
Theoretical Investigation on the ESIPT Process and Detection Mechanism for Dual-Proton Type Fluorescent Probe


College of Science, Northeast Forestry University, Harbin 150040, China
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(4), 2132; https://doi.org/10.3390/ijms23042132
Submission received: 30 December 2021
/
Revised: 7 February 2022
/
Accepted: 10 February 2022
/
Published: 15 February 2022
(This article belongs to the Section Physical Chemistry and Chemical Physics)
Abstract
:Recently, a new fluorescent probe AE-Phoswas reported to detect the activity of alkaline phosphatases (ALP) in different living cell lines. Here, we present an in-depth computational analysis of the mechanism and source of the fluorescence of the AE-Phos probe. There is an intermediate product (AE-OH-Phos) in the experiment as well as a different configuration of products that may emit fluorescence. It is essential to investigate the origin of fluorescence and the detection mechanism of the probe, which could help us eliminate the interference of other substances (including an intermediate product and possible isomers) on fluorescence during the experiment. According to the change of geometric parameters and Infrared spectra, we deduce that the dual intramolecular hydrogen bonds of salicylaldehyde azine (SA) were enhanced at the excited state, while AE-OH-Phos was attenuated. Considering the complex ESIPT behavior of the dual proton-type probe, the potential energy surfaces were further discussed. It can be concluded that the single proton transfer structure of SA (SA-SPT) is the most stable form. Both the concerted double proton transfer process and stepwise single proton transfer process of SA were forbidden. The fluorescence for SA was 438 nm, while that of SA-SPT was 521 nm, which agrees with the experimentally measured fluorescence wavelength (536 nm). The conclusion that single proton transfer occurs in SA is once again verified. In addition, the distribution of electron-hole and relative index was analyzed to investigate the intrinsic mechanism for the fluorescence quenching of the probe and the intermediate product. The identification of the origin of fluorescence sheds light on the design and use of dual-proton type fluorescent probes in the future.
1. Introduction
The excited-state intramolecular proton transfer process (ESIPT) is widely found in nature and is one of the most important processes in biology and chemistry [1,2,3]. This process is a photo-transfer isomerization, in which a proton is transferred to the adjacent N, O, S or other heteroatoms. The ESIPT molecules generally have a larger Stokes shift and could avoid self-absorption. The special photophysical property of ESIPT molecules has been widely studied by researchers working in the fields of materials, physics, chemistry, biology and medicine [4,5,6,7,8,9,10,11,12].
In recent years, fluorescent sensing technology has developed rapidly [13,14,15]. The excellent characteristics of fluorescence sensing have inspired explorations amongst research workers of fluorescence probes, which are widely used in the fields of biochemistry, medicine, environmental science, and industry as cogent tools to monitor molecular interactions. Considering that the ESIPT reactions tend to impose a great impact on the fluorescence emission spectra but cause almost no change to the absorption spectra, ESIPT has been considered as an ideal recognition mechanism to design ratiometric fluorescence probes, which possess a large Stokes shift. An increasing number of researchers have designed and synthesized ESIPT-based fluorescent probes for the selective detection of various anions, metal ions, and small organic molecules [16,17,18,19,20,21]. There are also many theoretical studies for the mechanistic elaboration of various fluorescence probes, as well as the design of novel probes [22,23,24,25,26,27,28,29,30,31,32]. Fluorescent probes for ALP detection generally feature phosphate as a recognition moiety, and after cleavage of the phosphate ester group, a phenolic hydroxyl group will be formed. In the case of ESIPT-based probes, the -OH moiety generated upon the cleavage of the phosphate will form an intramolecular hydrogen bond with another heteroatom, along which the proton-transferred reaction could occur. Before the cleavage, the ESIPT process is not possible.
Recently, Gao et al. [33] and Li [34] both designed a novel fluorescence probe for the detection of ALP activity and successfully applied it in living cells. He and colleagues also reported a new fluorescent probe, AE-Phos, for ALP activity detection, but the probe combined aggregation-induced emission and ESIPT and was able to image ALP well in different cell lines [35]. The detection mechanism of AE-Phos was shown in Scheme 1; the phosphate group was removed after AE-Phos reacted with ALP and the product SA and intermediate product AE-OH-Phos with ESIPT properties were generated. The authors suggested that the experimentally measured fluorescence spectra were emitted by SA. However, for such an ESIPT-based molecule, a single or double proton transfer process may happen at the excited state, which in turn emits fluorescence. In addition, the intermediate product may also emit fluorescence, so specific conclusions need to be studied in depth.
Herein, the methods of DFT and TD-DFT were used to investigate the probe AE-Phos and its possible products. In order to analyze the source of fluorescence and the possibility of single or double proton transfer processes occurring in SA, the potential energy surfaces for SA at the S0 and S1 states were scanned to ascertain their proton transfer process. Furthermore, the simulation of absorption and fluorescence spectra for the studied molecules not only verified the accuracy and reliability for the theoretical calculations, but also determined the concrete source of the experimental fluorescence. The causes of fluorescence quenching for the probe and its intermediate product were also investigated by electron-hole analysis. The possibility that all products and their corresponding isomers interfere with the fluorescence in the experimental process is excluded, and the specific source of fluorescence is determined, which will pave the way for experiments to understand and design better probe molecules.
2. Computational Details
All calculations in this work were performed employing Becke’s three-parameter hybrid exchange function with the Lee-Yang-Parr gradient-corrected correlation functional (B3LYP) and 6-311g(d,p) basis set [36,37,38,39,40] using density functional theory (DFT) [41,42,43] and time-dependent density functional theory (TD-DFT) [44,45,46,47,48] methods without any constraints under Gaussian 16 package [49]. To simulate the experimental environment, these calculations were accomplished in a Dimethyl sulfoxide (DMSO) solvent based on the integral equation formalism of the polarizable continuum model (IEFPCM). The geometrical structures of the ground and excited (S0 and S1) states for the probe and its products were separately optimized, and the relative geometric structure parameters were obtained. Furthermore, the infrared vibrations of the products were simulated based on the optimized structure, and the interaction region indicator (IRI) [50] isosurfaces combined with reduced density gradient (RDG) [51] scatter plots were plotted, applying Multiwfn 3.7 [52] and VMD [53] programs; all of these were used to analyze the change of hydrogen bond strength. The transition state theory is applied to calculate the energy change of excited-state isomerization. Additionally, the potential energy curves of AE-OH-PHOs were obtained by extending the distance of O2-H2, and the surfaces of SA were scanned by regulating the distance of O1-H1 and O2-H2. In addition, the spectra were simulated, employing a B3PW91 functional.
3. Results and Discussion
3.1. Intramolecular Hydrogen Bond
By analyzing the change in the strength of intramolecular hydrogen bonds (IHBs) upon photo-excitation, we can judge whether it is permissive for the ESIPT process and thus leads to isomerization. Therefore, the structures of all isomers for AE-OH-PHOs (enol and keto forms) and SA (named SA, SA-SPT, SA-DPT according to the proton transfer) have been optimized using the B3LYP/6-311G(d,p) basis set in the DMSO solvent, which is helpful to compare with the experiment. As shown in Figure 1, all structures of the probe products were included, and the atomic numbers associated with the hydrogen bonds were marked on the molecular structure of SA. The bond length and bond angle parameters related to molecules AE-OH-PHOs and SA were presented in Table 1 and Table 2, respectively.
In general, the longer the bond length (O-H distance) for the proton donor, the shorter the bond length (H…N distance) for the proton acceptor, and the closer the bond angle δ(O-H…N) is to 180°, the more stable the IHB [54,55,56]. For AE-OH-Phos, from the S0 state to S1 state (Table 1), the shortening of the O2-H2, the elongation of H2…N2, and the decrease of δ(O2-H2…N2) at the enol configuration indicate that the IHB is stronger and its structure is more stable at the S0 state, which is detrimental to the ESIPT reaction. In addition, the probe product SA had a completely symmetrical planar structure, with the same parameters for the two hydrogen bonds. Taking O2-H2…N2 as an example (Table 2), the O2-H2 length increased from 0.99 Å (S0) to 1.00 Å (S1), and the H2-N2 diminished from 1.76 Å to 1.70 Å, accompanied by the O2-H2-N2 angle, which enlarged from 146.1° to 148.3°. In addition, for SA-SPT that had undergone a single-step proton transfer process, it should be noted that the O1-H1, H1…N1 length and O1-H1…N1 angle were separately changed from 0.98 Å, 1.80 Å, and 144.7° (S0) to 0.99 Å, 1.77 Å, and 146.4° (S1), respectively. Although there is a trend of hydrogen bond enhancement, based on the slight changes of these structural parameters, we think it is not enough to support us to analyze whether the ESIPT process could occur. Therefore, the previously reported infrared spectra and the RDG scatter plots are used to further analyze the change of hydron bond strength. In addition, for O2…H2-N2 of which SA-SPT had undergone proton transfer, the change trend in the bond length and bond angle parameters indicates that the S1-state SA-SPT is more stable than the S0 state. However, the SA-DPT configuration is not stable in the S0 state. The detailed reasons will be discussed in the potential energy surfaces section.
Infrared (IR) spectra is a frequently used method to analyze the change of hydrogen bond; the degree of change for hydrogen bond strength can be judged by comparing the red-shift or blue-shift of hydroxyl stretching vibration peaks in the S0 and S1 states. The IR spectra for AE-OH-PHOs and SA are displayed in Figure 2. The calculated O2-H2 stretching vibration frequencies were separately located at 3229 cm−1 and 3620 cm−1 at the S0 and S1 states, respectively. Such a large blue-shift of 391 cm−1 demonstrates that the IHB at the S1 state is weaker than that at the S0 state, which is unfavorable for the ESIPT process. However, the stretching vibration frequencies of O1-H1 and O2-H2 for SA both change from 3260 cm−1 (S0) to 2957 cm−1 (S1), respectively, with a 303 cm−1 red-shift, which indicates that this is prone to ESIPT reaction. This result further validates our conjecture on the geometric structure.
For the sake of more intuitively showing whether the IHB is formed and the change of IHB strength, the IRI isosurfaces and RDG scatter plots were subjected to further analysis. The meaning of different colors in Figure 3 are: red, green, and blue represent the steric effect, van der Waals interaction and hydrogen bond, respectively. Additionally, the bluer the IRI surface, the more negative the blue spike of RDG, indicating a stronger IHB. As shown in Figure 3, the appearance of a blue isosurface between H2 and N2 atoms for the intermediate product AE-OH-Phos at the S0 and S1 states signifies that IHB interaction exists and its corresponding RDG blue spike was separately near −0.05 a.u. and −0.04 a.u.. This change in value clearly shows that the IHB strength in the S1 state is weaker, which is in full agreement with the conclusions of the previous structure parameters and IR analysis. Therefore, it can be speculated that the ESIPT process cannot occur in AE-OH-Phos. Furthermore, SA, being a completely symmetric structure, has two IHBs of the same strength with the blue spikes of the S0 and S1 states located at −0.05 a.u. < ρ <−0.04 a.u. and −0.06 a.u. < ρ < −0.05 a.u.. The enhancement of IHB is very favorable for the ESIPT process after photoexcitation. However, whether the SA molecule underwent stepwise single proton transfer or concerted double proton transfer requires further analysis.
3.2. ESIPT Process Analysis
To calculate the precise reaction energy barrier for the proton transfer process occurring in SA and AE-OH-Phos at the S1 state, the transition state (TS) structure of its different paths and the corresponding energies were calculated. The TS structure and energy for SA stepwise single proton transfer (path 1) and simultaneous double proton transfer (path 2), as well as AE-OH-Phos, are shown in Figure 4. The energy barrier of AE-OH-Phos is up to 13.75 kcal/mol; this indicates that the proton transfer process was forbidden at the S1 state. In addition, the reaction barrier of single proton transfer is 2.75 kcal/mol and the double proton transfer is 9.01 kcal/mol. This suggests that the SA is more inclined to undergo single proton transfer into the SA-SPT form than the process of double proton transfer. Moreover, the SA-SPT has the lowest energy, which means that its configuration is the most stable.
To be able to investigate the intrinsic mechanism of the ESIPT process and the possible proton transfer for AE-OH-Phos and SA, the potential energy surfaces (PESs) of SA and the potential energy curves (PECs) of AE-OH-Phos at the S0 and S1 states were scanned separately based on the optimized structure. The obtained PECs of AE-OH-Phos were as a function of O2-H2 distance and ranged from 0.99 Å(initial length)→1.99 Å and 0.97 Å(initial length)→1.97 Å at the S0 and S1 states, respectively. As depicted in Figure 5, the reverse potential barrier of 1.93 kcal/mol is lower than the forward potential barrier (7.16 kcal/mol), indicating that it is difficult for the proton transfer process to occur at the S0 state. This is consistent with the previously obtained result that weakening of the S1-state hydrogen bond is unfavorable for the ESIPT reaction to proceed.
The PESs of SA were obtained by extending the distance of O1-H1 and O2-H2, as shown in Figure 6. It is worth noting that the PESs of both the S0 and S1 states are symmetric, and the lowest energy points in the figures are marked for the convenience of analysis. In Figure 6a, the coordinates of these corresponding points are a (0.99 Å, 0.99 Å), b (1.99 Å, 0.99 Å), and c (1.99 Å, 1.99 Å). In addition, the energy of all points on the S0-state PES is higher than that of point a; this means that the structure of point a is the most stable at the S0 state. Furthermore, the potential energy of paths a→c and b→c increases monotonically, which indicates that these two paths do not actually exist at the S0 state. Moreover, the forward energy barrier of a→c is 8.03 kcal/mol, while the reverse energy barrier is only 0.31 kcal/mol, so it can be concluded that the isomerization process cannot happen at the S0 state either.
Interestingly, the calculated potential energies of the minima points are in order of EC > EA > EB at the S1 state, as present in Figure 6b. The coordinates of these corresponding points are A (1.00 Å, 1.00 Å), B (1.80 Å, 1.00 Å) and C (1.60 Å, 1.60 Å). It is clear that point B is the most stable configuration at the S1 state, and the path from A to B only needs to cross the energy barrier of 2.78 kcal/mol, which means that the S1-state SA molecule could easily undergo a single proton transfer process to form SA-SPT. Nevertheless, the energy barrier for the path A→C to cause a concerted double proton transfer process is 9.00 kcal/mol, and even though this energy barrier is not very high, the barrier of A→B is significantly lower than it, which demonstrates that the synchronous double proton transfer for SA is forbidden at the S1 state. After reaching point B, the SA-SPT needs to cross the potential barrier of 9.72 kcal/mol to reach point C. To analyze whether the proton transfer process in the second step can occur, the reverse proton transfer energy barrier for all paths was calculated: C→B, C→A, and B→A are 0.12 kcal/mol, 2.11 kcal/mol, and 5.49 kcal/mol, respectively. The results prove that even if the proton transfer from B to C occurred, it would return to point B with an almost no-barrier process. It can be concluded that the SA-SPT conformation is stable to exist at point B. Based on the analysis of the ESIPT behavior of the intermediate product AE-OH-Phos and product SA, the following conclusions can be drawn: due to the presence of phosphate group AE-OH-Phos, which is unable to experience ESIPT process, SA can neither undergo stepwise nor synergistic double proton transfer, but can only proceed with a single proton transfer process, which is stable in the form of SA-SPT.
3.3. Electronic Spectra and Fluorescence Mechanism
Absorption and fluorescence spectra are often used to fit theoretical calculations to experiments to verify the reliability and accuracy of a theory. Therefore, the spectra of ALP probe product SA were simulated separately, employing different functions in a DMSO solvent of IEFPCM at 6-311G(d,p) basis set, as listed in Table 3. The previous analysis can determine that the single proton transfer process of SA is definitely present, and the simulated fluorescence spectra are also the closest to the experimental value of SA-SPT. Hence, the Stokes-shift of SA-SPT was calculated using different functionals and found that the value (145 nm) of B3PW91 is the closest to the experimental value (180 nm). Therefore, the spectra of studied molecules were simulated by B3PW91 for subsequent discussion and the first three single excited state transition and fluorescence properties are separately gathered in Table 4 and Table 5.
To compare the spectral changes more visually, their spectra are plotted in Figure 7. It can be found that the fluorescence of probe AE-Phos and intermediate product AE-OH-Phos was quenched, which means that neither fluorescence can be observed experimentally. Moreover, the result is consistent with the experimentally measured spectra. As listed in Table 4, the oscillator strength of S1-state AE-Phos was 0.0010, which means that the fluorescence is almost unobservable experimentally. That is to say; the S1 state is a dark state. The electrons of AE-Phos cannot be directly excited to the S1 state, but are excited to the S2 state. According to Kasha’s rule, the electrons of the S2 state will return to the S1 state through internal conversion, resulting in fluorescence quenching. However, the electrons of AE-OH-Phos were excited to the S1 state but a fluorescence quenching also occurred, which we speculate may be due to the photo-induced electron transfer process; this will be further analyzed in the frontier molecular orbitals section. More importantly, the PEC shows that AE-OH-Phos is unable to undergo the ESIPT process into the keto form. As a consequence, we can conclude that AE-OH-Phos does not interfere with the experimental spectra by emitting fluorescence.
As shown in Figure 7c, the two peaks of the absorption band of SA are consistent with the experimentally measured peaks. The highest peak in the absorption band at 376 nm is produced by the electron transitions to the S1 state, which is in line with the experimental value (356 nm), and combined with Table 4, we can know that another peak at a smaller wavelength is emitted from the S0 state transitions to the S4 state. In addition, the calculated fluorescence peak of SA-SPT (521 nm) is also in line with the experiment (536 nm), which is consistent with the conclusion that the SA-SPT form is stable the S1 state obtained from our previous analysis. In a word, the probe AE-Phos produces the product SA when detecting the ALP activity, and the intermediate product AE-OH-PHOs may also be present, but since both AE-Phos and AE-OH-PHOs are fluorescence quenching, they will not affect the fluorescence spectra of the experiment. Moreover, the fluorescence of SA-SPT is almost identical to the experimental fluorescence; it is presumed that the fluorescence measured in the experiment is derived from SA-SPT, which is formed by SA undergoing a single proton transfer process.
3.4. Frontier Molecular Orbitals
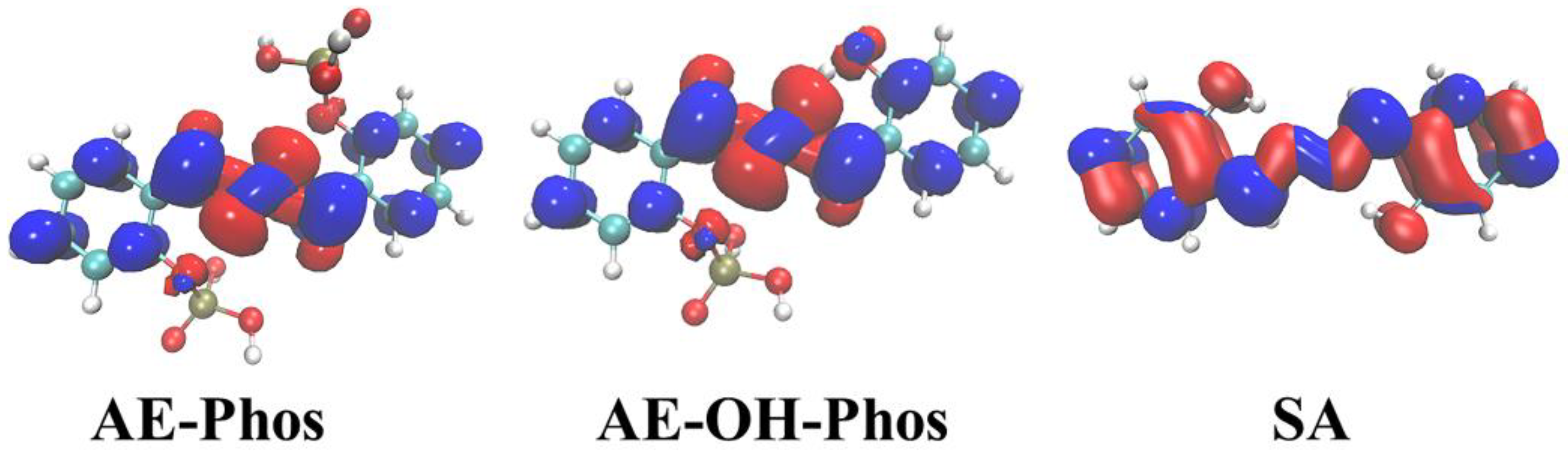
In the previous section, we mentioned that the fluorescence of AE-PHOs and AE-OH-PHOs is quenched, and this change in photophysical properties is attributed to the redistribution of electrons due to photoexcitation. To observe the change in charge distribution after photoexcitation, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) corresponding to their fluorescence emission are plotted in Figure 8. When the electrons of AE-PHOs and AE-OH-PHOs fluoresce from LUMO to HOMO, there is an obvious concentration for charge from the benzene ring on both sides to the middle near the N-N bond. Such a charge transfer process could cause the chromophore part to fail to emit fluorescence, and this process is called the photo-induced electron transfer process. However, SA has almost no significant change in electron cloud distribution due to local excitation.
In order to better visualize the distribution of electrons and holes, and thus discuss the intrinsic cause of the fluorescence quenching for the probe and its intermediate products, the electrons and holes distribution of three molecules was investigated using the electron-hole analysis method. The relevant parameters were gathered in Table 6. The D, t, H, and Δσ index reflect the distance between the center of mass for electrons and holes, the degree of separation, the overall average distribution breadth, and the difference between the overall spatial distribution breadth, respectively. Generally speaking, t > 0 and t < 0 separately indicate whether the charge transfer makes the electron-hole sufficiently separated. Combined with Table 6 and Figure 9, the t index of all three molecules is less than zero, but the figure shows an obvious separation of the electron-hole. In other words, the t index is not perfect enough to analyze multidirectional charge transfer. The Sr index means the degree of electron-hole overlap; the larger the value, the higher the degree of overlap. The smaller hole and electron delocalization index (HDI and EDI) illustrates the higher delocalization degree of the electron-hole, that is, the greater the distribution and uniformity.
As depicted in Figure 9, for AE-PHOs and AE-OH-PHOs, the holes are concentrated in the connecting part of the two benzene rings and the electrons are uniformly distributed throughout the framework of the molecule. The apparent movement of electrons from the middle part to the sides causes the fluorescence quenching in both molecules. The D index of the three molecules shows that the distance between their centers of mass is very short, again proving the opinion that the electrons are uniformly distributed on both sides. The Δσ index manifests that the electron distribution breadth for AE-PHOs and AE-OH-PHOs is larger than that of the holes, while SA is almost the same. The HDI of AE-PHOs and AE-OH-PHOs are approximately twice as high as the EDI, indicating that the electron delocalization of both molecules is greater than that of the holes. The electron-hole analysis verifies that AE-PHOs and AE-OH-PHOs produce a significant charge transfer, which is likely to lead to fluorescence quenching.
4. Conclusions
On the whole, theoretical calculations were performed to investigate the detection mechanism of the ALP probe and to analyze the origin of the fluorescence measured from the experiments. Geometric structure parameters, IR vibration, and RDG scatter plots indicate that the excited state IHB for SA is enhanced, while AE-OH-Phos is weakened. Combining the simulated spectra and potential energy curves (surfaces), AE-OH-Phos is unable to emit fluorescence, and the experimentally measured fluorescence is derived from the SA-SPT configuration formed by SA undergoing single proton transfer. The agreement between the simulated SA spectra and experimental spectra proves the accuracy and feasibility of the theoretical calculation. Finally, the distribution of frontier molecular orbital electron clouds and electron-hole analysis elucidates the endogenous causes of the fluorescence quenching for probe AE-Phos and intermediate product AE-OH-Phos. Our work based on the salicylaldehyde azine shows that the dual-proton type structure cannot undergo a double proton transfer process, but can only undergo a single proton transfer process. Additionally, the ESIPT-type intermediate AE-OH-Phos cannot in fact produce the ESIPT reaction. All of these could provide a theoretical reference for the designing and synthesizing of novel double proton transfer fluorescent probes.
Author Contributions
Formal analysis, Y.C.; data curation, X.Y.; writing-original draft preparation, Y.C.; writing-review and editing, C.S.; supervision, J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by [the Fundamental Research Funds for the Central Universities] grant number [2572020BC03] and [the National Training Program of Innovation and Entrepreneurship for Undergraduates] grant number [202110225159].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no known competing financial interest.
References
- He, L.; Dong, B.; Liu, Y.; Lin, W. Fluorescent chemosensors manipulated by dual/triple interplaying sensing mechanisms. Chem. Soc. Rev. 2016, 45, 6449–6461. [Google Scholar] [CrossRef]
- Kwon, J.E.; Park, S.Y. Advanced Organic Optoelectronic Materials: Harnessing Excited-State Intramolecular Proton Transfer (ESIPT) Process. Adv. Mater. 2011, 23, 3615–3642. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, W.; Ge, J.; Zhang, H.; Wang, P. New sensing mechanisms for design of fluorescent chemosensors emerging in recent years. Chem. Soc. Rev. 2011, 40, 3483–3495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Excited state intramolecular proton transfer (ESIPT): From principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. [Google Scholar] [CrossRef]
- Kim, T.-I.; Kang, H.J.; Han, G.; Chung, S.J.; Kim, Y. A highly selective fluorescent ESIPT probe for the dual specificity phospha-tase MKP-6. Chem. Commun. 2009, 39, 5895–5897. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, B.; Zhou, Q.; Zhang, X.; Li, B.; Su, X.; Shi, Y. Enhancing fluorescence of benzimidazole derivative via solvent-regulated ESIPT and TICT process: A TDDFT study. Spectrochim. Acta Part A 2021, 258, 119862. [Google Scholar] [CrossRef] [PubMed]
- Petdee, S.; Chaiwai, C.; Benchaphanthawee, W.; Nalaoh, P.; Kungwan, N.; Namuangruk, S.; Sudyoadsuk, T. Promarak, Imid-azole-based solid-state fluorophores with combined ESIPT and AIE features as self-absorption-free non-doped emitters for electroluminescent devices. Dyes Pigm. 2021, 193, 109488. [Google Scholar] [CrossRef]
- Baranowska, K.; Monka, M.; Bojarski, P.; Jozefowicz, M. Insight into Molecular Interactions of Two Methyl Benzoate Deriva-tives with Bovine Serum Albumin. Int. J. Mol. Sci. 2021, 22, 11705. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.W.; Luk, H.L.; Phillips, D.L. Anti-Kasha Behavior of 3-Hydroxyflavone and Its Derivatives. Int. J. Mol. Sci. 2021, 22, 11103. [Google Scholar] [CrossRef]
- Goswami, S.; Das, S.; Aich, K.; Pakhira, B.; Panja, S.; Mukherjee, S.K.; Sarkart, S. A Chemodosimeter for the Ratiometric Detec-tion of Hydrazine Based on Return of ESIPT and Its Application in Live-Cell Imaging. Org. Lett. 2013, 15, 5412–5415. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Zhang, G.; Bai, R.; Pang, Y. Flavone-Based ESIPT Ratiometric Chemodosimeter for Detection of Cysteine in Living Cells. ACS Appl. Mater. Interfaces 2014, 6, 4402–4407. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Sedgwick, A.C.; Sun, X.; Bull, S.D.; He, X.-P.; James, T.D. Reaction-Based Fluorescent Probes for the Detection and Imag-ing of Reactive Oxygen, Nitrogen, and Sulfur Species. Acc. Chem. Res. 2019, 52, 2582–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, T.; Ikeda, S.; Okamoto, A. Doubly Thiazole Orange-Labeled DNA for Live Cell RNA Imaging. Bull. Chem. Soc. Jpn. 2009, 82, 110–117. [Google Scholar] [CrossRef]
- Urano, Y. Novel live imaging techniques of cellular functions and in vivo tumors based on precise design of small mole-cule-based ‘Activatable’ fluorescence probes. Curr. Opin. Chem. Biol. 2012, 16, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.R.; Chen, H.Y.; Zhang, W.; Pan, J.X.; Dang, F.Q.; Zhang, Z.Q.; Zhang, J. Ratiometric fluorescence for sensitive and selec-tive detection of mitoxantrone using a MIP@rQDs@SiO2 fluorescence probe. Sens. Actuators B 2017, 244, 31–37. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, W. A new fluorescent probe for gasotransmitter H2S: High sensitivity, excellent selectivity, and a significant fluorescence off–on response. Chem. Commun. 2014, 50, 4214–4217. [Google Scholar] [CrossRef]
- Chen, W.-H.; Xing, Y.; Pang, Y. A Highly Selective Pyrophosphate Sensor Based on ESIPT Turn-On in Water. Org. Lett. 2011, 13, 1362–1365. [Google Scholar] [CrossRef]
- Sedgwick, A.C.; Wu, L.L.; Han, H.H.; Bull, S.D.; He, X.P.; James, T.D.; Sessler, J.L.; Tang, B.Z.; Tian, H.; Yoon, J. Excited-state in-tramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents. Chem. Soc. Rev. 2018, 47, 8842–8880. [Google Scholar] [CrossRef] [Green Version]
- Deng, B.; Ren, M.; Kong, X.; Zhou, K.; Lin, W. An ESIPT based fluorescent probe for imaging hydrogen sulfide with a large turn-on fluorescence signal. RSC Adv. 2016, 6, 62406–62410. [Google Scholar] [CrossRef]
- Lan, M.; Wu, J.; Liu, W.; Zhang, H.; Zhang, W.; Zhuang, X.; Wang, P. Highly sensitive fluorescent probe for thiols based on combination of PET and ESIPT mechanisms. Sens. Actuators B Chem. 2011, 156, 332–337. [Google Scholar] [CrossRef]
- Lv, H.-M.; Yuan, D.-H.; Liu, W.; Chen, Y.; Au, C.-T.; Yin, S.-F. A highly selective ESIPT-based fluorescent probe for cysteine sensing and its bioimaging application in living cells. Sens. Actuators B Chem. 2016, 233, 173–179. [Google Scholar] [CrossRef]
- Latha, V.; Annaraj, B.; Neelakantan, M. ESIPT inspired dual fluorescent probe (Z)-3-((4-(4-aminobenzyl) phenyl) amino)-1,3-diphenylprop-2-en-1-one: Experimental and DFT based approach to photophysical properties. Spectrochim. Acta Part A. 2014, 133, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.X.; Li, S.Y.; Wu, Y.B.; Lu, L.P.; Zhu, M.L. Zn(II)-selective and sensitive fluorescent chemosensor based on steric con-strains and inhibition of ESIPT. Sens. Actuators B 2017, 242, 1035–1042. [Google Scholar] [CrossRef]
- He, X.; Xiong, W.; Zhang, L.; Xu, C.; Fan, J.; Qian, Y.; Wen, J.; Ding, F.; Shen, J. ESIPT-based ratiometric fluorescent probe for highly selective and sensitive sensing and bioimaging of group IIIA ions in living cancer cells and zebrafish. Dye. Pigment. 2020, 174, 108059. [Google Scholar] [CrossRef]
- Karuppiah, K.; Muniyasamy, H.; Sepperumal, M.; Ayyanar, S. Design and synthesis of new salicylhydrazone tagged indole derivative for fluorometric sensing of Zn2+ ion and colorimetric sensing of F− ion: Applications in live cell imaging. Microchem. J. 2020, 159, 105543. [Google Scholar] [CrossRef]
- Kanlayakan, N.; Kungwan, N. Theoretical study of heteroatom and substituent effects on excited-state intramolecular proton transfers and electronic properties of amino-type hydrogen bonding molecules. J. Lumin. 2021, 238, 118260. [Google Scholar] [CrossRef]
- Chansen, W.; Yu, J.-S.K.; Kunggwan, N. A TD-DFT molecular screening for fluorescence probe based on excited-state intramo-lecular proton transfer of 2’-hydroxychalcone derivatives. J. Photochem. Photobiol. A 2021, 410, 113165. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Tsai, H.-Y. Synthesis, X-ray Structure, Spectroscopic Properties and DFT Studies of a Novel Schiff Base. Int. J. Mol. Sci. 2014, 15, 18706–18724. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Zhang, S.; Xie, Y.; Lei, C. A fluorescent probe for Gallium(III) ions based on 2-hydroxy-1-naphthaldehyde and L-serine. Dyes Pigm. 2020, 175, 108190. [Google Scholar] [CrossRef]
- Gwynne, L.; Williams, G.T.; Yan, K.-C.; Patenall, B.L.; Gardiner, J.E.; He, X.-P.; Maillard, J.-Y.; James, T.D.; Sedgwick, A.C.; Jenkins, A.T.A. TCF-ALP: A fluorescent probe for the selective detection of Staphylococcus bacteria and application in “smart” wound dressings. Biomater. Sci. 2021, 9, 4433–4439. [Google Scholar] [CrossRef]
- Liu, H.S.; Ma, C.B.; Wang, J.; Wang, K.M.; Wu, K.F. A turn-on fluorescent method for determination of the activity of alkaline phosphatase based on dsDNA-templated copper nanoparticles and exonuclease based amplification. Microchim. Acta 2017, 184, 2483–2488. [Google Scholar] [CrossRef]
- Mei, Y.Q.; Hu, Q.; Zhou, B.J.; Zhang, Y.H.; He, M.H.; Xu, T.; Li, F.; Kong, J.M. Fluorescence quenching based alkaline phospha-tase activity detection. Talanta 2018, 176, 52–58. [Google Scholar] [CrossRef]
- Gao, C.C.; Zang, S.P.; Nie, L.X.; Tian, Y.; Zhang, R.B.; Jing, J.; Zhang, X.L. A sensitive ratiometric fluorescent probe for quanti-tive detection and imaging of alkaline phosphatase in living cells. Anal. Chim. Acta 2019, 1066, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Li, C.-Y.; Li, Y.-F.; Fei, J.; Wu, P.; Yang, B.; Ou-Yang, J.; Nie, S.-X. Facile and Sensitive Near-Infrared Fluorescence Probe for the Detection of Endogenous Alkaline Phosphatase Activity In Vivo. Anal. Chem. 2017, 89, 6854–6860. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yu, J.; Hu, X.; Huang, S.; Cai, L.; Yang, L.; Zhang, H.; Jiang, Y.; Jia, Y.; Sun, H. An activity-based fluorescent probe and its application for differentiating alkaline phosphatase activity in different cell lines. Chem. Commun. 2020, 56, 13323–13326. [Google Scholar] [CrossRef]
- Atay Çiğdem, K.; Tilki, T.; Dede, B. Design and synthesis of novel ribofuranose nucleoside analogues as antiproliferative agents: A molecular docking and DFT study. J. Mol. Liq. 2018, 269, 315–326. [Google Scholar] [CrossRef]
- Swaminathan, J.; Ramalingam, M.; Sethuraman, V.; Sundaraganesan, N.; Sebastian, S. Vibrational spectroscopic studies and DFT calculations of 4-aminoantipyrine. Spectrochim. Acta Part A 2009, 73, 593–600. [Google Scholar] [CrossRef]
- Casanovas, J.; Namba, A.M.; da Silva, R.; Aleman, C. DFT-GIAO study of aryltetralin lignan lactones: Conformational anal-yses and chemical shifts calculations. Bioorg. Chem. 2005, 33, 484–492. [Google Scholar] [CrossRef]
- Makhlouf, M.M.; Radwan, A.S.; Ghazal, B. Experimental and DFT insights into molecular structure and optical properties of new chalcones as promising photosensitizers towards solar cell applications. Appl. Surf. Sci. 2018, 452, 337–351. [Google Scholar] [CrossRef]
- Ramos, J.M.; Cruz, M.T.D.; Costa, A.C.; Ondar, G.F.; Ferreira, G.B.; Raniero, L.; Martin, A.A.; Versiane, O.; Soto, C.A.T. Molecular structure, natural bond analysis, vibrational, and electronic spectra of aspartateguanidoacetatenickel (II), Ni(Asp)(GAA) cen-ter dot H2O: DFT quantum mechanical calculations. Spectrochim. Acta Part A 2012, 97, 1041–1051. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, X.; Diao, L.; Cao, B.; Yin, H.; Shi, Y. How the atomic electron-accepting ability affect the double ESIPT process of 2,5-bis(benzoxazol-2-yl)thiophene-3,4-diol? J. Lumin. 2020, 225, 117329. [Google Scholar] [CrossRef]
- Zhan, H.; Tang, Z.; Li, Z.; Chen, X.; Tian, J.; Fei, X.; Wang, Y. The influence of intermolecular hydrogen bonds on single fluorescence mechanism of 1-hydroxy-11H-benzo [b]fluoren-11-one and 10-hydroxy-11H-benzo [b]fluoren-11-one. Spectrochim. Acta Part A 2021, 260, 119993. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Yang, Y.; Zhai, H.; Zhang, Q.; He, Y.; Liu, Y.; Liu, Y. The mechanisms of a bifunctional fluorescent probe for detecting fluoride and sulfite based on excited-state intramolecular proton transfer and intramolecular charge transfer. Struct. Dyn. 2021, 8, 034103. [Google Scholar] [CrossRef]
- Shang, C.; Cao, Y.; Sun, C.; Li, Y. Tuning fluorescence behavior and ESIPT reaction of 2-(2-Hydroxy-phenyl)-4(3H)-quinazolinone by introducing different groups. J. Lumin. 2021, 235, 118059. [Google Scholar] [CrossRef]
- Liu, S.; Qin, M.; Lu, Q.; Lin, L.; Wang, C.-K.; Fan, J.; Song, Y. Sensing mechanism of fluorescent sensor to Cu2+ based on inhibiting ultra-fast intramolecular proton transfer process. Spectrochim. Acta Part A 2021, 254, 119685. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Yang, D.; Sui, X.; Wang, D. Time-dependent density functional theory (TD-DFT) study on the excited-state intramo-lecular proton transfer (ESIPT) in 2-hydroxybenzoyl compounds: Significance of the intramolecular hydrogen bonding. Spectrochim. Acta Part A 2013, 102, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, D.; Ma, C.; Liu, Y. DFT-TDDFT investigation of excited-state intramolecular proton transfer in 2-(2′-hydroxyphenyl)benzimidazole derivatives: Effects of electron acceptor and donor groups. J. Mol. Liq. 2016, 224, 83–88. [Google Scholar] [CrossRef]
- Sun, C.; Su, X.; Zhou, Q.; Shi, Y. Regular tuning of the ESIPT reaction of 3-hydroxychromone-based derivatives by substitution of functional groups. Org. Chem. Front. 2019, 6, 3093–3100. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, Q. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chem. Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rozas, I. On the nature of hydrogen bonds: An overview on computational studies and a word about patterns. Phys. Chem. Chem. Phys. 2007, 9, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liang, X.; Fang, H. The effect of ring aromaticity on ESIPT behavior and photophysical properties of 2-(2′-hydroxyphenyl)- 4-chloromethylthiazole derivatives: A TD-DFT study. J. Mol. Liq. 2021, 334, 116517. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Liu, J.; Hoffmann, M.R. Competitive excited-state single or double proton transfer mechanisms for bis-2,5-(2-benzoxazolyl)-hydroquinone and its derivatives. Phys. Chem. Chem. Phys. 2015, 17, 11990–11999. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
The experimental detection mechanism for ALP probe AE-Phos (Gray meaning no fluorescence, Green meaning green fluorescence).
Scheme 1.
The experimental detection mechanism for ALP probe AE-Phos (Gray meaning no fluorescence, Green meaning green fluorescence).
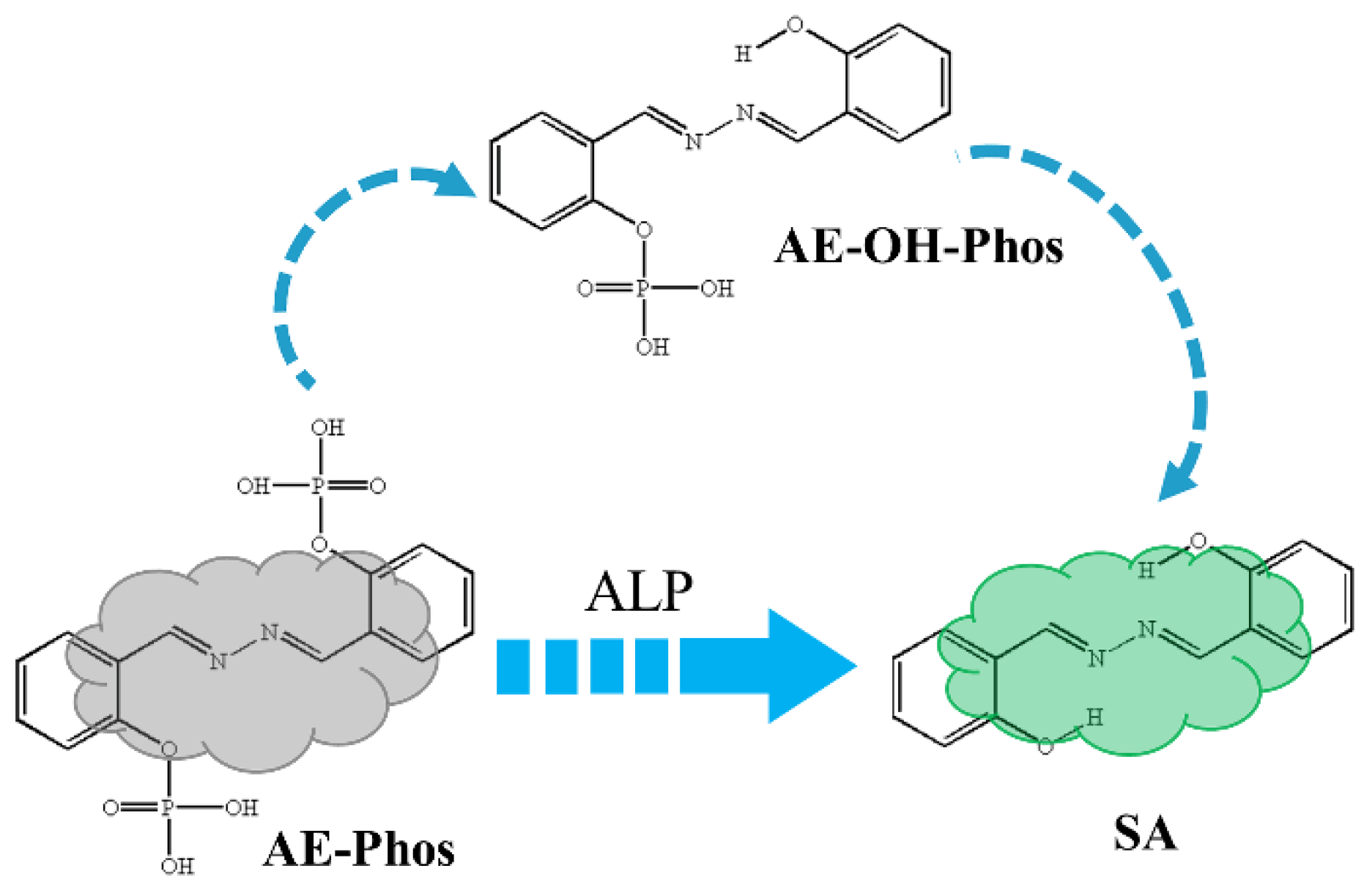
Figure 1.
Possible structures of probe product SA and intermediate product AE-OH-Phos (SPT: single proton transfer; DPT: double proton transfer).
Figure 1.
Possible structures of probe product SA and intermediate product AE-OH-Phos (SPT: single proton transfer; DPT: double proton transfer).

Figure 2.
(a,b) IR spectra of probe product SA and intermediate product AE-OH-Phos.
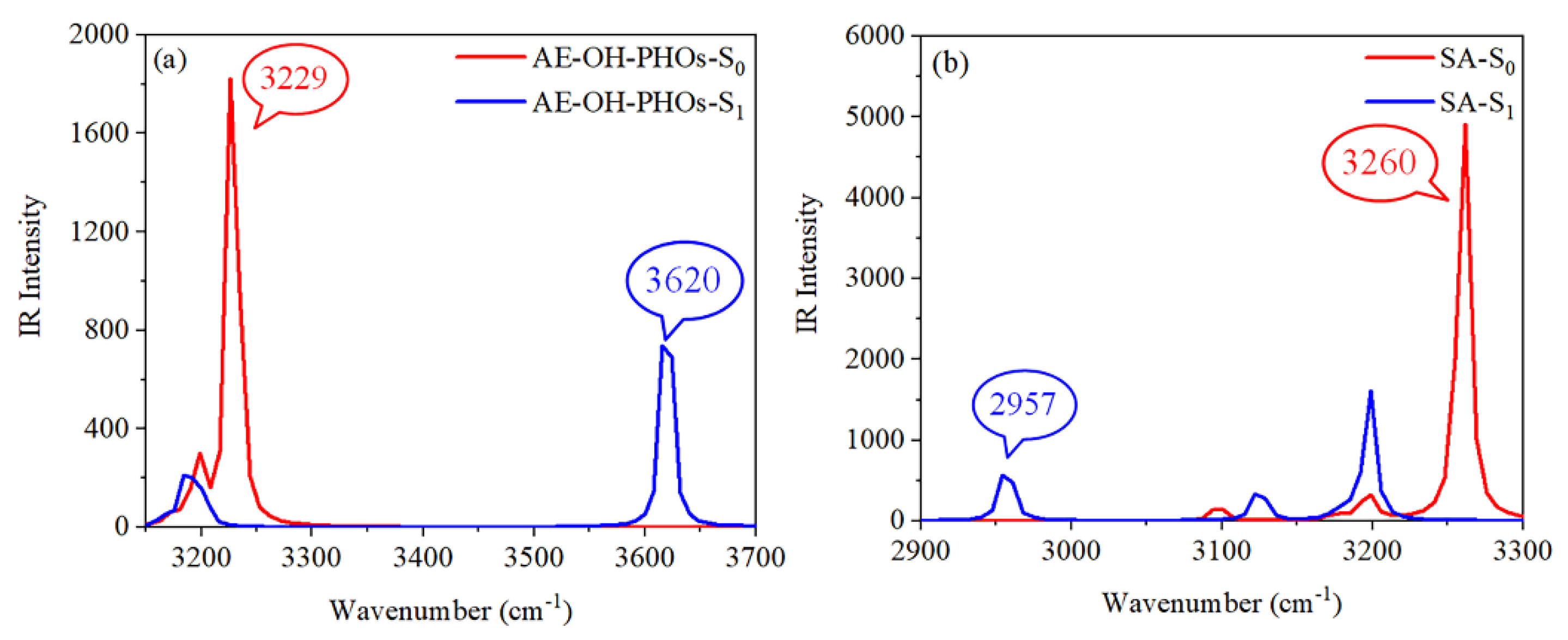
Figure 3.
RDG scatter plots and IRI isosurfaces of product SA and AE-OH-Phos.

Figure 4.
The transition state structure and energy of SA and AE-OH-Phos.

Figure 5.
The potential energy curves of AE-OH-Phos.

Figure 6.
The potential energy surfaces and contours of SA at (a) S0- and (b) S1- state.
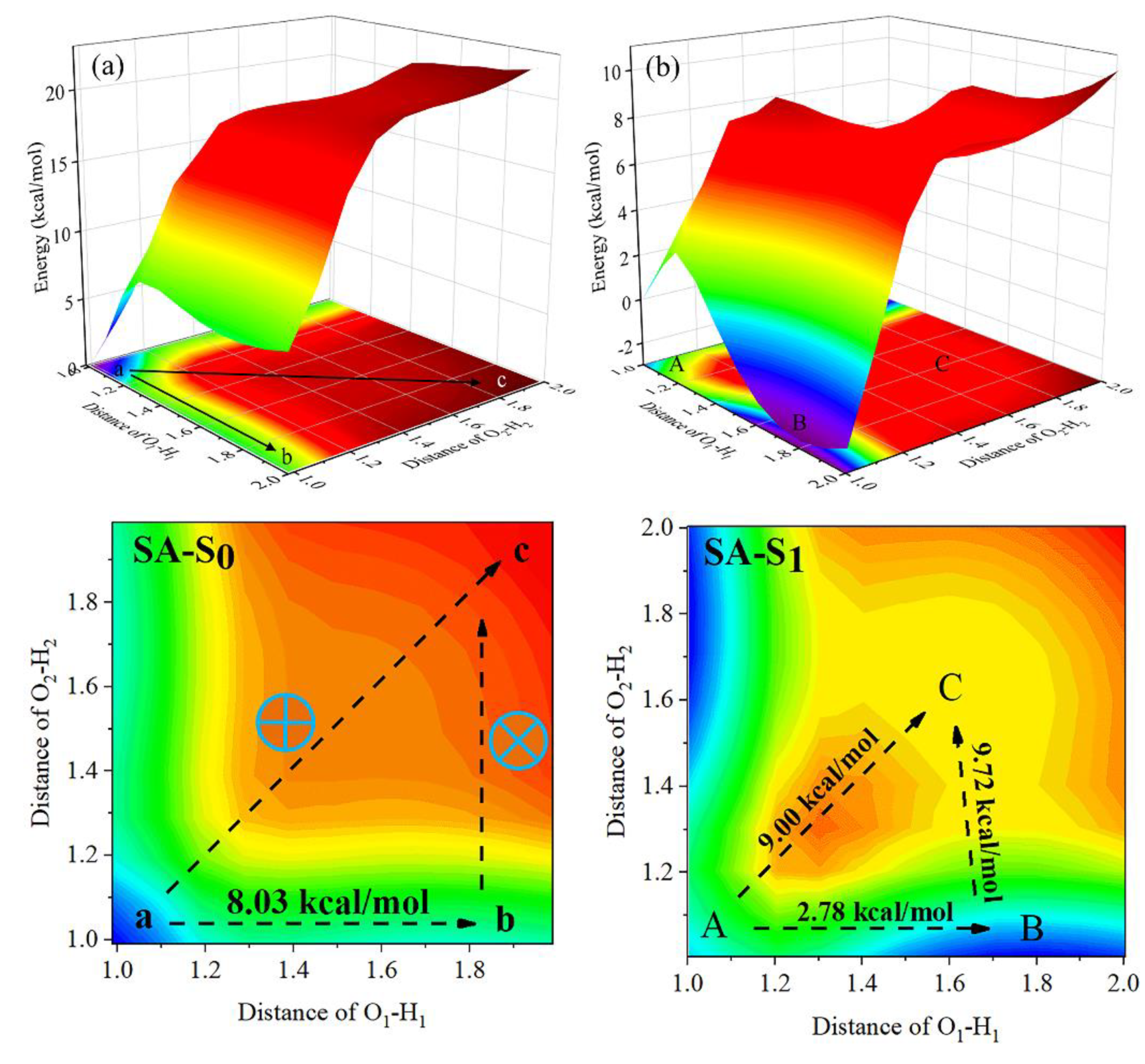
Figure 7.
The absorption and fluorescence spectra (a) probe AE-Phos, (b) intermediate product AE-OH-phos, (c) product SA (Experimental values are in parentheses).
Figure 7.
The absorption and fluorescence spectra (a) probe AE-Phos, (b) intermediate product AE-OH-phos, (c) product SA (Experimental values are in parentheses).
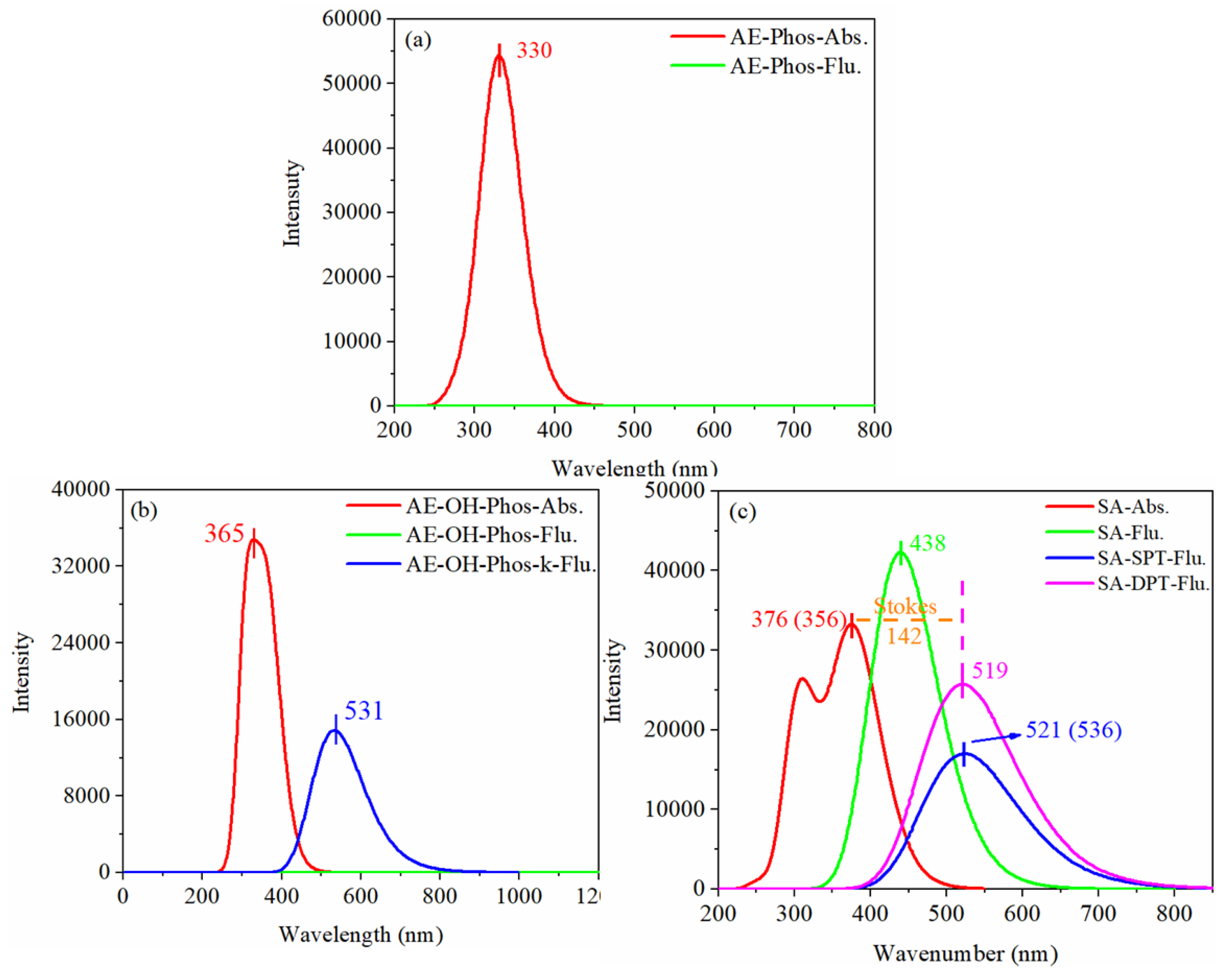
Figure 8.
Frontier molecular orbitals of ALP probe and its products.

Figure 9.
The electrons (blue) and holes (red) distribution for the studied molecules.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated relative bond lengths (Å) and bond angles (°) of the enol and keto forms for AE-OH-PHOs at the S0 and S1 states.
Table 1.
Calculated relative bond lengths (Å) and bond angles (°) of the enol and keto forms for AE-OH-PHOs at the S0 and S1 states.
| State | O2-H2 | H2-N2 | δ(O2-H2-N2) | |
|---|---|---|---|---|
| AE-OH-PHOs-e | S0 | 0.99 | 1.76 | 146.5 |
| S1 | 0.97 | 1.90 | 143.0 | |
| AE-OH-PHOs-k | S0 | 1.76 | 1.04 | 135.2 |
| S1 | 1.82 | 1.04 | 136.0 |
Table 2.
Calculated relative bond lengths (Å) and bond angles (°) of SA, SA-SPT, and SA-DPT at the S0 and S1 states.
Table 2.
Calculated relative bond lengths (Å) and bond angles (°) of SA, SA-SPT, and SA-DPT at the S0 and S1 states.
| SA | SA-SPT | SA-DPT | ||||
|---|---|---|---|---|---|---|
| State | S0 | S1 | S0 | S1 | S0 | S1 |
| O1-H1 | 0.99 | 1.00 | 0.98 | 0.99 | — | 1.64 |
| H1-N1 | 1.76 | 1.70 | 1.80 | 1.77 | — | 1.06 |
| O2-H2 | 0.99 | 1.00 | 1.73 | 1.79 | — | 1.64 |
| H2-N2 | 1.76 | 1.70 | 1.05 | 1.04 | — | 1.06 |
| δ(O1-H1-N1) | 146.1 | 148.3 | 144.7 | 146.4 | — | 140.9 |
| δ(O2-H2-N2) | 146.1 | 148.3 | 136.3 | 137.0 | — | 140.8 |
Table 3.
Performed functional correction by comparing the Stokes-shifts (nm) of the probe products SA.
Table 3.
Performed functional correction by comparing the Stokes-shifts (nm) of the probe products SA.
| B3PW91 | CAM-B3LYP | M06-2W | MPWPW91 | WB97XD | Exp. a | |
|---|---|---|---|---|---|---|
| SA-Abs. | 376 | 335 | 332 | 365 | 332 | 356 |
| SA-Flu. | 438 | 403 | 400 | 428 | 400 | 536 |
| SA-SPT-Flu. | 521 | 452 | 456 | 503 | 445 | |
| SA-DPT-Flu. | 519 | 461 | 468 | 504 | 456 | |
| Stokes-shifts | 145 | 117 | 124 | 138 | 113 | 180 |
a Represents the maximum absorption peak measured in the experiment.
Table 4.
The electron-excited properties of the probe and its products in DMSO solvent using the TD-DFT/B3PW91/6-311G(d,p) method.
Table 4.
The electron-excited properties of the probe and its products in DMSO solvent using the TD-DFT/B3PW91/6-311G(d,p) method.
| State | E (eV) | λabs (nm) | Contribution MO | f | |
|---|---|---|---|---|---|
| AE-PHOs | S1 | 3.1819 | 390 | (0.69920) H-1→L | 0.0010 |
| S2 | 3.7584 | 330 | (0.70193) H→L | 1.3290 | |
| S3 | 4.2759 | 290 | (0.67557) H-2→L | 0.0000 | |
| AE-OH-PHOs | S1 | 3.3963 | 365 | (0.69704) H→L | 0.6683 |
| S2 | 3.4735 | 357 | (0.70157) H-2→L | 0.0011 | |
| S3 | 3.9535 | 314 | (0.69131) H-1→L | 0.6960 | |
| SA | S1 | 3.2932 | 376 | (0.70025) H→L | 0.7985 |
| S2 | 3.6328 | 341 | (0.69900) H-1→L | 0.0000 | |
| S3 | 3.7808 | 328 | (0.70259) H-3→L | 0.0003 | |
| S4 | 4.0515 | 306 | (0.69207) H-2→L | 0.6351 |
Table 5.
The fluorescence properties of the probe and its products in DMSO solvent using the TD-DFT/B3PW91/6-311G(d,p) method.
Table 5.
The fluorescence properties of the probe and its products in DMSO solvent using the TD-DFT/B3PW91/6-311G(d,p) method.
| State | E (eV) | λflu (nm) | Contribution MO | f | |
|---|---|---|---|---|---|
| AE-PHOs | S1 | 0.6158 | 2013 | (0.70484) H→L | 0.0000 |
| AE-OH-PHOs | S1 | 1.1122 | 1115 | (0.70423) H→L | 0.0000 |
| AE-OH-PHOs-k | S1 | 2.3363 | 531 | (0.70719) H→L | 0.3710 |
| SA | S1 | 2.8331 | 438 | (0.70031) H→L | 1.0443 |
| SA-SPT | S1 | 2.3784 | 521 | (0.70706) H→L | 0.4243 |
| SA-DPT | S1 | 2.3882 | 519 | (0.70758) H→L | 0.6405 |
Table 6.
Indexes related to the distribution of electrons and holes for the probe and its products.
| D (Å) | t (Å) | H (Å) | Δσ (Å) | Sr (a.u.) | HDI | EDI | |
|---|---|---|---|---|---|---|---|
| AE -PHOs | 0.010 | −1.879 | 2.477 | 1.750 | 0.375 | 16.49 | 8.23 |
| AE-OH-PHOs | 0.303 | −1.537 | 2.504 | 1.696 | 0.381 | 16.48 | 8.20 |
| SA | 0.000 | −3.382 | 3.698 | −0.167 | 0.755 | 6.78 | 7.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cao, Y.; Yu, X.; Sun, C.; Cui, J. Theoretical Investigation on the ESIPT Process and Detection Mechanism for Dual-Proton Type Fluorescent Probe. Int. J. Mol. Sci. 2022, 23, 2132. https://doi.org/10.3390/ijms23042132
AMA Style
Cao Y, Yu X, Sun C, Cui J. Theoretical Investigation on the ESIPT Process and Detection Mechanism for Dual-Proton Type Fluorescent Probe. International Journal of Molecular Sciences. 2022; 23(4):2132. https://doi.org/10.3390/ijms23042132
Chicago/Turabian StyleCao, Yunjian, Xiangrui Yu, Chaofan Sun, and Jingang Cui. 2022. "Theoretical Investigation on the ESIPT Process and Detection Mechanism for Dual-Proton Type Fluorescent Probe" International Journal of Molecular Sciences 23, no. 4: 2132. https://doi.org/10.3390/ijms23042132
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.