
CD38–Cyclic ADP-Ribose Signal System in Physiology, Biochemistry, and Pathophysiology

Department of Biochemistry, Nara Medical University, 840 Shijo-cho, Kashihara 634-8521, Nara, Japan
Int. J. Mol. Sci. 2022, 23(8), 4306; https://doi.org/10.3390/ijms23084306
Submission received: 25 February 2022
/
Revised: 2 April 2022
/
Accepted: 12 April 2022
/
Published: 13 April 2022
(This article belongs to the Special Issue Signal Transduction 2.0: From Molecular Pathways to Translational Research)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Calcium (Ca2+) is a ubiquitous and fundamental signaling component that is utilized by cells to regulate a diverse range of cellular functions, such as insulin secretion from pancreatic β-cells of the islets of Langerhans. Cyclic ADP-ribose (cADPR), synthesized from NAD+ by ADP-ribosyl cyclase family proteins, such as the mammalian cluster of differentiation 38 (CD38), is important for intracellular Ca2+ mobilization for cell functioning. cADPR induces Ca2+ release from endoplasmic reticulum via the ryanodine receptor intracellular Ca2+ channel complex, in which the FK506-binding protein 12.6 works as a cADPR-binding regulatory protein. Recently, involvements of the CD38-cADPR signal system in several human diseases and animal models have been reported. This review describes the biochemical and molecular biological basis of the CD38-cADPR signal system and the diseases caused by its abnormalities.
1. Introduction
Changes in cytosolic Ca2+ are indispensable for normal cell function, such as secretion, proliferation, neuronal guidance, cell death, and development. Typically, cells respond to a Ca2+ signal that is generated inside the cell in response to the activation of a wide variety of extracellular signals, including those involved in nutrient, neurotransmitter, hormonal, and sensory signaling. In most cases, the initial Ca2+ signal generated in the cell is a specific increase in cytoplasmic Ca2+ concentrations resulting from the release of Ca2+ from internal Ca2+ stores (mainly the endoplasmic reticulum) or the entry of Ca2+ from the external spaces across the plasma membrane. Both routes involve the movement of Ca2+ through Ca2+ channels that are localized within these cellular membranes. While intracellular Ca2+ release from the endoplasmic reticulum occurs via channels activated by inositol 1,4,5-trisphosphate (IP3), cyclic ADP-ribose (cADPR), or Ca2+ itself (Ca2+-induced Ca2+ release), Ca2+ influx across the plasma membrane is achieved via numerous types of Ca2+ channels, including voltage-gated Ca2+ channels and store-operated Ca2+ channels as well as a variety of ligand-gated cation channels.
In this review, I focus specifically on the historical background, physiological significance, biochemical and molecular biological bases, and recent topics in the pathophysiology of the CD38-cADPR signal system in mammalian cells and human diseases.
2. cADPR as an Intracellular Ca2+-Releasing Messenger
Changes in cytosolic Ca2+ levels govern a multitude of cellular processes. Many extracellular stimuli mediate complex changes in Ca2+ through the production of second messengers [1]. These molecules effect the release of Ca2+ from the intracellular Ca2+ store. In addition to IP3, cADPR and nicotinic acid adenine dinucleotide phosphate (NAADP) play crucial roles in the generation of agonist-evoked Ca2+ signals [2,3]. IP3 is the best characterized of these molecules [1]. In addition, cADPR and NAADP, which were first discovered in sea urchin eggs [4], also play prominent roles in generating complex Ca2+ signals. cADPR induces Ca2+ release through the activation of the ryanodine receptors (RyRs) located on the endoplasmic reticulum [2,3].
Both cADPR and NAADP are synthesized by the same family of enzymes, the ADP-ribosyl cyclases (EC 3.2.2.6 and EC 2.4.99.20), which therefore play a central role in Ca2+ signaling and have been implicated in a variety of processes ranging from bacterial clearance to social behavior [5]. ADP-ribosyl cyclase (EC 3.2.2.6) activity was originally identified in sea urchin egg homogenates [4], but an enzyme with the activity was later purified [6,7] and cloned [8,9] from Aplysia ovotestes. The enzyme was shown to catalyze the cyclization of NAD+ to form cADPR and the replacement of the nicotinamide moiety in NADP with nicotinic acid to form NAADP. Based on amino acid sequence similarity, it became evident that the mammalian protein cluster of differentiation 38 (CD38; ADP-ribosyl cyclase 1; EC 3.2.2.6) [10,11,12,13] and cluster of differentiation 157 (CD157; ADP-ribosyl cyclase 2; EC 3.2.2.6) [14,15] were also ADP-ribosyl cyclase in mammalian cells.
3. cADPR in Insulin Secretion from Pancreatic β-Cells
Glucose induces an increase in intracellular Ca2+ concentration in pancreatic islet β-cells and induces the secretion of insulin. Ashcroft et al. first explained the importance of the increase in the Ca2+ concentration in 1984 [16]. The millimolar concentration of ATP is produced by the process of glucose metabolism in pancreatic β-cells. As a result, there is the closure of ATP-sensitive potassium (KATP) channels, which results in membrane depolarization, the influx of Ca2+ through voltage-dependent Ca2+ channels, and an increase in the cytosolic Ca2+ concentration that triggers the exocytosis of insulin granules. In 1993, another model of insulin secretion by glucose via cADPR-mediated Ca2+ mobilization from an intracellular Ca2+ pool, the endoplasmic reticulum, was proposed [12,17], as shown in Figure 1. According to this model, ATP inhibits the cADPR hydrolase of CD38, causing the accumulation of cADPR, which acts as a second messenger for Ca2+ mobilization from the endoplasmic reticulum for insulin secretion [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34].
The first important issue is whether the accumulation of cADPR is actually caused by glucose stimulation in pancreatic islets. In our laboratory, we assayed the cADPR content in islets isolated from normal rats (Wistar) and mice (C56BL/6J) that were incubated with low (2.8 mM) and high (20 mM) glucose by radioimmunoassay using an anti-cADPR antibody. The cADPR content in the high glucose-treated islets was rapidly increased within 5 min. In contrast, the cADPR content in the low glucose-treated islets was not [28].
Using the rat islet microsome Ca2+ releasing system, fluo 3 (2,2′-{[2-(2-{2-[Bis(caboxymethyl)amino]-5-}2,7-dichloro-6-hydroxy-3-oxo-3H-xanthen-9-yl)phenoxy]ethoxy}-4-methylphenyl]azanediyl}diacetic acid) fluorescence showed cADPR released Ca2+ [17,24,28]. IP3 did not cause the release of Ca2+, and after the addition of IP3, the islet microsomes were still responsive to cADPR. In contrast, IP3 caused a release of Ca2+ from cerebellum microsomes, and cADPR also caused a release of Ca2+. Heparin, an inhibitor of IP3 binding to its receptor, blocked the IP3-induced Ca2+ release from cerebellum microsomes but did not block the cADPR-induced Ca2+ release. These results indicate that islet microsomes respond to cADPR but not to IP3. In contrast, cerebellum microsomes respond to both cADPR and IP3, but cADPR induces the Ca2+ release via a different mechanism than that utilized by IP3.
The effect of cADPR on insulin secretion was examined using digitonin-permeabilized rat pancreatic islets. cADPR as well as Ca2+ induced insulin secretion, but IP3 did not. The combined addition of cADPR and Ca2+ did not induce significantly more insulin secretion than the addition of cADPR or Ca2+ alone. The cADPR-induced insulin secretion was inhibited by the addition of EGTA. These results indicated cADPR induced Ca2+ release from islet microsomes and the increment of Ca2+ resulted in insulin secretion from pancreatic islets [17].
From these results, glucose-induced insulin secretion via cADPR formation from NAD+ and cADPR-induced Ca2+ mobilization from the endoplasmic reticulum was proposed. Concerning the second messenger role of cADPR for glucose-induced insulin secretion, some researchers reported that cADPR-induced intracellular Ca2+ release was not observed in ob/ob mouse islets and rat insulinoma-derived RINm5F cells [19,35,36,37,38]. Malaisse et al. [39,40] measured the cADPR content in rat islets and reported that it appeared not to be significantly affected by glucose concentrations. On the other hand, the fasting of the rats before the isolation of islets and the usage of Hanks’ solution containing 2.8 mM glucose during the islet isolation may account for the rapid and significant increase in cADPR content in the islets in response to glucose stimulation. Furthermore we determined the cADPR content by assessing the recovery of cADPR in the extraction and concentration procedures, and they did not. Differences in the experimental conditions may be responsible for the different results [19,28]. Nevertheless, the reports that Balb/c mouse islets showed increases in glucose-induced cADPR production, the intracellular concentration of Ca2+, and insulin secretion [41] and that human insulin promoter-SV40 large T transgene-introduced C57BL/6 mouse pancreatic β-cell-derived MIN6 cells, which show glucose-induced insulin secretion, showed a dramatic Ca2+ mobilization in response to cADPR via the ryanodine receptor (RyR) [42,43] despite the lack of response to IP3 [43]. These results indicate that the CD38-cADPR signal system is functioning in glucose-induced insulin secretion in pancreatic β-cells.
4. CD38 as a Major Enzyme for the Synthesis of cADPR
CD38 is a 300-amino-acid protein and was first recognized as a human leukocyte surface antigen. CD38 was found to express in a variety of tissues and cells, including pancreatic β-cells [12,20,44]. We and others have found that CD38 has both ADP-ribosyl cyclase for cADPR synthesis from NAD+ and cADPR hydrolase for the hydrolysis of cADPR to form ADP-ribose [11,12,13,21,45]. Using purified human CD38 protein, it was found that millimolar concentrations of ATP inhibit the cADPR hydrolase activity of CD38, competing with the substrate, cADPR [12,26]. The competitive inhibition of the cADPR hydrolysis by ATP suggests that ATP and cADPR bind to the same site of CD38. We purified human recombinant CD38, incubated with an ATP analogue, 5′-p-fluorosulfonylbenzoyladenosine, and identified the binding site for ATP and/or cADPR as the lysine-129 of CD38 [26]. From these results and other available evidence, we proposed that CD38 catalyzes the formation of cADPR from NAD+ and also the hydrolysis of the cADPR to ADP-ribose. As shown in Figure 2, lysine-129 of CD38 is the cADPR binding site for hydrolysis to ADP-ribose and ATP competes with cADPR in the site, resulting in the inhibition of the hydrolysis of cADPR and then in the accumulation of cADPR by ATP [12,21,34].
The human CD38 gene was assigned to band p15 of chromosome four as a single copy gene by in situ fluorescence hybridization [22]. We then isolated the human CD38 gene and determined its primary structure [27]. The human CD38 gene as well as the Xenopus Cd38 gene extending ~70 kb [27,44] contained eight exons (Figure 3). The human and Xenopus Cd38 promoter regions have no TATA box but do have a CpG island, a methylation-controlled region more frequently associated with housekeeping than tissue-specific genes. Exon one encoded the 5′-untranslated region, translational start site, putative transmembrane domain and N-terminal region of Cd38. Exons two to eight encoded the remainder of Cd38. Cycsteines-119 and -201 of human CD38, which are indispensable for the hydrolysis of cADPR to form non-Ca2+ mobilizing ADPR [21], were encoded in exons two and five, respectively. Lysine-129, which is involved in cADPR binding to CD38 [26], was encoded in exon three. Glutamate-226 and Trp-185, which seem to play important roles in catalytic activities [46,47], were encoded in exons six and four, respectively. The 10 cysteine residues conserved among human, rat, mouse, chicken, and frog Cd38s; human, rat, mouse, chicken, and frog Cd157s; Aplysia ADP-ribosyl cyclases, purple sea urchin ADP-ribosyl cyclase, and blood fluke ADP-ribosyl cyclase were encoded into six exons [27,32,44]. The translational termination codon and 3′-untranslated region were located in exon eight.
Isolation and determination of the primary structure of the Aplysia kurodai ADP-ribosyl cyclase gene [9] and the frog (Xenopus laevis) Cd38 gene [44] as well as the human CD38 gene [27] were performed. The exon–intron organization of the Aplysia ADP-ribosyl cyclase gene and the frog Cd38 gene is very similar to that of the human CD38 gene, suggesting that they evolved from a common ancestral gene [27,44] (Figure 3).
Kaisho et al. [49] found that the amino acid sequence of bone marrow stromal antigen 1 (BST-1/CD157) had significant sequence homology (~30% identity) with those of CD38 and Aplysia ADP-ribosyl cyclase. They determined the gene structure of human CD157 [50]. The gene extended for ~30 kb, very close to its paralogue CD38, and consisted of nine exons. Comparison of the gene organization reveals that the human CD157 gene has an exon–intron organization similar to that of human CD38 and Aplysia ADP-ribosyl cyclase genes (Figure 3). However, the human CD157 gene has an additional exon, exon nine, that encodes a peptide that is removed upon attachment of the glycosyl phosphatidylinositol anchor. In addition to the similar exon–intron structures of CD38, CD157 and Aplysia ADP-ribosyl cyclase genes and human CD38 and CD157 genes are located on the subregion of the human chromosome 4p15 as a next neighbor; the fact that mouse Cd157 (known as BP-3) is very close to the map site of the Cd38 gene on mouse chromosome 5 [51] strongly suggests that the three genes (CD38, CD157, and Aplysia ADP-ribosyl cyclase) have evolved from a common ancestor, and CD38 and CD157 genes were created by gene duplication before human and rodent divergence (Figure 4).
The expression of Cd157 was reported in rat and mouse pancreatic β-cells [52,53]. However, no diabetic features were observed in Cd157 knockout mice [54]. Furthermore, CD157 exhibits neither cADPR-synthesizing (ADP-ribosyl cyclase) nor -hydrolyzing (cADPR hydrolase) activity in physiological conditions [14], suggesting that CD157 may mainly play a role as a surface antigen rather than a major enzyme for the synthesis of cADPR in response to glucose stimulation in pancreatic β-cells.
5. Physiological Significance of the CD38-cADPR Signal System in Mammalian Cells
In order to confirm the proposed mechanism of insulin secretion, in which the CD38-cADPR signal system plays a major role, CD38 transgenic, CD38 knockout, and FKBP12.6 knockout mice were produced [23,31,61]. In 19995, transgenic mice overexpressing human CD38 in pancreatic β-cells using rat insulin II promoter were produced [23]. Using immunohistochemistry, we demonstrated that the pancreatic islets of the two independent transgenic lines were densely stained for human CD38 [23]. The insulin secretion from the transgenic islets was significantly higher than that from the control islets at high concentrations of glucose. When islets were exposed to α-ketoisocaproate (4-methyl-2-ozopentanic acid), which, like glucose, is metabolized to form ATP, the insulin secretion from the transgenic islets was significantly higher than the control. However, with tolbutamide and KCl, the transgenic insulin secretion was not altered as compared to the control. Tolbutamide blocks the ATP-sensitive K+ channels, and KCl directly induces cell membrane depolarization resulting in Ca2+ influx from extracellular space [62]. In glucose-tolerance tests using living animals, the serum insulin level of CD38-transgenic mice in response to glucose charange was higher than that of control mice.
Furthermore, CD38 gene-disrupted mice were created [31]. CD38 knockout mice developed normally but showed no increase in their glucose-induced production of cADPR in pancreatic islets. Both the glucose-induced rise in intracellular Ca2+ concentration and insulin secretion were severely impaired in CD38 knockout islets, whereas CD38 knockout islets seemed normal compared to the typical extracellular Ca2+ influx stimulants such as tolbutamide and KCl. CD38 knockout mice showed impaired glucose tolerance, and the serum insulin level in response to glucose stimulation was significantly lower than the control. In an insulin tolerance test, the blood glucose levels of CD38 knockout mice 15–60 min after insulin injection were essentially similar to those of wild-type mice, suggesting that the glucose intolerance found in CD38 knockout mice is the result of an impaired glucose-induced insulin secretion rather than an increase in peripheral insulin resistance. Further experiments were performed regarding whether the observed phenotype (reduction in glucose-induced insulin secretion) could be rescued by the pancreatic β-cell-specific expression of human CD38 cDNA. Transgenic mice carrying a human CD38 cDNA under the control of the rat insulin II promoter described above were crossed with CD38 knockout mice. CD38 knockout mice carrying the human CD38 transgene were generated by intercrossing between CD38 knockout and CD38 transgenic mice. The resultant CD38 knockout mice having human CD38 transgene ameliorated the glucose intolerance and the decreased insulin secretion observed in CD38 knockout mice, suggesting that the observed phenotype in CD38 knockout mice is indeed caused by the disruption of the CD38 gene in pancreatic β-cells [31]. Thus, these results using CD38-transgenic and CD38 gene-disrupted mice provide further evidence for the contribution of the CD38-cADPR signal system to the glucose-induced Ca2+ release from intracellular Ca2+ stores for insulin secretion.
It is currently accepted that cADPR activates a ryanodine receptor (RyR) Ca2+ channel to release Ca2+ from the endoplasmic reticulum [63]. Type 2 RyR (RyR2) expression in rat pancreatic islets was confirmed [19,64]. Experiments using rat islet microsomes revealed that cADPR did not bind directly to the RyR channel but acted on the RyR channel through a mediator, such as the FK506 (tacrolimus)-binding protein 12.6 (FKBP12.6), to release Ca2+. The cellular target for FK506, one of the most widely used immunosuppressive agents in clinical situations, are FKBP12 (peptidyl-prolyl cis-trans isomerase FKBP1A, immunophilin FKBP12) and FKBP12.6 (peptidyl-prolyl cis-trans isomerase 1B, immunophilin FKBP12.6). Rat FKBP12 is 108-amino-acid protein and is highly conserved among human, mouse, bovine, and rabbit FKBP12. Rat FKBP12.6 is also a 108-amino-acid protein, and the amino acid sequence is completely conserved in human and bovine FKBP12.6. Noguchi et al. [25] isolated microsomes from rat islets and found that rat islet microsomes contained FKBP12.6 but did not contain FKBP12 by Western blot analysis. A binding experiment and its Scatchard plot analysis revealed that cADPR binds to FKBP12.6 at a Kd value of 35 nM. The cADPR-binding to FKBP12.6 was inhibited by FK506, but neither structurally nor functionally related analogues of cADPR (NAD+, ATP, ADPR, nicotinamide, IP3, and ryanodine) attenuated cADPR binding to FKBP12.6. These results indicate that FKBP12.6 functions as a cADPR-binding protein and strongly suggest that cADPR is a physiological ligand for FKBP12.6, since FK506 does not naturally exist in mammalian cells except for the use of FK506 as an immune-suppressant/immune-modulator. Noguchi et al. prepared rat microsomes and treated them with cADPR and reported that FKBP12.6, occurring in rat microsomes, was dissociated from the microsomes, releasing Ca2+ from them. When the microsomes had been treated with FK506 or cADPR, FKBP12.6 dissociated from the microsomes and the FKBP12.6-dissociated microsomes did not show Ca2+ release in response to cADPR. FKBP12.6, as a component of RyR-FKBP12.6 complex Ca2+ channel, binds to cADPR and dissociates from the complex and the RyR channel activity is increased to release Ca2+. Therefore, cADPR cannot act on the RyR complex to release Ca2+ in the presence of FKBP12.6, and the glucose-induced insulin secretion does not function. In fact, Noguchi et al. made FKBP12.6-deficient mice that showed glucose intolerance coupled to insufficient insulin secretion upon glucose challenge. Insulin secretion in response to glucose was markedly impaired in FKBP12.6-deficient mouse islets, while sulfonylurea- or KCl-induced insulin secretion was unaffected, indicating that FKBP12.6 plays a role in glucose-induced insulin secretion downstream of ATP production, independently of ATP-sensitive K+ channels, in pancreatic β-cells [61]. In the clinic, when FK506 (tacrolimus) was used as an immunosuppressant agent in kidney transplantation, hyperglycemia was observed in 20–35% of the transplantation recipients [65]. This diabetogenic side effect of FK506 in transplantation may be explained by the mechanism shown in Figure 1. It should also be noted that the islet microsome did not contain calmodulin (CaM), islet microsomes became sensitized to cADPR at much lower concentrations for Ca2+ release in the presence of CaM, and the Ca2+ release was greatly increased [24]. Inhibitors for CaM and CaM kinase II (W7, KN62, and KN-93) completely abolished the glucose-induced insulin secretion as well as the cADPR-mediated and CaM-activated Ca2+ mobilization, but non-inhibitory analogues (W5, KN-04, and KN-92). Furthermore, autocamtide-2-related inhibitory peptide (called as AIP), which is more specific CaM kinase II peptide inhibitor, inhibited the activation of cADPR-mediated Ca2+ release from islet microsomes. Immunoblot analyses revealed that rat microsomes contained CaM kinase IIα but did not contain CaM. When the active 30 kDa chymotryptic fragment of CaM kinase II, which lacks the autoinhibitory domain of CaM kinase II and is therefore activated in the absence of CaM, was added to the microsomes, fully activated cADPR-mediated Ca2+ release was observed in the absence of CaM [24]. These results suggest that CaM kinase II is required to phosphorylate and activate the RyR for the cADPR-mediated Ca2+ release. Cyclic AMP (cAMP)-dependent protein kinase (A-kinase) activated the cADPR-mediated Ca2+ release from islet microsomes [33]. In the absence of A-kinase, only a small amount of Ca2+ was released from the microsomes by low concentrations of cADPR. Alternatively, when the catalytic subunit of A-kinase was added to the islet microsome Ca2+ release system, the Ca2+ release from the microsomes was sensitized at much lower concentrations of cADPR [33]. As incretin peptide hormones, such as glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP, also known as glucose-dependent insulinotropic polypeptide), increase the intracellular cAMP concentrations to activate A-kinase and exchange protein activated by cAMP2 (EPAC2)/cAMP-guanine nucleotide exchange factor II [66], the cADPR-mediated Ca2+ mobilization for insulin secretion could be activated via the phosphorylation of RyR by CaM kinase II and/or A-kinase/EPAC2. Possibly, the activated kinases phosphorylate the RyR channel to sensitize the cADPR signal (Figure 5). Dr. Kim and his co-workers showed the role of cADPR in the insulin secretion by GLP-1 [67].
The CD38-cADPR signal system consists of two parts, the cADPR synthesis and metabolism, in which CD38 is a main player, and the Ca2+ release in response to cADPR from the RyR intracellular Ca2+ channel. There are three types of RyR in mammalian cells (type one, formerly called “skeletal type”), RyR2 (type two, formerly called “cardiac type”), and RyR3 (type three, formerly called “brain type”). The three types of RyR (RyR1, RyR2, and RyR3) are encoded by three unique genes and located in distinct chromosomal regions (19q13.2, 1q43, and 15q13.3-q14 in the human chromosome, respectively). RyR1 and RyR2 are predominantly expressed in skeletal muscles and cardiac muscles, respectively, and the names “skeletal type” and “cardiac type” were derived from the expressed tissues. In contrast, RyR3 was first isolated from brain and is now understood to express ubiquitously, including in the brain. Despite extensive physiological/pharmacological studies, the type of RyR that is the target for cADPR remains elusive. RyR2 appears to be expressed in pancreatic islets [19,28,63]. In order to determine the target RyR in glucose-induced insulin secretion from pancreatic β-cells, we screened the rat islet cDNA library and isolated a RyR cDNA clone using all the types of RyR cDNA as probe(s) and found it a novel RyR (islet-type RyR) that is generated from the RyR2 gene by the alternative splicing of exons 4 and 75 [64]. The deduced protein of the rat RyR2 cDNA isolated from islet cDNA library was 4947 amino acids with a calculated molecular weight of 562,291 daltons. These two regions, corresponding to the seven amino acids in exon 4 and the twelve amino acids in exon 75 of the authentic cardiac RyR2 gene, were not found in the RyR2 cDNA isolated from the rat islet cDNA library. Interestingly, neither exon 4 nor exon 75 were found in human and mouse RyR2 mRNA expressed in islets [19,64]. Further analyses of the alternative splicing patterns of RyR2 mRNA expressed in several rat and mouse tissues revealed that the islet-type RyR2 (lacking exons 4 and 75) was expressed not only in islets but also in other tissues such as cerebrum, cerebellum, pituitary gland, and adrenal gland. The cardiac-type (authentic) RyR2 was expressed in the atrium, ventriculum, and kidney. In many other tissues, such as those of the jejunum, ileum, colon, and liver, both types of RyR2 mRNA are expressed. In the human RYR2 gene, the tissue-specific alternative splicing pattern was essentially similar to that of the rat and mouse gene (see Figure 6), and the islet-type RYR2 mRNA (lacking exons 4 and 75) was also expressed in the human brain as well as in islets [19,64]. Interestingly, the splice donor consensus “gt” was substituted by “gg” in the intron 75 (1.0 kbp and 0.8 kbp introns in rat and human RyR2 genes, respectively), whereas all the other splice donor and acceptor sequences conformed to the consensus “gt/ag rule” for splicing [19,64].
By RT-PCR analyses of human and mouse RNAs, the expression of (alternatively spliced) pancreatic islet-type RyR2 mRNA was found not only in rat but also in human and mouse pancreatic islet RNAs. Moreover, the human and mouse islet-type RyR mRNAs expressed in islets and neuro-endocrine cells were also generated from the RyR2 gene by alternative splicing. The splice donor consensus “gt” was also substituted by “gg”. As shown in Figure 6, intron 75 of the RyR2 gene was spliced using “gg” as a donor for splicing instead of canonical “gt” in generating rat, mouse, and human authentic RyR2 mRNAs. The RyR2 gene for the CD38-cADPR signal system produced two different messenger RNAs by alternative splicing; one is for pancreatic islet β-cells and neuro-endocrine cells using canonical “gt/ag” splicing, and the other is for heart and blood vessels using not only the canonical “gt/ag” intron splicing site but also using the novel “gg/ag” site. Therefore, the heart/blood type RyR2 can contain exon 4 and exon 75 (Figure 6).
The islet-type RyR showed a greater increase in the Ca2+ release by caffeine when the RyR2-expression vectors (islet-type RyR2 and cardiac-type RyR2) were introduced and expressed in HEK293 cells pretreated with cADPR, suggesting that the novel islet-type RyR was an intracellular target for the CD38-cADPR signal system in mammalian cells, playing many important physiological roles in the functioning of the cADPR-sensitive Ca2+ release [64,68]. Most recently, it was reported that the replacement of “gg” with “gt” in intron 75 of the human insulin gene by gene-editing technology resulted not only in the reduction in glucose-induced insulin secretion but also glucose-induced proinsulin biosynthesis in 1.1B4 human insulin-producing cells [69], indicating that islets-type RyR2 was essential for insulin biosynthesis via Ca2+ homeostasis. The use of “gg” as a splice donor has not been reported in disease-free conditions [70,71]. The utilization of the “gg” as a splice donor was only reported in the cholesteryl ester transfer protein gene in a patient with cholesteryl ester protein deficiency [72].
6. Pathological Significance of the CD38-cADPR Signal System
6.1. Pathological Significance of the CD38-cADPR System in Diabetes
Yagui et al. analyzed the CD38 gene in 31 Japanese type 2 diabetes patients who had first and/or second-degree relative(s) with type 2 diabetes by single-stranded conformation polymorphism [29]. Two variant patterns were noted in exons three and four of the CD38 gene. Sequence analysis showed that one of the variants in exon four was a silent mutation in the codon for Ile-168 of CD38 from ATA to ATC. The second change in exon three resulted in amino acid substitution, in which Arg-140 (CGG) was replaced by Trp (TGG). Yagui et al. further studied the two variants in 90 non-diabetic controls who had no family history of diabetes and showed normal glucose tolerance. The frequency of the Ilw-168 (ATC) allele was 16% in type 2 diabetes patients as well as in controls. However, the Trp-140 (TGG) allele was observed in 4 of 31 type 2 diabetes patients but not at all in any controls. When the Arg140Trp mutant protein was expressed in COS-7 cells, both ADP-ribosyl cyclase and cADPR hydrolase activities were reduced to 40–50% of the normal CD38 enzymic activity. The decreased function of the CD38 mutant may have contributed to the impairment of glucose-stimulated insulin secretion in type 2 diabetes patients.
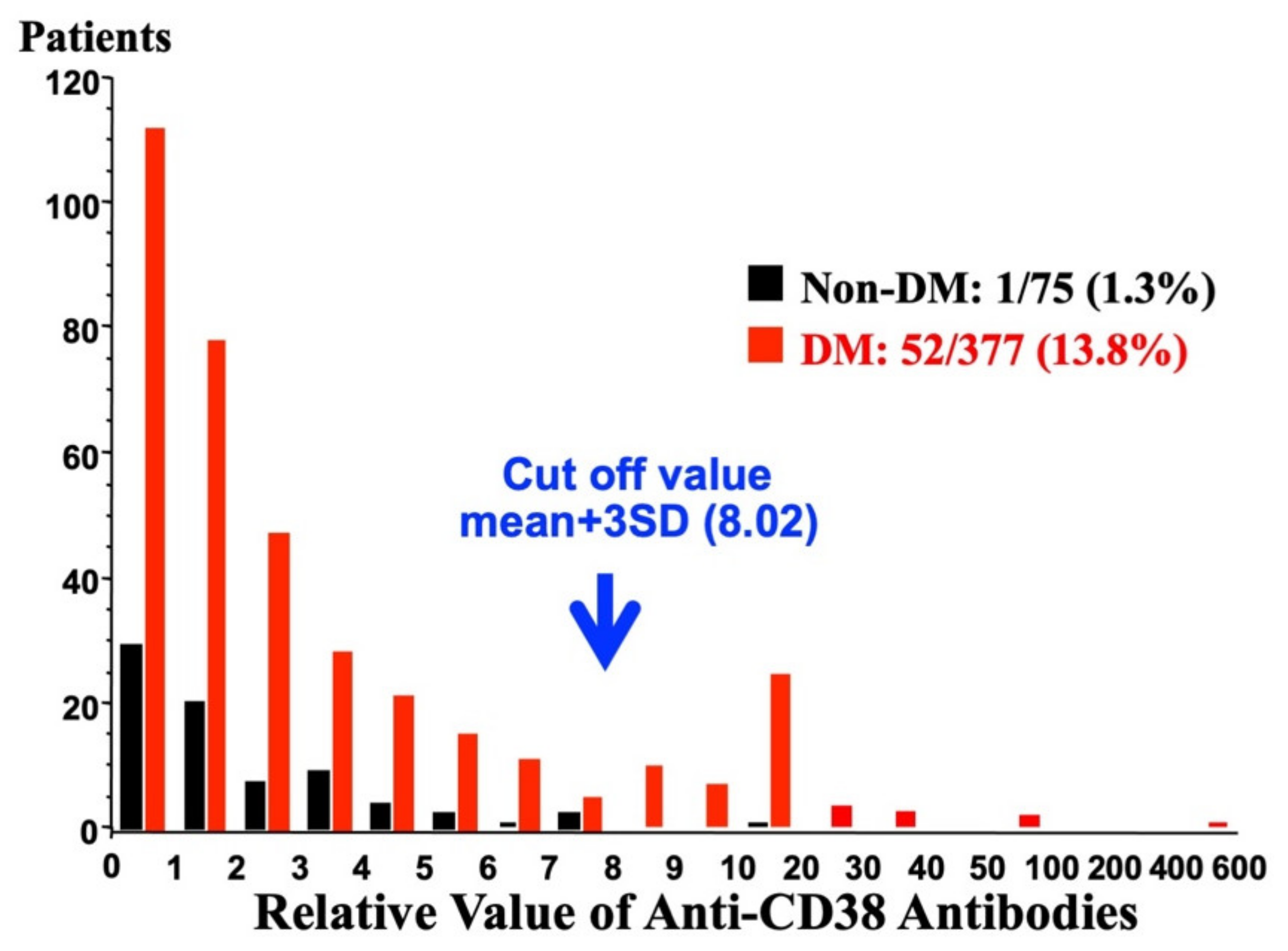
A monoclonal antibody against CD38 (T16) was reported to inhibit the enzymic activity of CD38 [12]. The antibody significantly inhibited the glucose-induced insulin secretion [30]. Therefore, when anti-CD38 antibodies are present in patients, glucose-induced insulin secretion could be impaired and result in type 2 diabetes. Ikehata et al. screened the existence of anti-CD38 antibodies in Japanese diabetes patients by Western blotting and found some type 2 diabetic sera showed a positive reaction with recombinant human CD38 [30]. When the diabetic serum was incubated with recombinant CD38 to absorb anti-CD38 autoantibodies, the signals in Western blot analysis were abolished, indicating that they contained anti-CD38 autoantibodies. The screening of sera for autoantibodies against CD38 was performed in 377 Japanese type 2 diabetes patients and 75 non-diabetic controls who had no family history of diabetes and showed normal fasting plasma glucose levels. In non-diabetic subjects, the relative antibody values were within the mean + 3 standard deviation (SD) of non-diabetic subjects except in one subject. In contrast, the distribution of relative anti-CD38 values showed two clear peaks in type 2 patients: one was below the mean + 3 SD of non-diabetic controls and the other was over the mean + 3 SD (Figure 7). Thus, Ikehata et al., set the cut of value as mean + 3 SD (8.02) of non-diabetic controls and revealed that 52 of 377 (13.8%) patients with type 2 diabetes had anti-CD38 autoantibodies [30].
Although the general characteristics such as gender, age, duration of diabetes, body mas index, fasting plasma glucose, HbA1c, and treatment of diabetes (diet, oral agents, or insulin) did not show significant differences between the anti-CD38 autoantibody positive and negative type 2 diabetes patients, there was an inverse correlation between the age of onset of diabetes and the relative value of the anti-CD38 autoantibody (p = 0.0002, r = −0.513) [30]. Of the 52 anti-CD38 autoantibody positive type 2 diabetic sera, seven diabetic sera showed a positive reaction for recombinant rat CD38. The examination of the effects of the diabetic sera that showed a positive reaction to rat CD38 on glucose-induced insulin secretion from isolated rat islets was carried out. All the seven diabetic sera with a positive reaction to recombinant rat CD38 significantly and dose-dependently inhibited glucose-induced insulin secretion from isolated rat islets, whereas the control sera did not show such an inhibitory effect on the glucose-induced insulin secretion [30]. The addition of recombinant rat CD38 nullified the inhibitory effect of the diabetic sera. Furthermore, the intracellular cADPR levels in rat islets incubated with the diabetic sera were significantly reduced, and the addition of recombinant CD38 in the medium attenuated the reduction [30], suggesting that the autoantibodies against CD38 inhibited the cADPR-synthesizing enzyme activity of CD38 and that the inhibition of glucose-induced insulin secretion is due to a change in the cADPR concentrations regulated by CD38. When CD38 enzymic activities were measured with the addition of diabetic sera in the enzyme assay mixture, the ADP-ribosyl cyclase activity of CD38 was significantly inhibited (p < 0.05 vs. the addition of non-diabetic sera), and the cADPR hydrolase activity of CD38 was instead activated (p < 0.05 vs. the addition of non-diabetic sera). These results well explain why the diabetic sera inhibited the glucose-induced insulin secretion from pancreatic β-cells, and the presence of anti-CD38 autoantibodies in diabetic patients is a major cause of impaired glucose-induced insulin secretion, which is frequently found in type 2 diabetes.
Anti-CD38 autoantibodies were later found in Caucasian diabetic patients, and some of their sera showed increased intracellular Ca2+ concentrations and glucose-independent insulin secretion from human islets [73,74,75]. In CD38 autoantibodies found in diabetes patients, some autoantibodies show agonistic effects on insulin secretion and others show antagonistic effects. Among the CD38 autoantibodies found in diabetic patients, differences in the effects between Japanese and Caucasian autoantibodies may be explained by the nature of the antibodies and the difference in islet species (rat and human) in the analyses of glucose-induced insulin secretion.
6.2. Pathological Significance of the CD38-cADPR System Other Than Diabetes
Although IP3 has been thought to be a second messenger for Ca2+ mobilization from intracellular stores, cADPR induces Ca2+ release from pancreatic islet microsomes but IP3 does not, as described above. In the microsomes of the cerebellum, Ca2+ release is induced by both cADPR and IP3. It is, therefore, apparent that cells can utilize two second-messengers, IP3 and cADPR, for Ca2+ mobilization, depending on the types of cells as well as differences in cellular conditions, physiological or pathological, performing a variety of cellular functions. Recently, various physiological phenomena in animal, plant, and bacterial cells have been found to utilize this novel signal system [5,19,32,44,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90]. In the pancreatic exocrine acinar cells of CD38 knockout mice, the acetylcholine-induced Ca2+ oscillation was greatly reduced or completely disappeared under a physiological concentration of acetylcholine (40 nM–4 µM) [83]. Furthermore, acetylcholine induced cADPR formation in normal (CD38+/+) exocrine acinar cells but not in CD38 knockout exocrine acinar cells. The IP3 formation was very small in the presence of a physiological concentration of acetylcholine (40 nM–4 µM) and showed no difference between normal and CD38 knockout pancreatic acinar cells. Acetylcholine appears to induce the cADPR formation via the G-protein-coupled CD38 system [79]. In pancreatic islet β-cells, glucose is metabolized to form ATP and increases cADPR in response to glucose stimulation through the CD38-cADPR signal system for insulin secretion. In other cells such as pancreatic acinar cells and neuronal cells, hormones and neurotransmitters regulate the CD38-cADPR-Ca2+ signaling system in a receptor-coupled manner, such as in a G-protein-coupled manner, for various types of physiological responses [33,34].
The possible involvement of the CD38-cADPR signal system in cardiovascular abnormalities has been reported. Myocardial hypertrophy was observed in male mice with null mutations of CD38 and FKBP12.6, a cADPR binding protein [84,85]. In addition, the altered stoichiometry of FKBP12.6 versus type 2 RyR as a cause of abnormal Ca2+ leak through RyR was reported in heart failure in humans [82]. As diabetic complications in the cardiovascular system are frequently observed, screening of the abnormalities in the CD38-cADPR signal system and its components may provide a clue as to the underlying molecular mechanism. Furthermore, the CD38-cADPR signal was recently revealed to be a regulator of renin expression in juxtaglomerular cells in intermittent hypoxia (IH) [91]. As hypertension is the most common complication in sleep apnea syndrome (SAS) patients, and cells and tissues in SAS patients are exposed to IH, the finding could well explain hypertension in SAS patients.
Jin et al., revealed that adult CD38 knockout female and male mice in CD-1 mouse background showed marked defects in maternal nurturing and social behavior [5]. The plasma level of oxytocin was significantly decreased in CD38 knockout mice and oxytocin injection or lentiviral-vector-mediated delivery of human CD38 in the hypothalamus rescued social memory and maternal care in CD38 knockout mice [5]. In contrast, the introduction of lentiviral-vector mediated delivery of human CD38 Arg140Trp (rs1800561), which had first been found in Japanese type 2 diabetic patients [29], failed to recover the social memory and maternal care. Depolarization-induced oxytocin secretion and Ca2+ elevation in oxytocinergic neurohypophysial axon terminals were disrupted in the CD38 knockout mice. These results indicate that CD38 has a key role in neuropeptide release, such as oxytocin release, thereby decisively regulating maternal and social behaviors, and may be a component in neurodevelopmental disorders. The CD38 mutation that caused tryptophan to replace arginine at amino acid residue 140 (R140W; [rs1800561, 4693C > T]) [29] was found not only in Japanese type 2 diabetes patients but also in 0.6–4.6% of the Japanese population and was associated with autism spectrum disorder in a smaller case-control study [92]. In addition, maternal CD38R140W (rs1800561[4693C > T]) polymorphism was reported to be associated with an increased risk of admission to the neonatal intensive care unit due to preterm birth in Nara Medical University, Japan [93]. These reports strongly suggest that the CD38R140W (rs1800561[4693C > T]) polymorphism is a disease-prone genotype although the polymorphism is found to be only in mongoloid subjects.
Most recently, Takeda et al. reported IH, which is a common feature of SAS patient cells, induced the upregulation of renin in juxtaglomerular cells via the upregulation of Cd38 [91]. The IH-induced upregulation of renin was attenuated by the introduction of small interfering RNA for Cd38 or the addition of the cADPR antagonist, 8-bromo-cADPR, in the juxtaglomerular cell culture medium.
Funding
This study was supported in part by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (08102003). I thank the Japan Society for the Promotion of Science and Japan Science and Technology Agency for grants with which this study was able to be conducted.
Acknowledgments
I thank all of my past and present colleagues for their contributions to the studies described herein, especially Asako Itaya-Hironaka, Akiyo Yamauchi, Mai Makino, Sumiyo Sakuramoto-Tsuchida, Keito Asai, Hiroyo Ota, Nobuko Enami, Ryogo Shobatake, Tomoko Uchiyama, Yoshinori Takeda, Koji Nata, Hideto Yonekura, Ichiro Kato, Akira Tohgo, Naoya Noguchi, Tetsuhiko Koguma, Takako Sugimoto, Yasuhito Furuya, Fumiko Ikehata, Takako Akiyama, Kei Nakagawa, Tetsuya Nakazawa, Michio Kuroki, Takayuki Ikeda, Toshinari Takamura, Kan-ichi Nakagawara, Tsukasa Sasaki, Tadahiro Karasawa, Ariki Matsuoka, Azuma Kanatsuka, Haruhiro Higashida, Uh-Hyun Kim, Yasuhiko Yamamoto, and Hiroshi Okamoto.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| A-kinase | Cyclic AMP-dependent protein kinase |
| BST-1 | Bone marrow stromal cell antigen 1 |
| cADPR | Cyclic ADP-ribose |
| cAMP | Cyclic AMP |
| CaM | Calmodulin |
| CaM kinase | Ca2+/calmodulin-dependent protein kinase |
| CD38 | Cluster of differentiation 38 |
| CD157 | Cluster of differentiation 157 |
| CICR | Ca2+-induced Ca2+ release |
| EPAC2 | Exchange protein activated by cAMP2 |
| FKBP | FK506 binding protein |
| GIP | Gastric inhibitory polypeptide |
| GLP-1 | Glucagon-like peptide-1 |
| IH | Intermittent hypoxia |
| IP3 | Inositol 1,4,5-trisphosphate |
| NAADP | Nicotinic acid adenine dinucleotide phosphate |
| RyR | Ryanodine receptor Ca2+ channel |
| SAS | Sleep apnea syndrome |
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility of calcium signalling. Nat. Rev. Mol Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H.; Lee, H.C. NAADP: A universal Ca2+ trigger. Sci. Signal. 2008, 1, re10. [Google Scholar] [CrossRef] [PubMed]
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar] [CrossRef]
- Jin, D.; Liu, H.X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 2007, 446, 41–45. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Strumwasser, F. Purification and characterization of a molluscan egg-specific NADase, a second-messenger enzyme. Cell Regul. 1991, 2, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Aarhus, R. ADP-ribosyl cyclase: An enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.L.; Hellmich, M.R.; Beushausen, S.; Tempst, P.; Bayley, H.; Strumwasser, F. Primary structure of a molluscan egg-specific NADase, a second-messenger enzyme. Cell Regul. 1991, 2, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Nata, K.; Sugimoto, T.; Tohgo, A.; Takamura, T.; Noguchi, N.; Matsuoka, A.; Numakunai, T.; Shikama, K.; Yonekura, H.; Takasawa, S.; et al. The structure of the Aplysia kurodai gene encoding ADP-ribosyl cyclase, a second-messenger enzyme. Gene 1995, 158, 213–218. [Google Scholar] [CrossRef]
- States, D.J.; Walseth, T.F.; Lee, H.C. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human leukocyte antigen CD38. Trends. Biochem. Sci. 1992, 17, 495. [Google Scholar] [CrossRef]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef]
- Takasawa, S.; Tohgo, A.; Noguchi, N.; Koguma, T.; Nata, K.; Sugimoto, T.; Yonekura, H.; Okamoto, H. Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J. Biol. Chem. 1993, 268, 26052–26054. [Google Scholar] [CrossRef]
- Summerhill, R.J.; Jackson, D.G.; Galione, A. Human leukocyte antigen CD38 catalyzes the production of cyclic ADP-ribose. FEBS Lett. 1993, 335, 231–233. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Kimura, N.; Sato, K.; Ohsugi, Y.; Takasawa, S.; Okamoto, H.; Ishikawa, J.; Kaisho, T.; Ishihara, K.; Hirano, T. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Lett. 1994, 356, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Chini, E.N.; Chini, C.C.; Kato, I.; Takasawa, S.; Okamoto, H. CD38 is the major enzyme responsible for synthesis of nicotinic acid-adenine dinucleotide phosphate in mammalian tissues. Biochem. J. 2002, 362, 125–130. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J.H. Glucose induced closure of single potassium channels in isolated rat pancreatic β-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef]
- Takasawa, S.; Nata, K.; Yonekura, H.; Okamoto, H. Cyclic ADP-ribose in insulin secretion from pancreatic β cells. Science 1993, 259, 370–373. [Google Scholar] [CrossRef]
- Okamoto, H.; Takasawa, S.; Tohgo, H. New aspects of the physiological significance of NAD, poly ADP-ribose and cyclic ADP-ribose. Biochimie 1995, 77, 356–363. [Google Scholar] [CrossRef]
- Okamoto, H.; Takasawa, S.; Nata, K. The CD38-cyclic ADP-ribose signalling system in insulin secretion: Molecular basis and clinical implications. Diabetologia 1997, 40, 1485–1491. [Google Scholar] [CrossRef] [Green Version]
- Koguma, T.; Takasawa, S.; Tohgo, A.; Karasawa, T.; Furuya, Y.; Yonekura, H.; Okamoto, H. Cloning and characterization of cDNA encoding rat ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase (homologue to human CD38) from islets of Langerhans. Biochim. Biophys. Acta 1994, 1223, 160–162. [Google Scholar] [CrossRef]
- Tohgo, A.; Takasawa, S.; Noguchi, N.; Koguma, T.; Nata, K.; Sugimoto, T.; Furuya, Y.; Yonekura, H.; Okamoto, H. Essential cysteine residues for cyclic ADP-ribose synthesis and hydrolysis by CD38. J. Biol. Chem. 1994, 269, 28555–28557. [Google Scholar] [CrossRef]
- Nakagawara, K.; Mori, M.; Takasawa, S.; Nata, K.; Takamura, T.; Berlova, A.; Tohgo, A.; Karasawa, T.; Yonekura, H.; Takeuchi, T.; et al. Assignment of CD38, the gene encoding human leukocyte antigen CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase), to chromosome 4p15. Cytogenet. Cell Genet. 1995, 69, 38–39. [Google Scholar] [CrossRef]
- Kato, I.; Takasawa, S.; Akabane, A.; Tanaka, O.; Abe, H.; Takamura, T.; Suzuki, Y.; Nata, K.; Yonekura, H.; Yoshimoto, T.; et al. Regulatory role of CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in insulin secretion by glucose in pancreatic β cells. Enhanced insulin secretion in CD38-expressing transgenic mice. J. Biol. Chem. 1995, 270, 30045–30050. [Google Scholar] [CrossRef] [Green Version]
- Takasawa, S.; Ishida, A.; Nata, K.; Nakagawa, K.; Noguchi, N.; Tohgo, A.; Kato, I.; Yonekura, H.; Fujisawa, H.; Okamoto, H. Requirement of calmodulin-dependent protein kinase II in cyclic ADP-ribose-mediated intracellular Ca2+ mobilization. J. Biol. Chem. 1995, 270, 30257–30259. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, N.; Takasawa, S.; Nata, K.; Tohgo, A.; Kato, I.; Ikehata, F.; Yonekura, H.; Okamoto, H. Cyclic ADP-ribose binds to FK506-binding protein 12.6 to release Ca2+ from islet microsomes. J. Biol. Chem. 1997, 272, 3133–3136. [Google Scholar] [CrossRef] [Green Version]
- Tohgo, A.; Munakata, H.; Takasawa, S.; Nata, K.; Akiyama, T.; Hayashi, N.; Okamoto, H. Lysine 129 of CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) participates in the binding of ATP to inhibit the cyclic ADP-ribose hydrolase. J. Biol. Chem. 1997, 272, 3879–3882. [Google Scholar] [CrossRef] [Green Version]
- Nata, K.; Takamura, T.; Karasawa, T.; Kumagai, T.; Hashioka, W.; Tohgo, A.; Yonekura, H.; Takasawa, S.; Nakamura, S.; Okamoto, H. Human gene encoding CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase): Organization, nucleotide sequence and alternative splicing. Gene 1997, 186, 285–292. [Google Scholar] [CrossRef]
- Takasawa, S.; Akiyama, T.; Nata, K.; Kuroki, M.; Tohgo, A.; Noguchi, N.; Kobayashi, S.; Kato, I.; Katada, T.; Okamoto, H. Cyclic ADP-ribose and inositol 1,4,5-trisphosphate as alternate second messengers for intracellular Ca2+ mobilization in normal and diabetic β-cells. J. Biol. Chem. 1998, 273, 2497–2500. [Google Scholar] [CrossRef] [Green Version]
- Yagui, K.; Shimada, F.; Miura, M.; Hashimoto, N.; Suzuki, Y.; Tokuyama, Y.; Nata, K.; Tohgo, A.; Ikehata, F.; Takasawa, S.; et al. A missense mutation in the CD38 gene, a novel factor for insulin secretion: Association with Type II diabetes mellitus in Japanese subjects and evidence of abnormal function when expressed in vitro. Diabetologia 1998, 41, 1024–1028. [Google Scholar] [CrossRef] [Green Version]
- Ikehata, F.; Satoh, J.; Nata, K.; Tohgo, A.; Nakazawa, T.; Kato, I.; Kobayashi, S.; Akiyama, T.; Takasawa, S.; Toyota, T.; et al. Autoantibodies against CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) that impair glucose-induced insulin secretion in noninsulin- dependent diabetes patients. J. Clin. Investig. 1998, 102, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Kato, I.; Yamamoto, Y.; Fujimura, M.; Noguchi, N.; Takasawa, S.; Okamoto, H. CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J. Biol. Chem. 1999, 274, 1869–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, H.; Takasawa, S.; Nata, K.; Kato, I.; Tohgo, A.; Noguchi, N. Physiological and pathological significance of the CD38-cyclic ADP-ribose signaling system. Chem. Immunol. 2000, 75, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, S.; Okamoto, H. Pancreatic β-cell death, regeneration and insulin secretion: Roles of poly(ADP-ribose) polymerase and cyclic ADP-ribose. Int. J. Diabetes Res. 2002, 3, 79–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, H.; Takasawa, S. Recent advances in the Okamoto model: The CD38-cyclic ADP-ribose signal system and the regenerating gene protein (Reg)-Reg receptor system in β-cells. Diabetes 2002, 51 (Suppl. S3), S462–S473. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Larsson, O.; Berggren, P.O.; Takasawa, S.; Nata, K.; Yonekura, H.; Okamoto, H.; Galione, A. Cyclic ADP-ribose in β cells. Science 1993, 262, 584–586. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Theler, J.-M.; Wollheim, C.B. Ca2+ stores in insulin-secreting cells: Lack of the effect of cADP ribose. Cell Calcium 1994, 16, 71–80. [Google Scholar] [CrossRef]
- Webb, D.-L.; Islam, M.S.; Efanov, A.M.; Brown, G.; Köhler, M.; Larsson, O.; Berggren, P.-O. Insulin exocytosis and glucose-mediated increase in cytoplasmic free Ca2+ concentration in the pancreatic β-cell are independent of cyclic ADP-ribose. J. Biol. Chem. 1996, 271, 19074–19079. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Berggren, P.O. Cyclic ADP-ribose and the pancreatic beta cell: Where do we stand? Diabetologia 1997, 40, 1480–1484. [Google Scholar] [CrossRef] [Green Version]
- Malaisse, W.J.; Kanda, Y.; Inageda, K.; Scruel, O.; Sener, A.; Katada, T. Cyclic ADP-ribose measurements in rat pancreatic islets. Biochem. Biophys. Res. Commun. 1997, 231, 546–548. [Google Scholar] [CrossRef]
- Scruel, O.; Wada, T.; Kontani, K.; Sener, A.; Katada, T.; Malaisse, W.J. Effects of D-glucose and starvation upon the cyclic ADP-ribose content of rat pancreatic islets. Biochem. Mol. Biol. Int. 1998, 45, 783–790. [Google Scholar] [CrossRef] [Green Version]
- An, N.H.; Han, M.K.; Um, C.; Park, B.H.; Park, B.J.; Kim, H.K.; Kim, U.H. Significance of ecto-cyclase activity of CD38 in insulin secretion of mouse pancreatic islet cells. Biochem. Biophys. Res. Commun. 2001, 282, 781–786. [Google Scholar] [CrossRef]
- Varadi, A.; Rutter, G.A. Dynamic imaging of endoplasmic reticulum Ca2+ concentration in insulin-secreting MIN6 cells using recombinant targeted cameleons: Roles of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2 and ryanodine receptors. Diabetes 2002, 51, S190–S201. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, K.J.; Pinton, P.; Varadi, A.; Tacchetti, C.; Ainscow, E.K.; Pozzan, T.; Rizzuto, R.; Rutter, G.A. Dense core secretory vesicles revealed as a dynamic Ca2+ store in neuroendocrine cells with a vesicle-associated membrane protein aequorin chimaera. J. Cell Biol. 2001, 155, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Takasawa, S.; Noguchi, N.; Nata, K.; Yamauchi, A.; Takahashi, I.; Yoshikawa, T.; Sugawara, A.; Yonekura, H.; Okamoto, H. Identification of a major enzyme for the synthesis and hydrolysis of cyclic ADP-ribose in amphibian cells and evolutional conservation of the enzyme from human to invertebrate. Mol. Cell. Biochem. 2012, 366, 69–80. [Google Scholar] [CrossRef]
- Zocchi, E.; Franco, L.; Guida, L.; Benatti, U.; Bagellesi, A.; Malavasi, F.; Lee, H.C.; De Flora, A. A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem. Biophys. Res. Commun. 1993, 196, 1459–1465. [Google Scholar] [CrossRef]
- Munshi, C.; Thiel, D.J.; Mathews, I.I.; Aarhus, R.; Walseth, T.F.; Lee, H.C. Characterization of the active site of ADP-ribosyl cyclase. J. Biol. Chem. 1999, 274, 30770–30777. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, H.; Takasawa, S. CD38. In Encyclopedia of Molecular Medicine; Creighton, T.E., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2002; pp. 601–604. [Google Scholar]
- Ferrero, E.; Lo Buono, N.; Morone, S.; Parrotta, R.; Mancini, C.; Brusco, A.; Giancomino, A.; Augeri, S.; Rosal-Vela, A.; García-Rodríguez, S.; et al. Human canonical CD157/Bst1 is an alternatively spliced isoform masking a previously unidentified primate-specific exon included in a novel transcript. Sci. Rep. 2017, 7, 15923. [Google Scholar] [CrossRef] [Green Version]
- Kaisho, T.; Ishikawa, J.; Oritani, K.; Inazawa, J.; Muraoka, O.; Ochi, T.; Hirano, T. BST-1, a surface molecule of bone marrow stromal cell lines that facilitates pre-B-cell growth. Proc. Natl. Acad. Sci. USA 1994, 91, 5325–5329. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, O.; Tanaka, H.; Itoh, M.; Ishihara, K.; Hirano, T. Genomic structure of human BST-1. Immunol. Lett. 1996, 54, 1–4. [Google Scholar] [CrossRef]
- Dong, C.; Willerford, D.; Alt, F.W.; Cooper, M.D. Genomic organization and chromosomal localization of the mouse BP3 gene, a member of the CD38/ADP-ribosy cyclase family. Immunogenetics 1996, 45, 35–43. [Google Scholar] [CrossRef]
- Furuya, Y.; Takasawa, S.; Yonekura, H.; Tanaka, T.; Takehara, J.; Okamoto, H. Cloning of a cDNA encoding rat bone marrow stromal cell antigen 1 (BST-1) from the islets of Langerhans. Gene 1995, 165, 329–330. [Google Scholar] [CrossRef]
- Kajimoto, Y.; Miyagawa, J.; Ishihara, K.; Okuyama, Y.; Fujitani, Y.; Itoh, M.; Yoshida, H.; Kaisho, T.; Matsuoka, T.; Watada, H.; et al. Pancreatic islets cells express BST-1, a CD38-like surface molecule having ADP-ribosyl cyclase activity. Biochem. Biophys. Res. Commun. 1996, 219, 941–946. [Google Scholar] [CrossRef]
- Itoh, M.; Ishihara, K.; Hiroi, T.; Lee, B.O.; Maeda, H.; Iijima, H.; Yanagita, M.; Kiyono, H.; Hirano, H. Deletion of bone marrow stromal cell antigen-1 (CD157) gene impaired systemic thymus independent-2 antigen-induced IgG3 and mucosal TD antigen-elicited IgA responses. J. Immunol. 1998, 161, 3974–3983. [Google Scholar] [PubMed]
- Jackson, D.G.; Bell, J.I. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J. Immunol. 1990, 144, 2811–2815. [Google Scholar] [PubMed]
- Harada, N.; Santos-Arugumedo, L.; Chang, R.; Grimaldi, J.C.; Lund, F.E.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; Heath, A.W.; Parkhouse, R.M. Expression cloning of a cDNA encoding a novel murine B cell activation marker. Homology to human CD38. J. Immunol. 1993, 151, 3111–3118. [Google Scholar]
- Itoh, M.; Ishihara, K.; Tomizawa, H.; Tanaka, H.; Kobune, Y.; Ishikawa, J.; Kaisho, T.; Hirano, T. Molecular cloning of murine BST-1 having homology with CD38 and Aplysia ADP-ribosyl cyclase. Biochem. Biophys. Res. Commun. 1994, 203, 1309–1317. [Google Scholar] [CrossRef]
- Churamani, D.; Boulware, M.J.; Geach, T.J.; Martin, A.C.R.; Moy, G.W.; Su, Y.-H.; Vacquier, V.D.; Marchant, J.S.; Dale, L.; Patel, S. Molecular characterization of a novel intracellular ADP-ribosyl cyclase. PLoS ONE 2007, 2, e797. [Google Scholar] [CrossRef] [Green Version]
- Goodrich, S.P.; Muller-Steffner, M.; Osman, A.; Moutin, M.-J.; Kusser, K.; Roberts, A.; Woodland, D.L.; Randall, T.D.; Kellenberger, E.; LoVerde, P.T.; et al. Production of calcium-mobilizing metabolites by a novel member of the ADP-ribosyl cyclase family expressed in Schistosoma mansoni. Biochemistry 2005, 44, 11082–11097. [Google Scholar] [CrossRef]
- Kelu, J.J.; Web, S.E.; Galione, A.; Miller, A.L. Characterization of ADP-ribosyl cyclase 1-like (ARC1-like) activity and NAADP signaling during slow muscle cell development in zebrafish embryos. Dev. Biol. 2019, 445, 211–225. [Google Scholar] [CrossRef]
- Noguchi, N.; Yoshikawa, T.; Ikeda, T.; Takahashi, I.; Shervani, N.J.; Uruno, A.; Yamauchi, A.; Nata, K.; Takasawa, S.; Okamoto, H.; et al. FKBP12.6 disruption impairs glucose-induced insulin secretion. Biochem. Biophys. Res. Commun. 2008, 371, 735–740. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Ashcroft, S.J.H. (Eds.) Insulin: Molecular Biology to Pathology; IRL Press: Oxford, UK, 1992; pp. 97–150. [Google Scholar]
- Galione, A. Cyclic ADP-ribose: A new way to control calcium. Science 1993, 259, 325–326. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, S.; Kuroki, M.; Nata, K.; Noguchi, N.; Ikeda, T.; Yamauchi, A.; Ota, H.; Itaya-Hironaka, A.; Sakuramoto-Tsuchida, S.; Takahashi, I.; et al. A novel ryanodine receptor expressed in pancreatic islets by alternative splicing from type 2 ryanodine receptor gene. Biochem. Biophys. Res. Commun. 2010, 397, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Pirsch, J.D.; Miller, J.; Deierhoi, M.H.; Vincenti, F.; Filo, R.S. A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression after cadaveric renal transplantation: FK506 Kidney Transplant Study Group. Transplantation 1997, 63, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Park, K.H.; Yim, C.Y.; Takasawa, S.; Okamoto, H.; Im, M.J.; Kim, U.H. Generation of nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose by glucagon-like peptide-1 evokes Ca2+ signal that is essential for insulin secretion in mouse pancreatic islets. Diabetes 2008, 57, 868–878. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, H.; Takasawa, S.; Yamamoto, Y. From insulin synthesis to secretion: Alternative splicing of type 2 ryanodine receptor gene is essential for insulin secretion in pancreatic β cells. Int. J. Biochem. Cell Biol. 2017, 91, 176–183. [Google Scholar] [CrossRef]
- Makino, M.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Takasawa, S. Tissue-specific alternative splicing of type 2 ryanodine receptor gene affects insulin biosynthesis in pancreatic β cells. Diabetologia 2020, 63 (Suppl. S1), 385. Available online: https://www.easd.org/virtualmeeting/home.html#!resources/tissue-specific-alternative-splicing-of-type-2-ryanodine-receptor-gene-affects-insulin-biosynthesis-in-pancreatic-beta-cells-f6bbb60b-8e31-40c9-ab74-c0702c5f27f6 (accessed on 22 September 2020).
- Mount, S.M. A catalog of splice junction sequences. Nucleic Acids Res. 1982, 10, 459–472. [Google Scholar] [CrossRef]
- Nakai, K.; Sakamoto, H. Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene 1994, 141, 171–177. [Google Scholar] [CrossRef]
- Sakai, N.; Santamarina-Fojo, S.; Yamashita, S.; Matsuzawa, Y.; Brewer, H.B., Jr. Exon 10 skipping caused by intron 10 splice donor site mutation in cholesterol ester transfer protein gene results in abnormal downstream splice site selection. J. Lipid Res. 1996, 37, 2065–2073. [Google Scholar] [CrossRef] [PubMed]
- Pupilli, C.; Giannini, S.; Marchetti, P.; Lupi, R.; Antonelli, A.; Malavasi, F.; Takasawa, S.; Okamoto, H.; Ferrannini, E. Autoantibodies to CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in Caucasian patients with diabetes: Effects on insulin release from human islets. Diabetes 1999, 48, 2309–2315. [Google Scholar] [CrossRef]
- Mallone, R.; Ortolan, E.; Baj, G.; Funaro, A.; Giunti, S.; Lillaz, E.; Saccucci, F.; Cassader, M.; Cavallo-Perin, P.; Malavasi, F. Autoantibody response to CD38 in Caucasian patients with type 1 and type 2 diabetes: Immunological and genetic characterization. Diabetes 2001, 50, 752–762. [Google Scholar] [CrossRef]
- Antonelli, A.; Baj, G.; Marchetti, P.; Fallahi, P.; Surico, N.; Pupilli, C.; Malavasi, F.; Ferrannini, E. Human anto-CD38 autoantibodies raise intracellular calcium and stimulate insulin release in human pancreatic islets. Diabetes 2001, 50, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.Y.; Tokimasa, T.; Takasawa, S.; Furuya, Y.; Nohmi, M.; Okamoto, H.; Kuba, K. Cyclic ADP-ribose modulates Ca2+ release channels for activation by physiological Ca2+ entry in bullfrog sympathetic neurons. Neuron 1994, 12, 1073–1079. [Google Scholar] [CrossRef]
- Karasawa, T.; Takasawa, S.; Yamakawa, K.; Yonekura, H.; Okamoto, H.; Nakamura, S. NAD+-glycohydrolase from Streptococcus pyogenes shows cyclic ADP-ribose forming activity. FEMS Microbiol. Lett. 1995, 130, 201–204. [Google Scholar] [CrossRef]
- Ebihara, S.; Sasaki, T.; Hida, W.; Kikuchi, Y.; Ohshiro, T.; Shimura, S.; Takasawa, S.; Okamoto, H.; Nishiyama, A.; Akaike, N.; et al. Role of cyclic ADP-ribose in ATP-activated potassium currents in alveolar macrophages. J. Biol. Chem. 1997, 272, 16023–16029. [Google Scholar] [CrossRef] [Green Version]
- Higashida, H.; Yokoyama, S.; Hashii, M.; Taketo, M.; Higashida, M.; Takayasu, T.; Ohshima, T.; Takasawa, S.; Okamoto, H.; Noda, M. Muscarinic receptor-mediated dual regulation of ADP-ribosyl cyclase in NG108-15 neuronal cell membranes. J. Biol. Chem. 1997, 272, 31272–31277. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Kuzma, J.; Maréchal, E.; Graeff, R.; Lee, H.C.; Foster, R.; Chua, N.-H. Abscisic acid signalling through cyclic ADP-ribose in plats. Science 1997, 278, 2126–2130. [Google Scholar] [CrossRef]
- Leckie, C.P.; McAinsh, M.R.; Allen, G.J.; Sanders, D.; Hetherington, A.M. Abscisic acid-induced stomatal closure mediated by cyclic ADP-ribose. Proc. Natl. Acad. Sci. USA 1998, 95, 15837–15842. [Google Scholar] [CrossRef] [Green Version]
- Yano, M.; Ono, K.; Ohkusa, T.; Suetsugu, M.; Kohno, M.; Hisaoka, T.; Kobayashi, S.; Hisamatsu, Y.; Yamamoto, T.; Kohno, M.; et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circilation 2000, 102, 2131–2136. [Google Scholar] [CrossRef] [Green Version]
- Fukushi, Y.; Kato, I.; Takasawa, S.; Sasaki, T.; Ong, B.H.; Sato, M.; Ohsaga, A.; Sato, K.; Shirato, K.; Okamoto, H.; et al. Identification of cyclic ADP-ribose-dependent mechanisms in pancreatic muscarinic Ca2+ signaling using CD38 knockout mice. J. Biol. Chem. 2001, 276, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.-B.; Senbonmatsu, T.; Cheng, D.-S.; Wang, Y.-X.; Copello, J.A.; Ji, G.-J.; Collier, M.L.; Deng, Y.-X.; Jeyakumar, L.H.; Magnuson, M.A.; et al. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature 2002, 416, 334–337. [Google Scholar] [CrossRef]
- Takahashi, J.; Kagaya, Y.; Kato, I.; Ohta, J.; Isoyama, S.; Miura, M.; Sugai, Y.; Hirose, M.; Wakayama, Y.; Ninomiya, M.; et al. Deficit of CD38/cyclic ADP-ribose is differentially compensated in hearts by gender. Biochem. Biophys. Res. Commun. 2003, 312, 434–440. [Google Scholar] [CrossRef]
- Mitsui-Saito, M.; Kato, I.; Takasawa, S.; Okamoto, H.; Yanagisawa, T. CD38 gene disruption inhibits the contraction induced by α-adrenoceptor stimulation in mouse aorta. J. Vet. Med. Sci. 2003, 65, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Sasamori, K.; Sasaki, T.; Takasawa, S.; Tamada, T.; Nara, M.; Irokawa, T.; Shimura, S.; Shirato, K.; Hattori, T. Cyclic ADP-ribose, a putative Ca2+-mobilizing second messenger, operates in submucosal gland acinar cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L69–L78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellner, S.K.; Parker, L. Endothelin-1, superoxide and adeninediphosphate ribose cyclase in shark vascular smooth muscle. J. Exp. Biol. 2005, 208, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Dodd, A.N.; Gardner, M.J.; Hotta, C.T.; Hubbard, K.E.; Delchau, N.; Love, J.; Assie, J.M.; Robertson, F.C.; Jakobsen, M.K.; Gonçalves, J.; et al. The Arabidopsis circadian clock incorporates a cADPR-based feedback loop. Science 2007, 318, 1789–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, H.; Tamaki, S.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Morioka, T.; Takasawa, S.; Kimura, H. Attenuation of glucose-induced insulin secretion by intermittent hypoxia via down-regulation of CD38. Life Sci. 2012, 90, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Itaya-Hironaka, A.; Yamauchi, A.; Makino, M.; Sakuramoto-Tsuchida, S.; Ota, H.; Kawaguchi, R.; Takasawa, S. Intermittent hypoxia upregulates the Renin and Cd38 mRNAs in renin-producing cells via the downregulation of miR-203. Int. J. Mol. Sci. 2021, 22, 10127. [Google Scholar] [CrossRef]
- Munesue, T.; Yokoyama, S.; Nakamura, K.; Anitha, A.; Yamada, K.; Hayashi, K.; Asaka, T.; Liu, H.X.; Jin, D.; Koizumi, K.; et al. Two genetic variants of CD38 in subjects with autism spectrum disorder and controls. Neurosci. Res. 2010, 67, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Enami, N.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Takasawa, S.; Takahashi, Y. The CD38 genotype (rs1800561 (4693C>T): R140W) is associated with an increased risk of admission to the neonatal intensive care unit. Early Hum. Dev. 2015, 91, 467–470. [Google Scholar] [CrossRef]
Figure 1.
Insulin secretion by glucose stimulation in pancreatic β-cells (adapted from [33]). The insulin secretion via the CD38-cADPR signal system is shown in red. cADPR, produced from NAD+ by the ADP-ribosyl cyclase of CD38, binds to FKBP12.6 to release Ca2+, dissociating FKBP12.6 from RyR [25]. CaM kinase II phosphorylates RyR to sensitize and activate the Ca2+ channel (Pi, phosphorylation of RyR by CaM kinase II) [24]. Ca2+, released from intracellular stores and/or supplied from extracellular sources, further activates CaM kinase II and amplifies the process. In this way, Ca2+-induced Ca2+ release (CICR) can be explained in glucose-induced insulin secretion in pancreatic β-cells. The conventional insulin secretion mechanism by Ca2+ influx from extracellular sources [16] is shown in black.
Figure 1.
Insulin secretion by glucose stimulation in pancreatic β-cells (adapted from [33]). The insulin secretion via the CD38-cADPR signal system is shown in red. cADPR, produced from NAD+ by the ADP-ribosyl cyclase of CD38, binds to FKBP12.6 to release Ca2+, dissociating FKBP12.6 from RyR [25]. CaM kinase II phosphorylates RyR to sensitize and activate the Ca2+ channel (Pi, phosphorylation of RyR by CaM kinase II) [24]. Ca2+, released from intracellular stores and/or supplied from extracellular sources, further activates CaM kinase II and amplifies the process. In this way, Ca2+-induced Ca2+ release (CICR) can be explained in glucose-induced insulin secretion in pancreatic β-cells. The conventional insulin secretion mechanism by Ca2+ influx from extracellular sources [16] is shown in black.

Figure 2.
Schematic representation of human CD38 in enzyme activities for the synthesis and hydrolysis of cADPR (adapted from [26]). Glu-226 in human CD38 is essential for cADPR synthesis from NAD+ (ADP-ribosyl cyclase activity) [46]. Cys-119 and Cys-201 are essential for cADPR hydrolysis to form ADP-ribose (cADPR hydrolase activity). Lys-129 is the cADPR binding site and is indispensable for cADPR hydrolysis (cADPR hydrolase) [21]. ATP, produced by glucose metabolism, competes with cADPR for the binding site (Lys-129), inhibiting the cADPR hydrolysis by cADPR hydrolase of CD38, which causes the accumulation of cADPR in the cell [12]. [Enzyme-cADPR*] is proposed as an enzyme-stabilized ADP-ribosyl oxocarbonium ion intermediate.
Figure 2.
Schematic representation of human CD38 in enzyme activities for the synthesis and hydrolysis of cADPR (adapted from [26]). Glu-226 in human CD38 is essential for cADPR synthesis from NAD+ (ADP-ribosyl cyclase activity) [46]. Cys-119 and Cys-201 are essential for cADPR hydrolysis to form ADP-ribose (cADPR hydrolase activity). Lys-129 is the cADPR binding site and is indispensable for cADPR hydrolysis (cADPR hydrolase) [21]. ATP, produced by glucose metabolism, competes with cADPR for the binding site (Lys-129), inhibiting the cADPR hydrolysis by cADPR hydrolase of CD38, which causes the accumulation of cADPR in the cell [12]. [Enzyme-cADPR*] is proposed as an enzyme-stabilized ADP-ribosyl oxocarbonium ion intermediate.
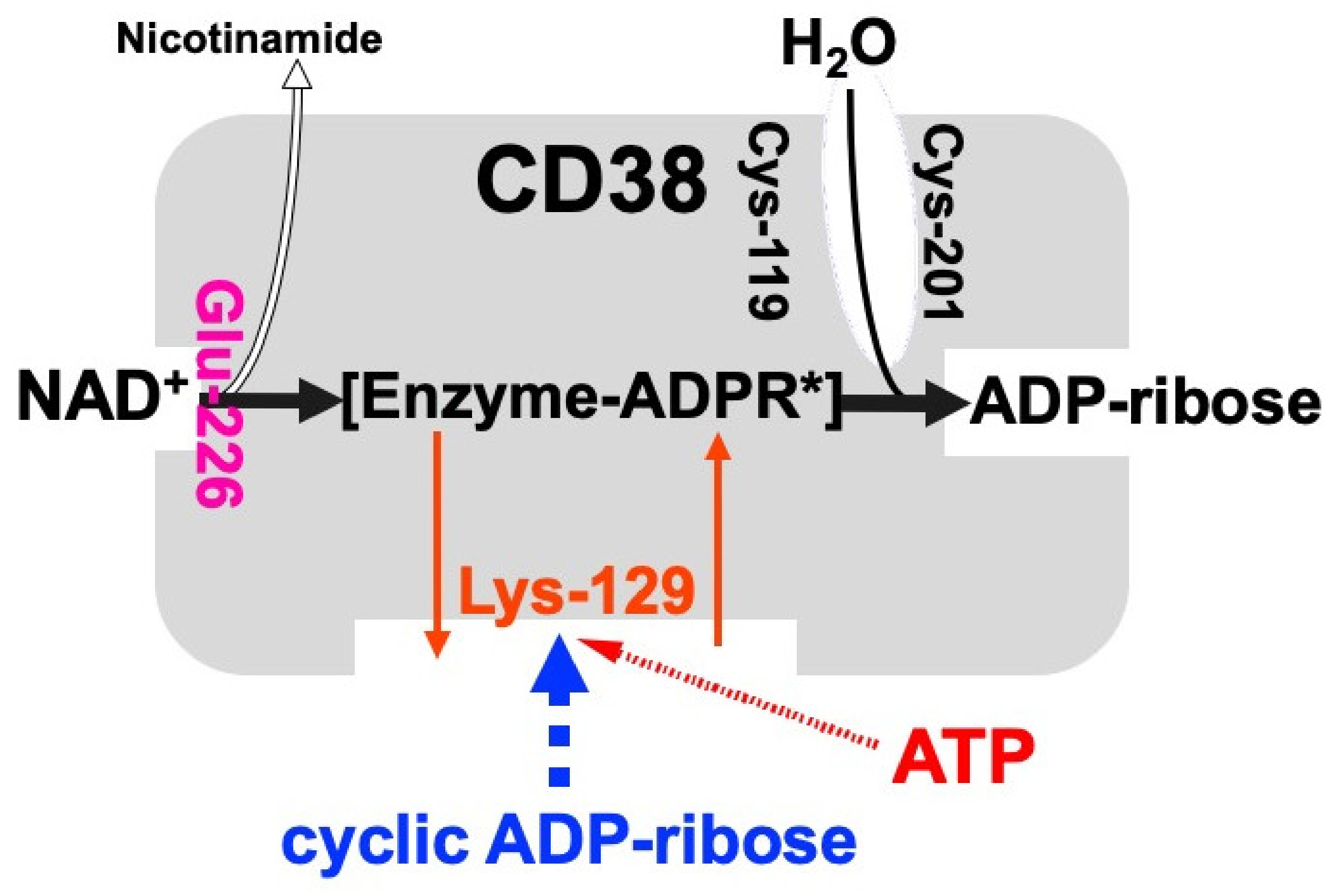
Figure 3.
Structural organization of human CD38, frog Cd38, human CD157, and Aplysia kurodai ADP-ribosyl cyclase genes (adapted from [9,27,44]). The exons are depicted as boxes; filled and open boxes represent protein-coding regions and untranslated regions, respectively. Recently, primate-specific exon 1b, located between exon 1 and 2 of human CD157, that encodes 15 additional amino acids, was reported [48]. Hatched boxes represent transmembrane (CD38)- or signal peptide (CD157 and Aplysia ADP-ribosyl cyclase)-coding regions. The corresponding exon-intron junctions are indicated as broken lines. Exons are numbered.
Figure 3.
Structural organization of human CD38, frog Cd38, human CD157, and Aplysia kurodai ADP-ribosyl cyclase genes (adapted from [9,27,44]). The exons are depicted as boxes; filled and open boxes represent protein-coding regions and untranslated regions, respectively. Recently, primate-specific exon 1b, located between exon 1 and 2 of human CD157, that encodes 15 additional amino acids, was reported [48]. Hatched boxes represent transmembrane (CD38)- or signal peptide (CD157 and Aplysia ADP-ribosyl cyclase)-coding regions. The corresponding exon-intron junctions are indicated as broken lines. Exons are numbered.
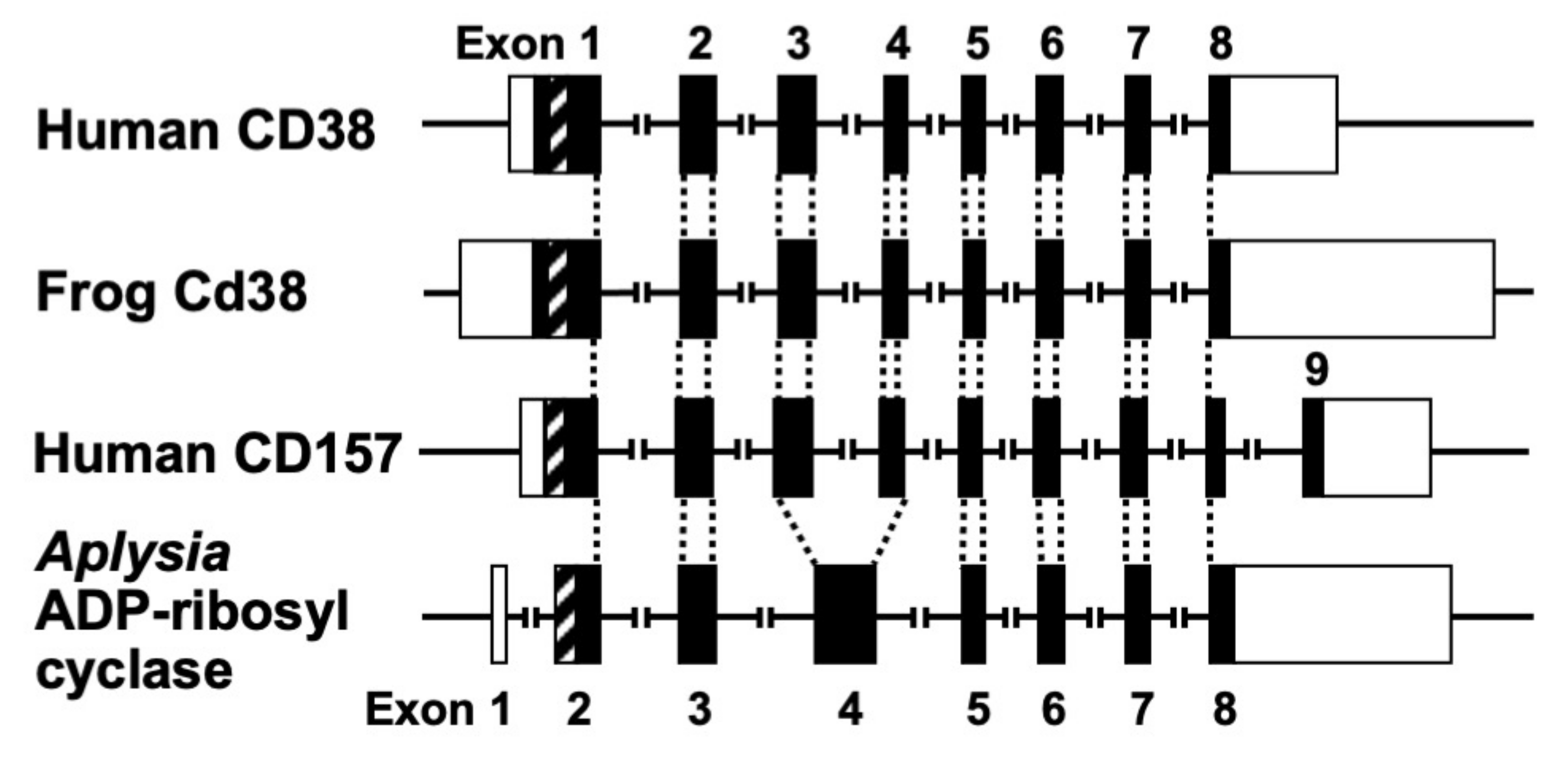
Figure 4.
Alignment of the amino acid sequences of the CD38/CD157/ADP-ribosyl cyclase family based on the primary structures of the encoded proteins (adapted from [44]). Dots indicate amino acids identical to human CD38. Dashes indicate gaps for maximal alignment. Ten cysteine and five amino acid residues conserved among all the proteins are highlighted in red and gray, respectively. The underlined amino acid residues are a putative transmembrane domain in CD38 and signal sequence in CD157 and ADP-ribosyl cyclase. The sequences used in alignment are #1: human CD38 (AAA68482 [55]), #2: rat CD38 (BAA06129 [20]), #3: mouse CD38 (AAA03163 [56]), #4: chicken CD38 (NM_001201388), #5: Xenopus laevis CD38 (AB194899 [44]), #6: human BST1 (CD157) (BAA04885 [47]), #7: rat BST1 (CD157) (Q63072 [51]), #8: mouse BST1 (CD157) (BAA06597 [57]), #9: chicken CD157 (NM_001200043), #10: Xenopus laevis CD157 (AB194901 [44]), #11: Aplysia californica ADP-ribosyl cyclase (AAA65698 [8]), #12: Aplysia kurodai ADP-ribosyl cyclase (BAA06284 [9]), #13: Strongylocentrotus purpuratus ADP-ribosyl cyclase (AM494973 [58]), and #14: Schistoma mansoni ADP-ribosyl cyclase (AAX35328 [59]). Although XP_005162637 (Danio rerio ADP-ribosyl cyclase 1-like [60]) showed significant homology with CD38/CD157/ADP-ribosyl cyclase family, the 8th and 10th cysteines in the conserved 10 cysteine residues (red), which are essential for the enzyme activities, are not conserved. It may be important to measure ADP-ribosyl cyclase activity in zebrafish ADP-ribosyl cyclase 1-like for understanding the involvement of the two cysteines in ADP-ribosyl cyclase activity.
Figure 4.
Alignment of the amino acid sequences of the CD38/CD157/ADP-ribosyl cyclase family based on the primary structures of the encoded proteins (adapted from [44]). Dots indicate amino acids identical to human CD38. Dashes indicate gaps for maximal alignment. Ten cysteine and five amino acid residues conserved among all the proteins are highlighted in red and gray, respectively. The underlined amino acid residues are a putative transmembrane domain in CD38 and signal sequence in CD157 and ADP-ribosyl cyclase. The sequences used in alignment are #1: human CD38 (AAA68482 [55]), #2: rat CD38 (BAA06129 [20]), #3: mouse CD38 (AAA03163 [56]), #4: chicken CD38 (NM_001201388), #5: Xenopus laevis CD38 (AB194899 [44]), #6: human BST1 (CD157) (BAA04885 [47]), #7: rat BST1 (CD157) (Q63072 [51]), #8: mouse BST1 (CD157) (BAA06597 [57]), #9: chicken CD157 (NM_001200043), #10: Xenopus laevis CD157 (AB194901 [44]), #11: Aplysia californica ADP-ribosyl cyclase (AAA65698 [8]), #12: Aplysia kurodai ADP-ribosyl cyclase (BAA06284 [9]), #13: Strongylocentrotus purpuratus ADP-ribosyl cyclase (AM494973 [58]), and #14: Schistoma mansoni ADP-ribosyl cyclase (AAX35328 [59]). Although XP_005162637 (Danio rerio ADP-ribosyl cyclase 1-like [60]) showed significant homology with CD38/CD157/ADP-ribosyl cyclase family, the 8th and 10th cysteines in the conserved 10 cysteine residues (red), which are essential for the enzyme activities, are not conserved. It may be important to measure ADP-ribosyl cyclase activity in zebrafish ADP-ribosyl cyclase 1-like for understanding the involvement of the two cysteines in ADP-ribosyl cyclase activity.

Figure 5.
Possible model for glucose-induced insulin secretion via the CD38-cADPR signal system and its enhancement by CaM kinase II and A-kinase. CaM kinase II and A-kinase phosphorylate and activate RyR in pancreatic β-cells to secrete insulin much more. Peptide hormones such as vasoactive intestinal peptide (VIP), pituitary adenylate cyclase-activating peptide (PACAP), glucagon-like peptide-1 (GLP-1), and glucose-dependent insulinotropic polypeptide/gastric inhibitory polypeptide (GIP) activate adenylate cyclase (A-kinase) to increase cAMP. As a result, RyR Ca2+ channel is phosphorylated and sensitized/activated for cADPR-induced Ca2+ release.
Figure 5.
Possible model for glucose-induced insulin secretion via the CD38-cADPR signal system and its enhancement by CaM kinase II and A-kinase. CaM kinase II and A-kinase phosphorylate and activate RyR in pancreatic β-cells to secrete insulin much more. Peptide hormones such as vasoactive intestinal peptide (VIP), pituitary adenylate cyclase-activating peptide (PACAP), glucagon-like peptide-1 (GLP-1), and glucose-dependent insulinotropic polypeptide/gastric inhibitory polypeptide (GIP) activate adenylate cyclase (A-kinase) to increase cAMP. As a result, RyR Ca2+ channel is phosphorylated and sensitized/activated for cADPR-induced Ca2+ release.
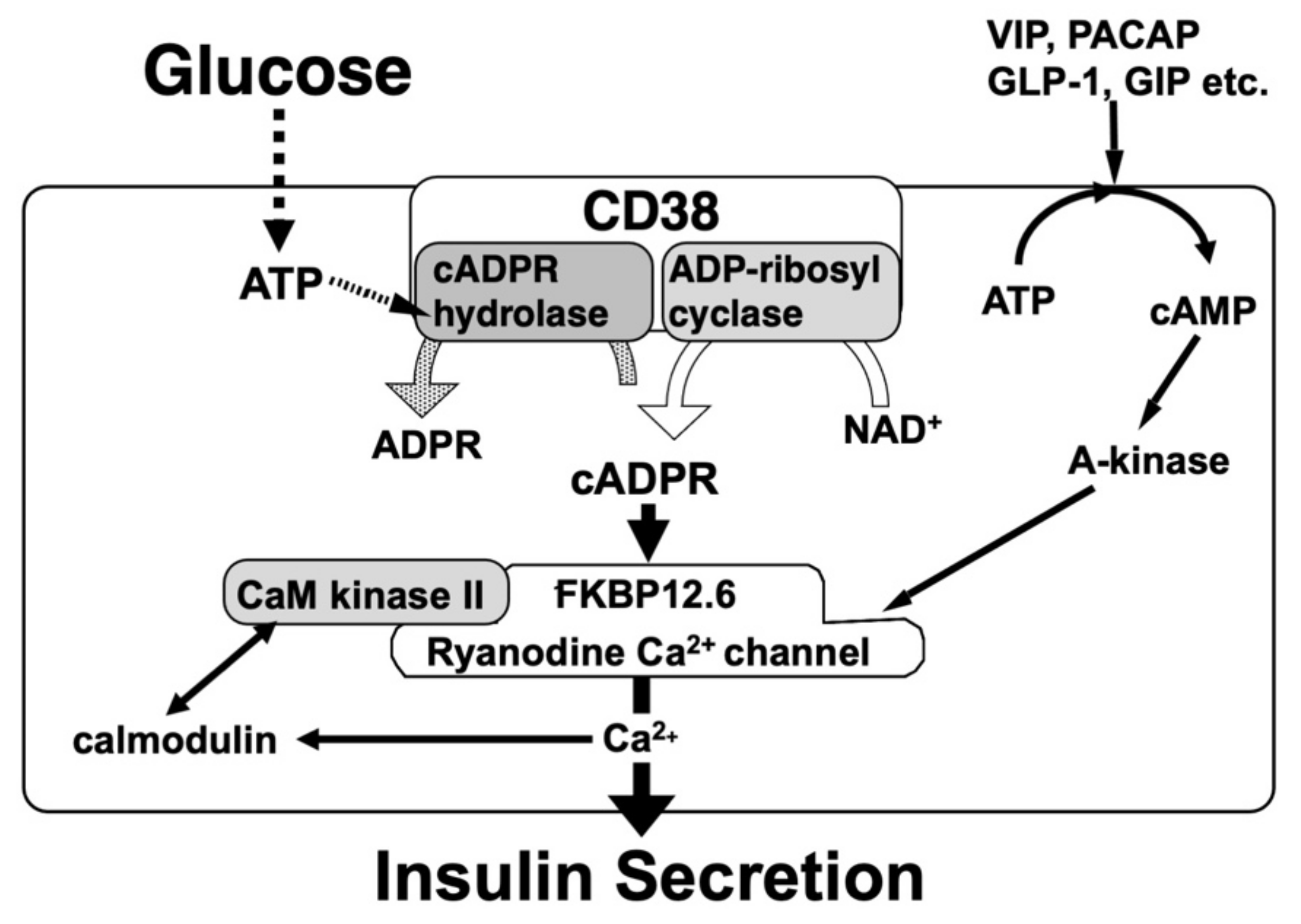
Figure 6.
Usage of “gg” in splice donor sites of rat, mouse, and human RyR2 intron 75 (adapted from [64,68]). RNA splicing in eukaryote follows the “GT-AG” rule by both cis-elements and regulatory trans-acting factors. Donor site dinucleotides of RyR2 intron 75 in rat, mouse, and human are “gg” but not consensus “gt.” In consensus, “M”, “r”, “y”, and “n” represent A or C, a or c, c or t, and a, c, g, or t, respectively.
Figure 6.
Usage of “gg” in splice donor sites of rat, mouse, and human RyR2 intron 75 (adapted from [64,68]). RNA splicing in eukaryote follows the “GT-AG” rule by both cis-elements and regulatory trans-acting factors. Donor site dinucleotides of RyR2 intron 75 in rat, mouse, and human are “gg” but not consensus “gt.” In consensus, “M”, “r”, “y”, and “n” represent A or C, a or c, c or t, and a, c, g, or t, respectively.

Figure 7.
Distribution of relative values of anti-CD38 autoantibodies in 377 Japanese diabetic patients (adapted from [30]). The relative levels of CD38 antibodies are expressed as an index value relative to a standard non-diabetic serum. Values above the mean + 3 SD of the control samples (non-DM) were regarded as antibody positive. The arrow indicates a mean + 3 SD of non-diabetic control. Anti-CD38 positive patients were observed in diabetic patients.
Figure 7.
Distribution of relative values of anti-CD38 autoantibodies in 377 Japanese diabetic patients (adapted from [30]). The relative levels of CD38 antibodies are expressed as an index value relative to a standard non-diabetic serum. Values above the mean + 3 SD of the control samples (non-DM) were regarded as antibody positive. The arrow indicates a mean + 3 SD of non-diabetic control. Anti-CD38 positive patients were observed in diabetic patients.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Takasawa, S. CD38–Cyclic ADP-Ribose Signal System in Physiology, Biochemistry, and Pathophysiology. Int. J. Mol. Sci. 2022, 23, 4306. https://doi.org/10.3390/ijms23084306
AMA Style
Takasawa S. CD38–Cyclic ADP-Ribose Signal System in Physiology, Biochemistry, and Pathophysiology. International Journal of Molecular Sciences. 2022; 23(8):4306. https://doi.org/10.3390/ijms23084306
Chicago/Turabian StyleTakasawa, Shin. 2022. "CD38–Cyclic ADP-Ribose Signal System in Physiology, Biochemistry, and Pathophysiology" International Journal of Molecular Sciences 23, no. 8: 4306. https://doi.org/10.3390/ijms23084306
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.