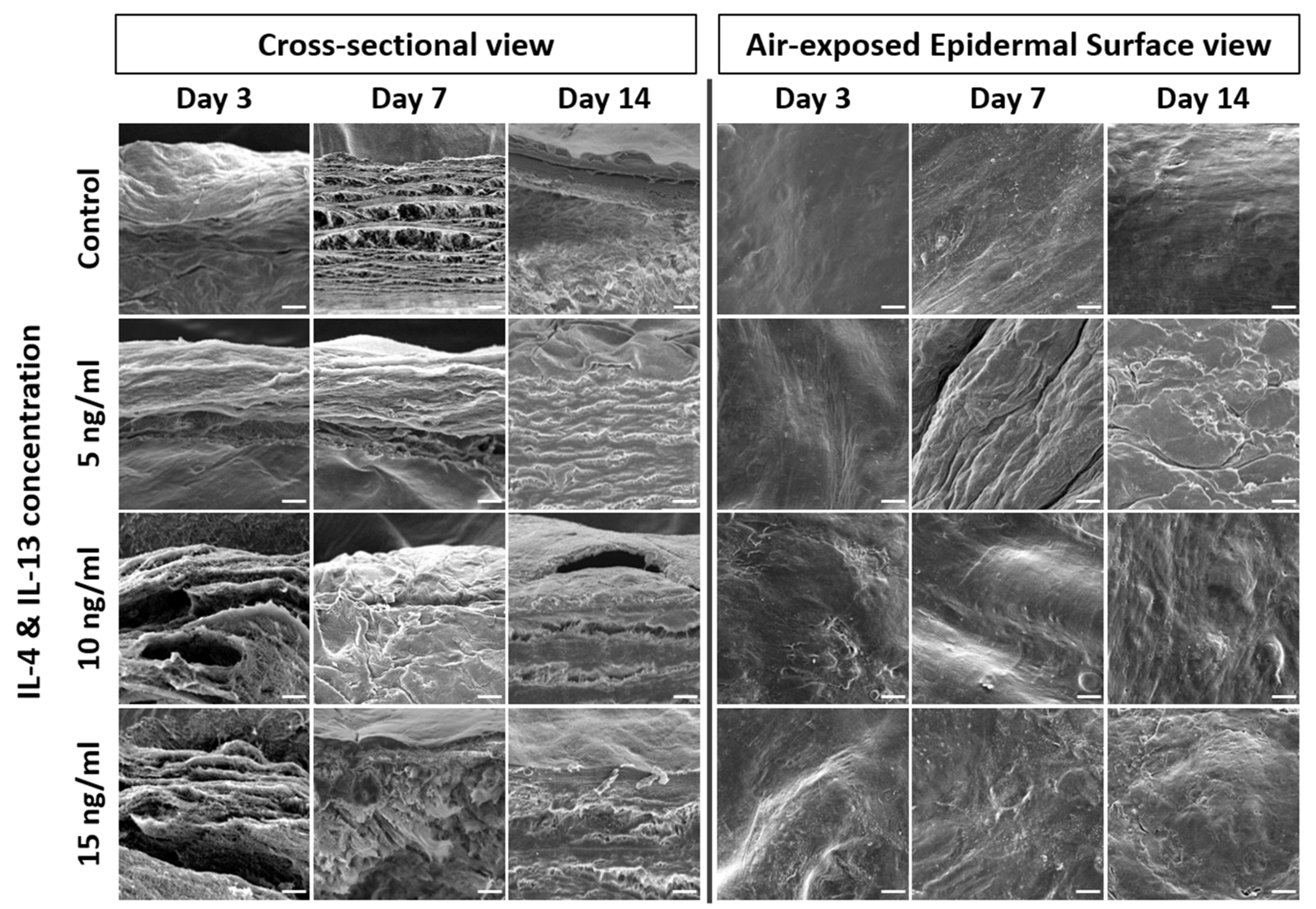
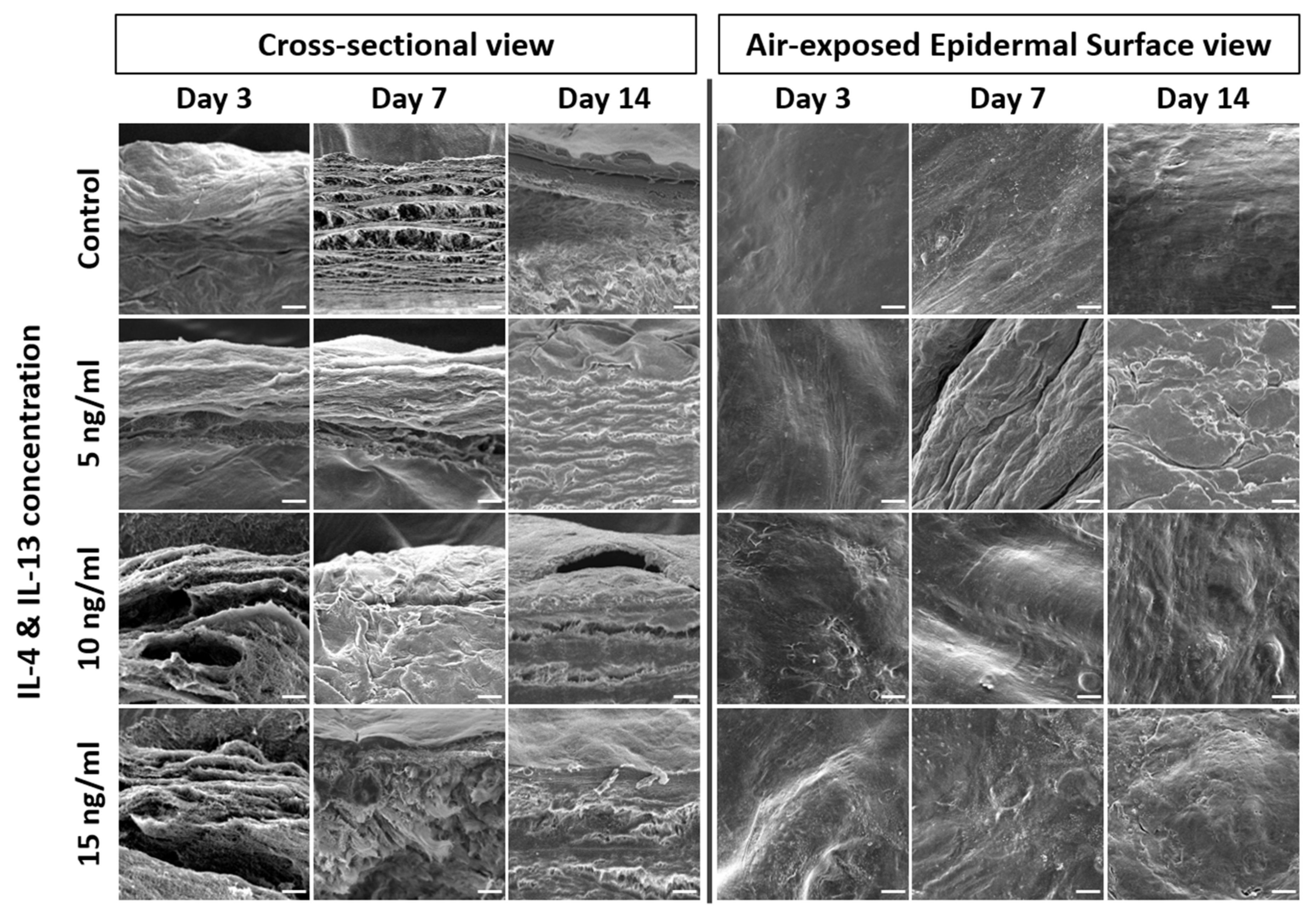
2.3. Alteration of Epidermal Morphologies in AD-HSE Stimulated with IL-4 and IL-13
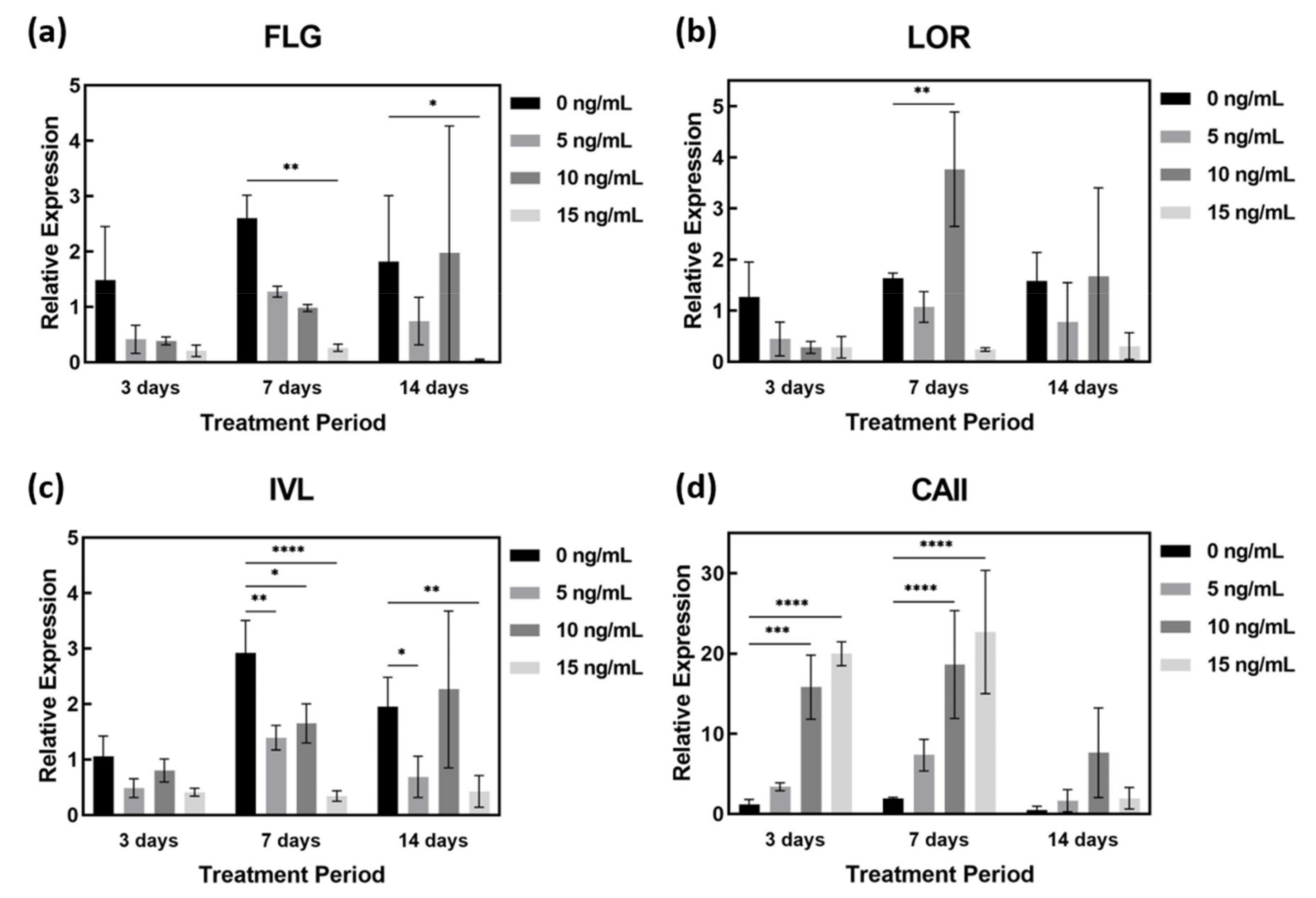
2.4. AD-Related Gene Expression in AD-HSE Stimulated with IL-4 and IL-13
2.5. Secreted Cytokines in AD-HSE Stimulated with IL-4 and IL-13
3. Discussion
4. Materials and Methods
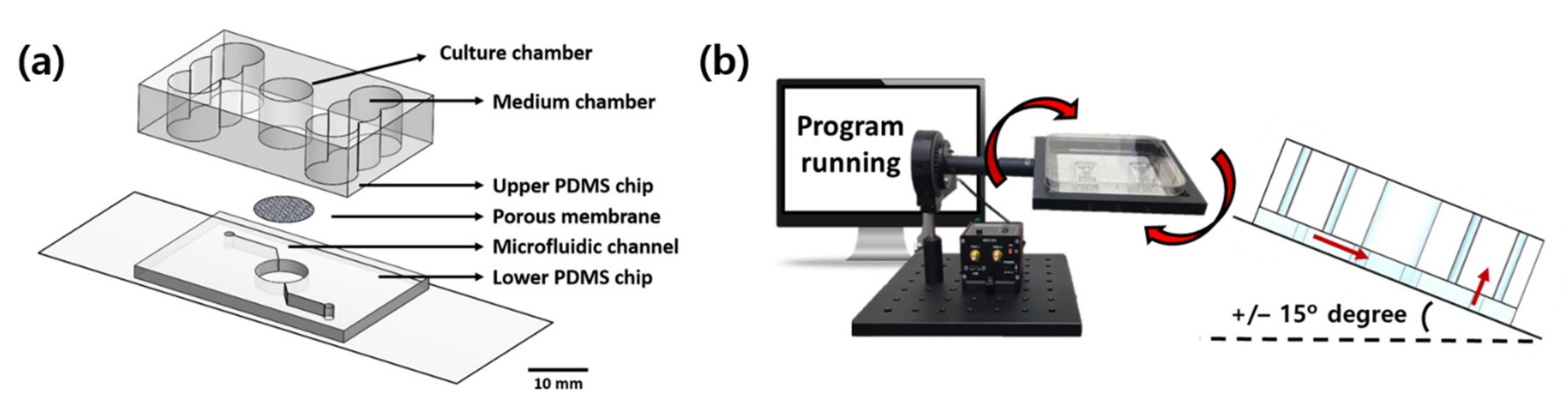
4.1. Fabrication of Pumpless Microfluidic Chip and Microphysiological System
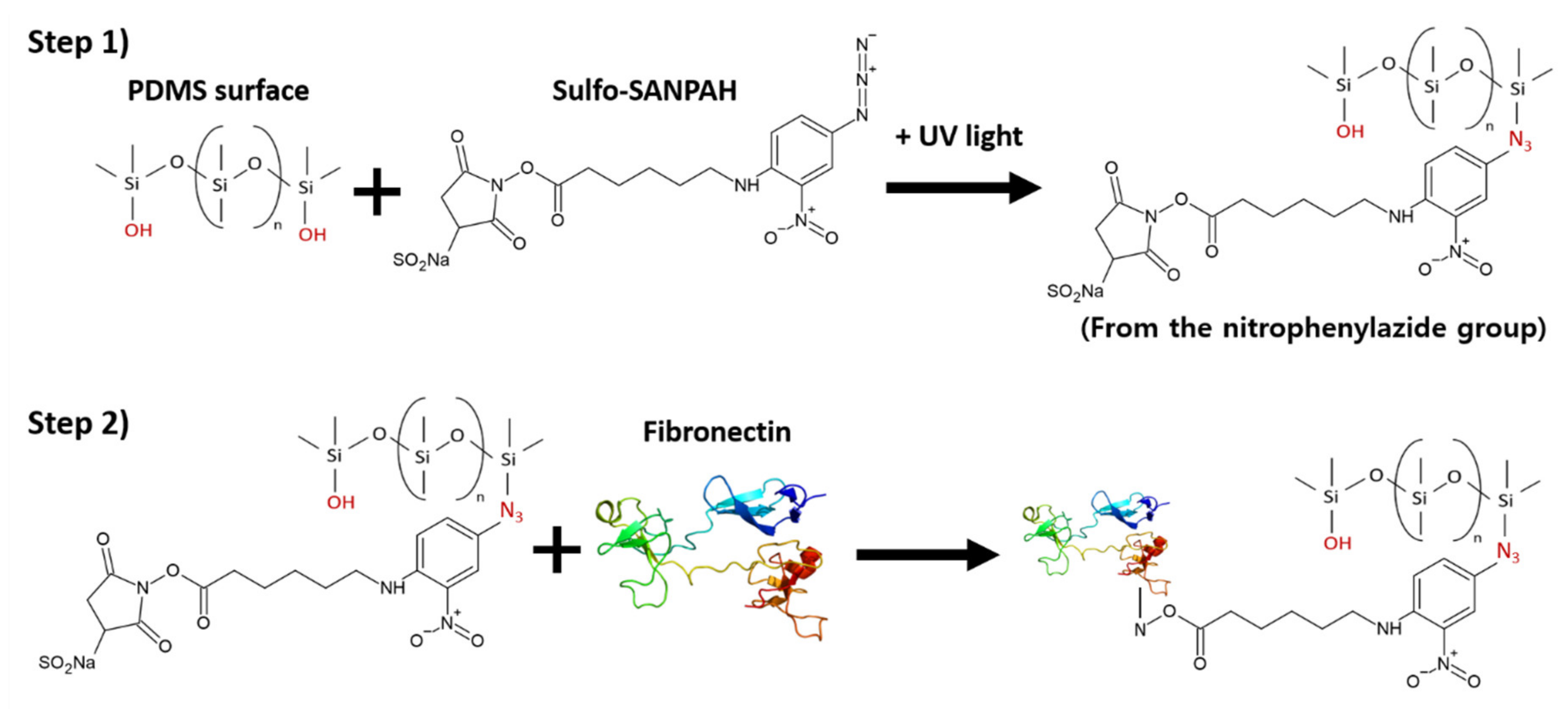
4.2. Surface Functionalization
4.3. Cell Culture
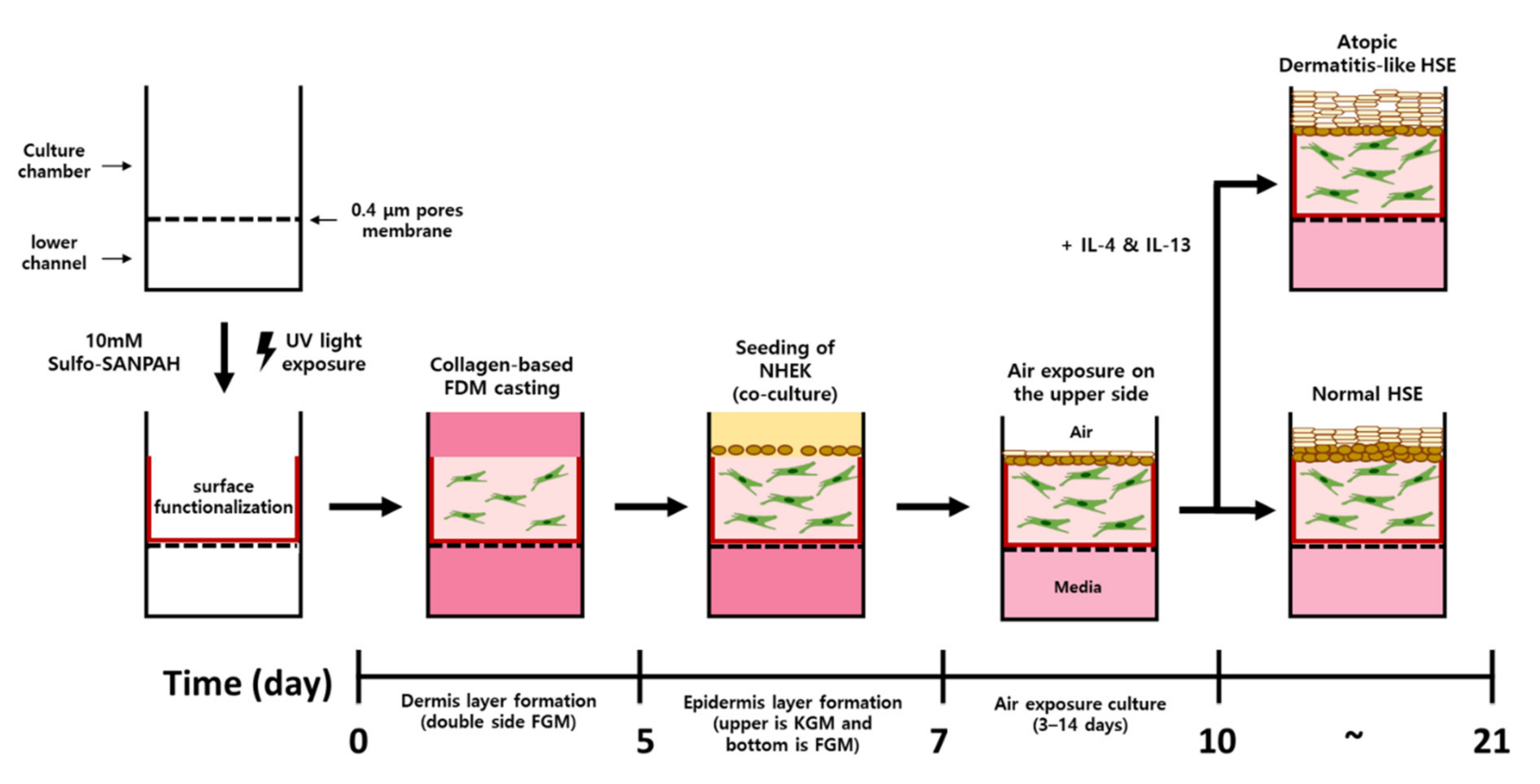
4.4. Construction of Atopic Dermatitis Skin Equivalent Model
4.5. Measurement of Contraction of Human Skin Equivalents
4.6. Hematoxylin and Eosin (H&E) Staining
4.7. Immunohistochemistry Staining (IHC) Staining
4.8. RNA Extraction from Human Skin Equivalent
4.9. Quantitative Real-Time PCR
4.10. Analysis of mRNA Expression by RT-PCR and Gel Electrophoresis
4.11. Scanning Electron Microscopy
4.12. Enzyme-Linked Immunosorbent Assay Measurement
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Ulzii, D.; Vu, Y.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T. Pathogenesis of atopic dermatitis: Current paradigm. Iran. J. Immunol. 2019, 16, 97–107. [Google Scholar] [PubMed]
- Morelli, P.; Gaspari, M.; Gabriele, C.; Dastoli, S.; Bennardo, L.; Pavel, A.B.; Patruno, C.; Del Duca, E.; Nisticò, S.P. Proteomic analysis from skin swabs reveals a new set of proteins identifying skin impairment in atopic dermatitis. Exp. Dermatol. 2021, 30, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Nettis, E.; Ortoncelli, M.; Pellacani, G.; Foti, C.; Di Leo, E.; Patruno, C.; Rongioletti, F.; Argenziano, G.; Ferrucci, S.; Macchia, L. A multicenter study on the prevalence of clinical patterns and clinical phenotypes in adult atopic dermatitis. J. Investig. Allergol. Clin. Immunol 2020, 448–450. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, E.; Salmon, M.; Evrard, C.; Lambert de Rouvroit, C.; Poumay, Y. Atopic dermatitis studies through in vitro models. Front. Med. 2017, 4, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman-Yassky, E.; Suárez-Fariñas, M.; Chiricozzi, A.; Nograles, K.E.; Shemer, A.; Fuentes-Duculan, J.; Cardinale, I.; Lin, P.; Bergman, R.; Bowcock, A.M. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J. Allergy Clin. Immunol. 2009, 124, 1235–1244.e58. [Google Scholar] [CrossRef]
- Totsuka, A.; Omori-Miyake, M.; Kawashima, M.; Yagi, J.; Tsunemi, Y. Expression of keratin 1, keratin 10, desmoglein 1 and desmocollin 1 in the epidermis: Possible downregulation by interleukin-4 and interleukin-13 in atopic dermatitis. Eur. J. Dermatol. 2017, 27, 247–253. [Google Scholar] [CrossRef]
- Jensen, J.-M.; Fölster-Holst, R.; Baranowsky, A.; Schunck, M.; Winoto-Morbach, S.; Neumann, C.; Schütze, S.; Proksch, E. Impaired sphingomyelinase activity and epidermal differentiation in atopic dermatitis. J. Investig. Dermatol. 2004, 122, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Cork, M.J.; Danby, S.G.; Vasilopoulos, Y.; Hadgraft, J.; Lane, M.E.; Moustafa, M.; Guy, R.H.; MacGowan, A.L.; Tazi-Ahnini, R.; Ward, S.J. Epidermal barrier dysfunction in atopic dermatitis. J. Investig. Dermatol. 2009, 129, 1892–1908. [Google Scholar] [CrossRef]
- Boguniewicz, M.; Leung, D.Y. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol. Rev. 2011, 242, 233–246. [Google Scholar] [CrossRef]
- Hänel, K.H.; Cornelissen, C.; Lüscher, B.; Baron, J.M. Cytokines and the skin barrier. Int. J. Mol. Sci. 2013, 14, 6720–6745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kypriotou, M.; Huber, M.; Hohl, D. The human epidermal differentiation complex: Cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp. Dermatol. 2012, 21, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Higuchi, K.; Wakao, H.; Hamasaki, N.; Izuhara, K. Identification of the Critical Portions of the Human IL-4 Receptor α Chain for Activation of STAT6. Biochem. Biophys. Res. Commun. 1998, 246, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, C.; Trivedi, S.; Wijesundara, D.K.; Jackson, R.J. IL-4 and IL-13 receptors: Roles in immunity and powerful vaccine adjuvants. Cytokine Growth Factor Rev. 2014, 25, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Furue, M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic implications in atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 5382. [Google Scholar] [CrossRef]
- Sneddon, L.U.; Halsey, L.G.; Bury, N.R. Considering aspects of the 3Rs principles within experimental animal biology. J. Exp. Biol. 2017, 220, 3007–3016. [Google Scholar] [CrossRef] [Green Version]
- Rusche, B. The 3Rs and animal welfare-conflict or the way forward? ALTEX-Altern. Anim. Exp. 2003, 20 (Supp. 1), 63–76. [Google Scholar]
- Serpell, J. In the Company of Animals: A Study of Human-Animal Relationships; Cambridge University Press: Philadelphia, PA, USA, 1996. [Google Scholar]
- Maeno, S.; Niki, Y.; Matsumoto, H.; Morioka, H.; Yatabe, T.; Funayama, A.; Toyama, Y.; Taguchi, T.; Tanaka, J. The effect of calcium ion concentration on osteoblast viability, proliferation and differentiation in monolayer and 3D culture. Biomaterials 2005, 26, 4847–4855. [Google Scholar] [CrossRef]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Advanced cell culture techniques for cancer drug discovery. Biology 2014, 3, 345–367. [Google Scholar] [CrossRef] [Green Version]
- Huet, F.; Severino-Freire, M.; Chéret, J.; Gouin, O.; Praneuf, J.; Pierre, O.; Misery, L.; Le Gall-Ianotto, C. Reconstructed human epidermis for in vitro studies on atopic dermatitis: A review. J. Dermatol. Sci. 2018, 89, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Ataç, B.; Wagner, I.; Horland, R.; Lauster, R.; Marx, U.; Tonevitsky, A.G.; Azar, R.P.; Lindner, G. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 2013, 13, 3555–3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, N.; Morimoto, Y.; Takeuchi, S. Skin integrated with perfusable vascular channels on a chip. Biomaterials 2017, 116, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Song, H.J.; Lim, H.Y.; Chun, W.; Choi, K.C.; Sung, J.H.; Sung, G.Y. Fabrication of a pumpless, microfluidic skin chip from different collagen sources. J. Ind. Eng. Chem. 2017, 56, 375–381. [Google Scholar] [CrossRef]
- Song, H.J.; Lim, H.Y.; Chun, W.; Choi, K.C.; Lee, T.-y.; Sung, J.H.; Sung, G.Y. Development of 3D skin-equivalent in a pump-less microfluidic chip. J. Ind. Eng. Chem. 2018, 60, 355–359. [Google Scholar] [CrossRef]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schröder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Investig. 2013, 123. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Henry, J.; Hsu, C.-Y.; Balica, S.; Jean-Decoster, C.; Méchin, M.-C.; Hansmann, B.; Rodriguez, E.; Weindinger, S.; Schmitt, A.-M. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 2013, 131, 1094–1102. [Google Scholar] [CrossRef]
- Ishida-Yamamoto, A.; Iizuka, H. Differences in involucrin immunolabeling within cornified cell envelopes in normal and psoriatic epidermis. J. Investig. Dermatol. 1995, 104, 391–395. [Google Scholar] [CrossRef] [Green Version]
- Novak, N.; Bieber, T. Allergic and nonallergic forms of atopic diseases. J. Allergy Clin. Immunol. 2003, 112, 252–262. [Google Scholar] [CrossRef]
- Omori-Miyake, M.; Yamashita, M.; Tsunemi, Y.; Kawashima, M.; Yagi, J. In vitro assessment of IL-4-or IL-13-mediated changes in the structural components of keratinocytes in mice and humans. J. Investig. Dermatol. 2014, 134, 1342–1350. [Google Scholar] [CrossRef] [Green Version]
- Grewe, M.; Walther, S.; Gyufko, K.; Czech, W.; Schöpf, E.; Krutmann, J. Analysis of the cytokine pattern expressed in situ in inhalant allergen patch test reactions of atopic dermatitis patients. J. Investig. Dermatol. 1995, 105, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, T.; Memezawa, A.; Okuyama, R.; Sayo, T.; Sugiyama, Y.; Inoue, S.; Aiba, S. Increased hyaluronan production and decreased E-cadherin expression by cytokine-stimulated keratinocytes lead to spongiosis formation. J. Investig. Dermatol. 2009, 129, 1412–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, Y.; Adachi, Y.; Elias, P.M.; Crumrine, D.; Sakai, T.; Kurahashi, R.; Katagiri, K.; Fujiwara, S. The T h2 cytokine, interleukin-4, abrogates the cohesion of normal stratum corneum in mice: Implications for pathogenesis of atopic dermatitis. Exp. Dermatol. 2013, 22, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.; Roberts-Thompson, L.; Reichelt, J. Deletion of K1/K10 does not impair epidermal stratification but affects desmosomal structure and nuclear integrity. J. Cell Sci. 2012, 125, 1750–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinin, A.; Marekov, L.N.; Steinert, P.M. Assembly of the epidermal cornified cell envelope. J. Cell Sci. 2001, 114, 3069–3070. [Google Scholar] [CrossRef]
- Yuspa, S.H.; Kilkenny, A.E.; Steinert, P.M.; Roop, D.R. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J. Cell Biol. 1989, 109, 1207–1217. [Google Scholar] [CrossRef] [Green Version]
- McAleer, M.A.; Irvine, A.D. The multifunctional role of filaggrin in allergic skin disease. J. Allergy Clin. Immunol. 2013, 131, 280–291. [Google Scholar] [CrossRef]
- Kezic, S.; O’regan, G.; Yau, N.; Sandilands, A.; Chen, H.; Campbell, L.; Kroboth, K.; Watson, R.; Rowland, M.; Irwin McLean, W. Levels of filaggrin degradation products are influenced by both filaggrin genotype and atopic dermatitis severity. Allergy 2011, 66, 934–940. [Google Scholar] [CrossRef] [Green Version]
- Elias, P.M. Lipid abnormalities and lipid-based repair strategies in atopic dermatitis. Biochimica et Biophysica Acta BBA-Mol. Cell Biol. Lipids 2014, 1841, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.M.; Yosipovitch, G. Skin pH: From basic science to basic skin care. Acta Derm.-Venereol. 2013, 93, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Howell, M.D.; Fairchild, H.R.; Kim, B.E.; Bin, L.; Boguniewicz, M.; Redzic, J.S.; Hansen, K.C.; Leung, D.Y. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J. Investig. Dermatol. 2008, 128, 2248–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thyssen, J.P.; Kezic, S. Causes of epidermal filaggrin reduction and their role in the pathogenesis of atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.; Hardman, M.; Nield, K.; Byrne, C.; McCarroll, S.; Hadnott, T.; Perry, G.; Sabeti, P.; Steinert, P.; Parry, D. Increased Expression of Carbonic Anhydrase II (CA II) in Lesional Skin of Atopic Dermatitis: Regulation by Th2 Cytokines. Nat. Genet 1786, 38, 86–92. [Google Scholar]
- Kamsteeg, M.; Bergers, M.; de Boer, R.; Zeeuwen, P.L.; Hato, S.V.; Schalkwijk, J.; Tjabringa, G.S. Type 2 helper T-cell cytokines induce morphologic and molecular characteristics of atopic dermatitis in human skin equivalent. Am. J. Pathol. 2011, 178, 2091–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glück, J.; Rogala, B. Chemokine RANTES in atopic dermatitis. Archivum immunologiae et therapiae experimentalis 1999, 47, 367–372. [Google Scholar]
- Nomura, I.; Ohya, Y.; Gao, B.; Boguniewicz, M.; Darst, M.; Travers, J.; Leung, D. Distinct patterns of chemokine gene expression in the skin lesions of atopic dermatitis and psoriasis: A gene microarray analysis. J. Allergy Clin. Immunol. 2004, 113, S54. [Google Scholar] [CrossRef]
- Renne, J.; Schäfer, V.; Werfel, T.; Wittmann, M. Interleukin-1 from epithelial cells fosters T cell-dependent skin inflammation. Br. J. Dermatol. 2010, 162, 1198–1205. [Google Scholar] [CrossRef]
- Sumimoto, S.; Kawai, M.; Kasajima, Y.; Hamamoto, T. Increased plasma tumour necrosis factor-alpha concentration in atopic dermatitis. Arch. Dis. Child. 1992, 67, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Ohmen, J.D.; Hanifin, J.M.; Nickoloff, B.J.; Rea, T.H.; Wyzykowski, R.; Kim, J.; Jullien, D.; McHugh, T.; Nassif, A.S.; Chan, S.C. Overexpression of IL-10 in atopic dermatitis. Contrasting cytokine patterns with delayed-type hypersensitivity reactions. J. Immunol. 1995, 154, 1956–1963. [Google Scholar]
- Shao, X.; Chen, S.; Yang, D.; Cao, M.; Yao, Y.; Wu, Z.; Li, N.; Shen, N.; Li, X.; Song, X. FGF2 cooperates with IL-17 to promote autoimmune inflammation. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Huang, W.; Chen, L.; Jin, M.; Wang, Q.; Gao, Z.; Jin, Z. Topical application of JAK1/JAK2 inhibitor momelotinib exhibits significant anti-inflammatory responses in DNCB-induced atopic dermatitis model mice. Int. J. Mol. Sci. 2018, 19, 3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, K.M.; Jaunin, F.; Masouyé, I.; Saurat, J.-H.; Hauser, C. Th2 cells mediate IL-4-dependent local tissue inflammation. J. Immunol. 1993, 150, 5576–5584. [Google Scholar] [PubMed]
- Gros, E.; Bussmann, C.; Bieber, T.; Förster, I.; Novak, N. Expression of chemokines and chemokine receptors in lesional and nonlesional upper skin of patients with atopic dermatitis. J. Allergy Clin. Immunol. 2009, 124, 753–760.e1. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Yun, J.W.; Shin, T.H.; Lee, S.H.; Lee, B.C.; Yu, K.R.; Seo, Y.; Lee, S.; Kang, T.W.; Choi, S.W. Human Umbilical Cord Blood Mesenchymal Stem Cell-Derived PGE 2 and TGF-β1 Alleviate Atopic Dermatitis by Reducing Mast Cell Degranulation. Stem Cells 2015, 33, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Xue, K.; Liu, H.; Jian, Q.; Liu, B.; Zhu, D.; Zhang, M.; Gao, L.; Li, C. Leptin induces secretion of pro-inflammatory cytokines by human keratinocytes in vitro–a possible reason for increased severity of psoriasis in patients with a high body mass index. Exp. Dermatol. 2013, 22, 406–410. [Google Scholar] [CrossRef]
- Gillitzer, R.; Berger, R.; Mielke, V.; Müller, C.; Wolff, K.; Stingl, G. Upper keratinocytes of psoriatic skin lesions express high levels of NAP-1/IL-8 mRNA in situ. J. Investig. Dermatol. 1991, 97, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, A.B.; Luster, A.D.; Posnett, D.N.; Carter, D. Detection of a gamma interferon-induced protein IP-10 in psoriatic plaques. J. Exp. Med. 1988, 168, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Niwa, Y. Elevated RANTES levels in plasma or skin and decreased plasma IL-10 levels in subsets of patients with severe atopic dermatitis. Arch. Dermatol. 2000, 136, 125–126. [Google Scholar] [CrossRef]
- Zweiman, B.; Kaplan, A.P.; Tong, L.; Moskovitz, A.R. Cytokine levels and inflammatory responses in developing late-phase allergic reactions in the skin. J. Allergy Clin. Immunol. 1997, 100, 104–109. [Google Scholar] [CrossRef]
- Kapp, A.; Neuner, P.; Krutmann, J.; Luger, T.; Schöpf, E. Production of interleukin-2 by mononuclear cells in vitro in patients with atopic dermatitis and psoriasis. Comparison with serum interleukin-2 receptor levels. Acta Derm.-Venereol. 1991, 71, 403–406. [Google Scholar]
- Bhushan, M.; McLaughlin, B.; Weiss, J.; Griffiths, C. Levels of endothelial cell stimulating angiogenesis factor and vascular endothelial growth factor are elevated in psoriasis. Br. J. Dermatol. 1999, 141, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Detmar, M.; Brown, L.F.; Claffey, K.P.; Yeo, K.-T.; Kocher, O.; Jackman, R.W.; Berse, B.; Dvorak, H.F. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J. Exp. Med. 1994, 180, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Bracci-Laudiero, L.; Pincelli, C. Nerve growth factor is increased in psoriatic skin. J. Investig. Dermatol. 1995, 105, 854–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raychaudhuri, S.P.; Jiang, W.-Y.; Farber, E.M. Psoriatic keratinocytes express high levels of nerve growth factor. Acta Derm.-Venereol. 1998, 78, 84–86. [Google Scholar] [PubMed] [Green Version]
- Pastore, S.; Mascia, F.; Mariani, V.; Girolomoni, G. The epidermal growth factor receptor system in skin repair and inflammation. J. Investig. Dermatol. 2008, 128, 1365–1374. [Google Scholar] [CrossRef] [Green Version]
- Pine, G.M.; Batugedara, H.M.; Nair, M.G. Here, there and everywhere: Resistin-like molecules in infection, inflammation, and metabolic disorders. Cytokine 2018, 110, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Lederle, W.; Stark, H.-J.; Skobe, M.; Fusenig, N.E.; Mueller, M.M. Platelet-derived growth factor-BB controls epithelial tumor phenotype by differential growth factor regulation in stromal cells. Am. J. Pathol. 2006, 169, 1767–1783. [Google Scholar] [CrossRef] [Green Version]
- Maas-Szabowski, N.; Shimotoyodome, A.; Fusenig, N.E. Keratinocyte growth regulation in fibroblast cocultures via a double paracrine mechanism. J. Cell Sci. 1999, 112, 1843–1853. [Google Scholar] [CrossRef]
- Harada, M.; Kamimura, D.; Arima, Y.; Kohsaka, H.; Nakatsuji, Y.; Nishida, M.; Atsumi, T.; Meng, J.; Bando, H.; Singh, R. Temporal expression of growth factors triggered by epiregulin regulates inflammation development. J. Immunol. 2015, 194, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Miura, H.; Sano, S.; Higashiyama, M.; Yoshikawa, K.; Itami, S. Involvement of insulin-like growth factor-I in psoriasis as a paracrine growth factor: Dermal fibroblasts play a regulatory role in developing psoriatic lesions. Arch. Dermatol. Res. 2000, 292, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.C.; Tang, B.; Guo, D.; Zhang, J.; Liang, Y.Y.; Ma, D.; Zhu, J.Y. Expression of insulin-like growth factor-1 receptor in keloid and hypertrophic scar. Clin. Exp. Dermatol. 2014, 39, 822–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanath, L.; Bindhu, J.; Krishnamurthy, B.; Suresh, K. Granulocyte-Colony Stimulating Factor (G-CSF) accelerates healing of radiation induced moist desquamation of the skin. Klin. Onkol. 2012, 25, 199–205. [Google Scholar] [PubMed]
- Lee, S.; Jin, S.-P.; Kim, Y.K.; Sung, G.Y.; Chung, J.H.; Sung, J.H. Construction of 3D multicellular microfluidic chip for an in vitro skin model. Biomed. Microdevices 2017, 19, 22. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| GAPDH | 5′-CTCCTCTGACTTCAACAGCG-3′ | 5′-GCCAAATTCGTTGTCATACCAG-3′ |
| FLG | 5′-GGAGTCACGTGGCAGTCCTCACA-3′ | 5′-GGTGTCTAAACCCGGATTCACC-3′ |
| IVL | 5′-CCGCAAATGAAACAGCCAACTCC-3′ | 5′-GGATTCCTCATGCTGTTCCCAG-3′ |
| LOR | 5′-CCGGAGATGGTGGCCTTCTCTCT-3′ | 5′-GGCCTGATGTGAGTTGCCATGCT-3′ |
| CA II | 5′-CAAGAGAGCTGAAGACTATCCCA-3′ | 5′-TGAAGTCCGAAGTAATCCTCCT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Kim, H.; Sung, G.Y. An Interleukin-4 and Interleukin-13 Induced Atopic Dermatitis Human Skin Equivalent Model by a Skin-On-A-Chip. Int. J. Mol. Sci. 2022, 23, 2116. https://doi.org/10.3390/ijms23042116
Kim K, Kim H, Sung GY. An Interleukin-4 and Interleukin-13 Induced Atopic Dermatitis Human Skin Equivalent Model by a Skin-On-A-Chip. International Journal of Molecular Sciences. 2022; 23(4):2116. https://doi.org/10.3390/ijms23042116
Chicago/Turabian StyleKim, Kyunghee, Hyeju Kim, and Gun Yong Sung. 2022. "An Interleukin-4 and Interleukin-13 Induced Atopic Dermatitis Human Skin Equivalent Model by a Skin-On-A-Chip" International Journal of Molecular Sciences 23, no. 4: 2116. https://doi.org/10.3390/ijms23042116