Author Contributions
Conceptualization, Z.Z. and N.Y.; investigation, Z.Z. and N.Y.; formal analysis, Z.Z., N.Y. and Y.F.; validation, X.Y., K.M., R.S., Z.W., Q.P. and Z.X.; writing—original draft preparation, Z.Z.; writing—review and editing, Z.Z., R.C. and Y.F.; supervision, R.C. and Y.F.; project administration, Z.X.; funding acquisition: X.Y., K.M., R.C. and Y.F. All authors have read and agreed to the published version of the manuscript.
Figure 1.
Expression of NUMB E12-skipping p65/p66 isoforms promotes cancer cell migration. (A) Schematic diagrams of NUMB splice variants. Constitutive exons are shown as grey boxes, whereas green and red boxes stand for alterative exons 6 and 12 (E6 and E12), respectively. (B) Overexpression of NUMB isoforms in HeLa cells. HeLa cells were transiently transfected with indicated expression plasmids. Whole-cell lysates were extracted and analyzed by Western blot. β-actin was used as a loading control. (C) Cell migration assay was performed with HeLa cells, as described in (B). Representative images of migratory cells stained with crystal violet are shown. Scale bar is 200 μm. Quantification was based on the three independent experiments, and results are shown as mean ± SD on the right. (D) Diagrams for detection of NUMB variants including or lacking E12 using RT-PCR. Primer pairs and product sizes for the two variants E12+ and E12− are shown. (E) Inclusion of NUMB E12 and E6 was examined in the six cancer cell lines by RT-PCR. Ratio for E12+/E12− is listed below the panel. (F) Whole-cell lysates were extracted from the six cancer cell lines described in (E) and subject to Western blot analysis using indicated antibodies. Qualification of NUMB isoforms is shown in the right bar graph. ** p < 0.01.
Figure 1.
Expression of NUMB E12-skipping p65/p66 isoforms promotes cancer cell migration. (A) Schematic diagrams of NUMB splice variants. Constitutive exons are shown as grey boxes, whereas green and red boxes stand for alterative exons 6 and 12 (E6 and E12), respectively. (B) Overexpression of NUMB isoforms in HeLa cells. HeLa cells were transiently transfected with indicated expression plasmids. Whole-cell lysates were extracted and analyzed by Western blot. β-actin was used as a loading control. (C) Cell migration assay was performed with HeLa cells, as described in (B). Representative images of migratory cells stained with crystal violet are shown. Scale bar is 200 μm. Quantification was based on the three independent experiments, and results are shown as mean ± SD on the right. (D) Diagrams for detection of NUMB variants including or lacking E12 using RT-PCR. Primer pairs and product sizes for the two variants E12+ and E12− are shown. (E) Inclusion of NUMB E12 and E6 was examined in the six cancer cell lines by RT-PCR. Ratio for E12+/E12− is listed below the panel. (F) Whole-cell lysates were extracted from the six cancer cell lines described in (E) and subject to Western blot analysis using indicated antibodies. Qualification of NUMB isoforms is shown in the right bar graph. ** p < 0.01.
![Ijms 23 04363 g001]()
Figure 2.
Knockdown of p71/p72 isoforms enhanced cancer cell migration by promoting EMT, whereas knockdown of p65/p66 isoforms inhibited cell migration by reversing EMT. (A) Diagram of siRNA targeted against the p71/p72 isoforms (top). Knockdown efficiency was assessed by RT-PCR analysis in HepG2 and RKO cells treated with control siRNA (siNC) and p71/p72 siRNAs (#1 and #2), respectively (bottom). (B) Transwell migration assays were performed using cells described in (A). Representative images of migratory cells stained with crystal violet were shown. Scale bar is 200 μm. The number of migratory cells was quantified, and results are shown as mean ± SD on the right bar graph. (C) Diagram of siRNA targeted against the p65/p66 isoforms (top). Knockdown efficiency was assessed by RT-PCR analysis in SK-Hep-1 and HeLa cells treated with siNC and p65/p66 siRNAs (#1 and #2), respectively (middle and bottom). (D) Transwell migration assays were performed with cells described in (C). Representative images of migratory cells stained with crystal violet were shown on the top. Scale bar is 200 μm. The number of migratory cells was quantified, and results are shown as mean ± SD on the bottom. (E) Western blot analysis was performed using whole-cell lysates isolated from RKO and HeLa cells described in (A,C). ** p < 0.01, *** p < 0.001.
Figure 2.
Knockdown of p71/p72 isoforms enhanced cancer cell migration by promoting EMT, whereas knockdown of p65/p66 isoforms inhibited cell migration by reversing EMT. (A) Diagram of siRNA targeted against the p71/p72 isoforms (top). Knockdown efficiency was assessed by RT-PCR analysis in HepG2 and RKO cells treated with control siRNA (siNC) and p71/p72 siRNAs (#1 and #2), respectively (bottom). (B) Transwell migration assays were performed using cells described in (A). Representative images of migratory cells stained with crystal violet were shown. Scale bar is 200 μm. The number of migratory cells was quantified, and results are shown as mean ± SD on the right bar graph. (C) Diagram of siRNA targeted against the p65/p66 isoforms (top). Knockdown efficiency was assessed by RT-PCR analysis in SK-Hep-1 and HeLa cells treated with siNC and p65/p66 siRNAs (#1 and #2), respectively (middle and bottom). (D) Transwell migration assays were performed with cells described in (C). Representative images of migratory cells stained with crystal violet were shown on the top. Scale bar is 200 μm. The number of migratory cells was quantified, and results are shown as mean ± SD on the bottom. (E) Western blot analysis was performed using whole-cell lysates isolated from RKO and HeLa cells described in (A,C). ** p < 0.01, *** p < 0.001.
![Ijms 23 04363 g002]()
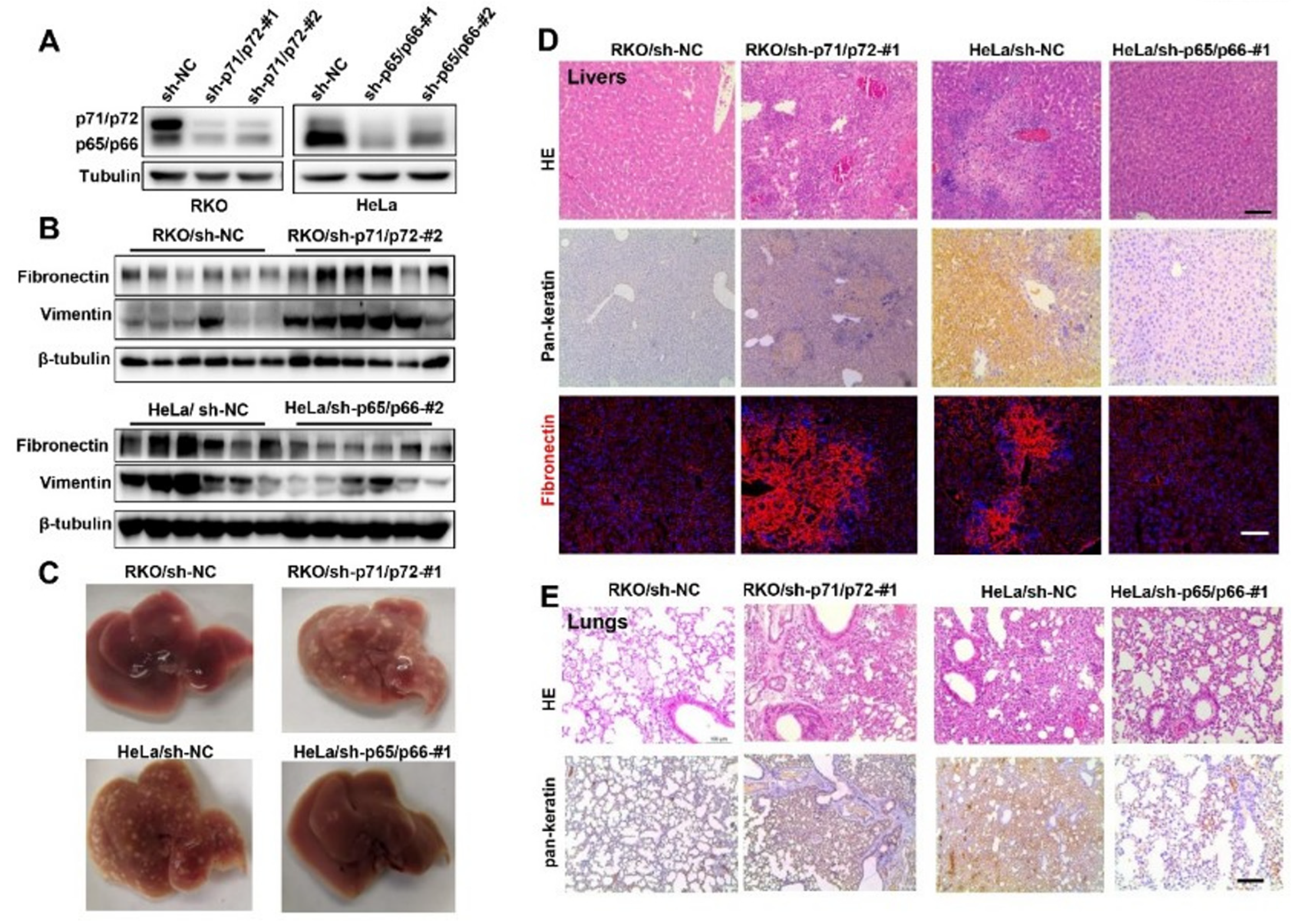
Figure 3.
NUMB isoforms distinctly controlled tumor migration and metastasis in mice. (A) Western blot analysis of NUMB isoform knockdown in stable RKO cells or HeLa cells expressing sh-p71/72 (#1 and #2) or sh-p65/p66 (#1 and #2), respectively. (B) Stable RKO cells (sh-NC and sh-p71/p72-#2) and HeLa cells (sh-NC and sh-p65/p66-#2) were laterally abdominally injected into nude mice. Three weeks after the injection, livers were harvested, and protein extracts were examined by Western blotting with indicated antibodies. (C) Stable RKO cells (sh-p71/p72-#1 and sh-NC) and stable HeLa cells (sh-NC and sh-p65/p66-#1) were subcutaneously injected into nude mice. Mice were killed at four weeks after the injection, and representative images of livers were shown from the indicated groups. (D) Representative HE staining, IHC staining of pan-keratin and immunostaining of fibronectin were shown from indicated groups. Scale bar is 100 μm. (E) Representative HE staining and immunohistochemical staining images of lung sections prepared from the indicated groups. Scale bar is 100 μm.
Figure 3.
NUMB isoforms distinctly controlled tumor migration and metastasis in mice. (A) Western blot analysis of NUMB isoform knockdown in stable RKO cells or HeLa cells expressing sh-p71/72 (#1 and #2) or sh-p65/p66 (#1 and #2), respectively. (B) Stable RKO cells (sh-NC and sh-p71/p72-#2) and HeLa cells (sh-NC and sh-p65/p66-#2) were laterally abdominally injected into nude mice. Three weeks after the injection, livers were harvested, and protein extracts were examined by Western blotting with indicated antibodies. (C) Stable RKO cells (sh-p71/p72-#1 and sh-NC) and stable HeLa cells (sh-NC and sh-p65/p66-#1) were subcutaneously injected into nude mice. Mice were killed at four weeks after the injection, and representative images of livers were shown from the indicated groups. (D) Representative HE staining, IHC staining of pan-keratin and immunostaining of fibronectin were shown from indicated groups. Scale bar is 100 μm. (E) Representative HE staining and immunohistochemical staining images of lung sections prepared from the indicated groups. Scale bar is 100 μm.
![Ijms 23 04363 g003]()
Figure 4.
Notch1 activation is required for p65/p66-induced EMT and tumor cell migration. (A) RKO cells were transiently transfected with siNC and p71/p72 siRNA (#1 or #2), and HeLa cells were transiently transfected with siNC and p65/p66 siRNA (#1 or #2), followed by Western blotting analysis using indicated antibodies. GAPDH was used as loading controls. (B) Nuclear and cytoplasmic proteins were separately isolated from cells described in (A) and subjected to Western blotting analysis. β-actin and histone3 were used as loading controls. (C) RKO cells were transiently transfected with increasing amounts of a p66-expression plasmid, and HeLa cells were transfected with a p72-expression plasmid, followed by Western blot analysis with indicated antibodies. (D) HeLa cells were co-transfected with p65/p66 siRNA-#1 and an N1ICD-expression plasmid as indicated, followed by the cell migration assay. Representative images of migratory cells stained with crystal violet are shown (top). Scale bar is 200 μm. Quantification of migratory cells is shown as mean ± SD on the left bottom, and Western blot analysis with indicated antibodies is shown on the right bottom. (E) HeLa cells were co-transfected with anti-Notch1 siRNA (siNotch1) and a p66-expression plasmid as indicated, followed by the cell migration assay. Representative images of migratory cells stained with crystal violet are shown (top). Scale bar is 200 μm. Quantification of migratory cells is shown as mean ± SD in the bottom left, and Western blot analysis is shown in the bottom right. ** p < 0.01, *** p < 0.001, NS p > 0.05.
Figure 4.
Notch1 activation is required for p65/p66-induced EMT and tumor cell migration. (A) RKO cells were transiently transfected with siNC and p71/p72 siRNA (#1 or #2), and HeLa cells were transiently transfected with siNC and p65/p66 siRNA (#1 or #2), followed by Western blotting analysis using indicated antibodies. GAPDH was used as loading controls. (B) Nuclear and cytoplasmic proteins were separately isolated from cells described in (A) and subjected to Western blotting analysis. β-actin and histone3 were used as loading controls. (C) RKO cells were transiently transfected with increasing amounts of a p66-expression plasmid, and HeLa cells were transfected with a p72-expression plasmid, followed by Western blot analysis with indicated antibodies. (D) HeLa cells were co-transfected with p65/p66 siRNA-#1 and an N1ICD-expression plasmid as indicated, followed by the cell migration assay. Representative images of migratory cells stained with crystal violet are shown (top). Scale bar is 200 μm. Quantification of migratory cells is shown as mean ± SD on the left bottom, and Western blot analysis with indicated antibodies is shown on the right bottom. (E) HeLa cells were co-transfected with anti-Notch1 siRNA (siNotch1) and a p66-expression plasmid as indicated, followed by the cell migration assay. Representative images of migratory cells stained with crystal violet are shown (top). Scale bar is 200 μm. Quantification of migratory cells is shown as mean ± SD in the bottom left, and Western blot analysis is shown in the bottom right. ** p < 0.01, *** p < 0.001, NS p > 0.05.
![Ijms 23 04363 g004]()
Figure 5.
NUMB p71/p72 isoforms promoted ubiquitylation of N1ICD and its degradation. (A) RKO cells were transiently transfected with si-p71/72-#1 for 48 h, then incubated in the presence of cycloheximide (CHX, 50 μg/mL) for the indicated time course, followed by Western blotting analysis. Quantification of Notch1 proteins is shown at the bottom. (B) HeLa cells were transiently transfected with si-p65/p66-#1 for 48 h, then incubated in the presence of cycloheximide (CHX, 50 μg/mL) for the indicated time course, followed by Western blotting analysis. Quantification of Notch1 proteins is shown on the bottom. (C) 293T cells were co-transfected with Flag-N1ICD, Ubiquitin (Ub) and NUMB isoform expression plasmids. Whole-cell lysates were extracted and immunoprecipitated with anti-Flag antibodies followed by Western blotting analysis. Overall, 10% of whole cell lysates were used as input. * p < 0.05, ** p < 0.01.
Figure 5.
NUMB p71/p72 isoforms promoted ubiquitylation of N1ICD and its degradation. (A) RKO cells were transiently transfected with si-p71/72-#1 for 48 h, then incubated in the presence of cycloheximide (CHX, 50 μg/mL) for the indicated time course, followed by Western blotting analysis. Quantification of Notch1 proteins is shown at the bottom. (B) HeLa cells were transiently transfected with si-p65/p66-#1 for 48 h, then incubated in the presence of cycloheximide (CHX, 50 μg/mL) for the indicated time course, followed by Western blotting analysis. Quantification of Notch1 proteins is shown on the bottom. (C) 293T cells were co-transfected with Flag-N1ICD, Ubiquitin (Ub) and NUMB isoform expression plasmids. Whole-cell lysates were extracted and immunoprecipitated with anti-Flag antibodies followed by Western blotting analysis. Overall, 10% of whole cell lysates were used as input. * p < 0.05, ** p < 0.01.
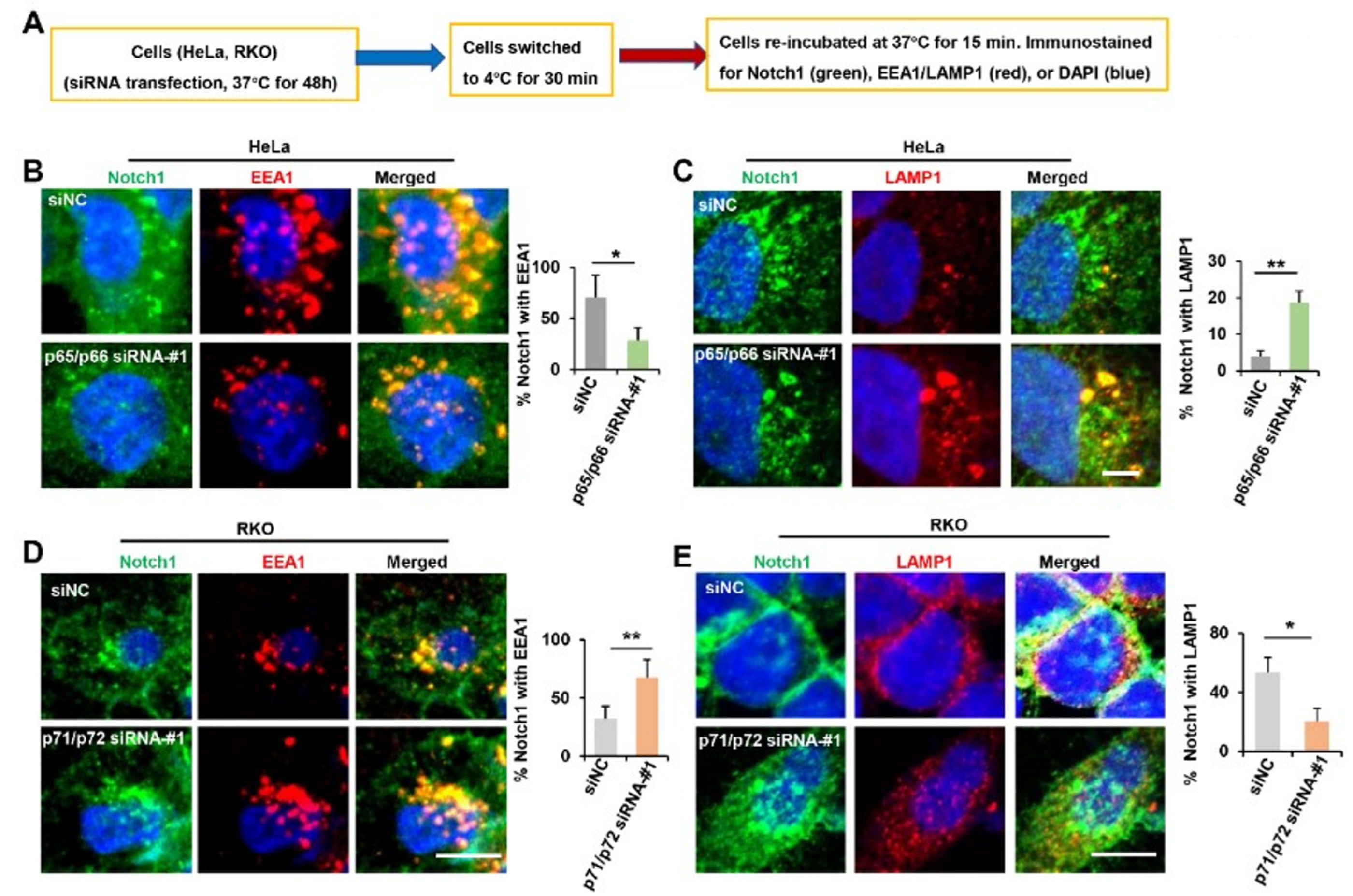
Figure 6.
NUMB isoforms distinctly influence the post-endocytic trafficking of Notch1. (A) Diagram of cell treatment before immunostaining. (B,C) HeLa were treated with siNC and p65/p66 siRNA-#1, as described in (A), and stained with Notch1 (green), EEA1 (red) or LAMP1 (red). Representative confocal images were shown. Scale bar is 10 μm. Percentage of Notch1 colocalized with EEA1 or with LAMP1among the total amount of Notch1 is shown as mean ± SD in the right panel. (D,E) RKO cells were treated with siNC and p71/p72 siRNA-#1, as described in (A), and stained with Notch1 (green), EEA1 (red) or LAMP1 (red). Representative confocal images are shown. Scale bar is 10 μm. Percentage of Notch1 colocalized with EEA1 or with LAMP1 with the total amount of Notch1 is shown as mean ± SD on the right panel. * p < 0.05, ** p < 0.01.
Figure 6.
NUMB isoforms distinctly influence the post-endocytic trafficking of Notch1. (A) Diagram of cell treatment before immunostaining. (B,C) HeLa were treated with siNC and p65/p66 siRNA-#1, as described in (A), and stained with Notch1 (green), EEA1 (red) or LAMP1 (red). Representative confocal images were shown. Scale bar is 10 μm. Percentage of Notch1 colocalized with EEA1 or with LAMP1among the total amount of Notch1 is shown as mean ± SD in the right panel. (D,E) RKO cells were treated with siNC and p71/p72 siRNA-#1, as described in (A), and stained with Notch1 (green), EEA1 (red) or LAMP1 (red). Representative confocal images are shown. Scale bar is 10 μm. Percentage of Notch1 colocalized with EEA1 or with LAMP1 with the total amount of Notch1 is shown as mean ± SD on the right panel. * p < 0.05, ** p < 0.01.
Figure 7.
Notch1 and SMAD3 interactions medicated NUMB-induced EMT and cancer cell migration. (A) HeLa cells were transfected with the N1ICD expression plasmid, following by immunostaining with indicated antibodies. Representative confocal images are shown. Scale bar is 10 μm. (B) 293T cells were co-transfected with HA-SMAD3, Flag-N1ICD expression plasmids or control vector, as indicated. Whole-cell lysates were extracted and immunoprecipitated with anti-HA antibodies (lanes 1–4) or anti-Flag antibodies (lanes 5–8), followed by Western blot analysis. (C) 293T cells were co-transfected with a HA-SMAD3-expression plasmid and increasing amounts of Flag-N1ICD expression plasmids (lanes 1–3), or with a Flag-N1ICD expression plasmid and increasing amounts of HA-SMAD3 expression plasmids (lanes 4–6). Whole-cell lysates were extracted and subjected to Western blotting with indicated antibodies. Overall, 10% of whole cell lysates were used as input. (D) 293T cells were transiently transfected with the N1ICD expression plasmid for 48 h, followed by the addition of cycloheximide (CHX, 50 μg/mL), and Western blotting analysis. Quantification of SMAD3 proteins is shown at the bottom. (E) 293T cells were transiently transfected with the SMAD3 expression plasmid for 48 h, followed by the addition of cycloheximide (CHX, 50 μg/mL) and Western blotting analysis. Quantification of N1ICD proteins is shown at the bottom. (F) 293T cells were transiently co-transfected with indicated plasmids for 48 h. Whole-cell lysates were extracted and immunoprecipitated with anti-Flag antibodies followed by Western blotting analysis. Overall, 10% of whole cell lysates were used as input. (G) HeLa cells were co-transfected with p65/p66 siRNA-#1 and a SMAD3-expression plasmid, followed by the cell migration assay. Representative images of migratory cells stained with crystal violet were shown on the top. Scale bar is 360 μm. Quantification of migratory cells as mean ± SD was shown on the left bottom. Western blot analysis was shown on the right bottom. * p < 0.05, ** p < 0.01, *** p < 0.001, NS p > 0.05.
Figure 7.
Notch1 and SMAD3 interactions medicated NUMB-induced EMT and cancer cell migration. (A) HeLa cells were transfected with the N1ICD expression plasmid, following by immunostaining with indicated antibodies. Representative confocal images are shown. Scale bar is 10 μm. (B) 293T cells were co-transfected with HA-SMAD3, Flag-N1ICD expression plasmids or control vector, as indicated. Whole-cell lysates were extracted and immunoprecipitated with anti-HA antibodies (lanes 1–4) or anti-Flag antibodies (lanes 5–8), followed by Western blot analysis. (C) 293T cells were co-transfected with a HA-SMAD3-expression plasmid and increasing amounts of Flag-N1ICD expression plasmids (lanes 1–3), or with a Flag-N1ICD expression plasmid and increasing amounts of HA-SMAD3 expression plasmids (lanes 4–6). Whole-cell lysates were extracted and subjected to Western blotting with indicated antibodies. Overall, 10% of whole cell lysates were used as input. (D) 293T cells were transiently transfected with the N1ICD expression plasmid for 48 h, followed by the addition of cycloheximide (CHX, 50 μg/mL), and Western blotting analysis. Quantification of SMAD3 proteins is shown at the bottom. (E) 293T cells were transiently transfected with the SMAD3 expression plasmid for 48 h, followed by the addition of cycloheximide (CHX, 50 μg/mL) and Western blotting analysis. Quantification of N1ICD proteins is shown at the bottom. (F) 293T cells were transiently co-transfected with indicated plasmids for 48 h. Whole-cell lysates were extracted and immunoprecipitated with anti-Flag antibodies followed by Western blotting analysis. Overall, 10% of whole cell lysates were used as input. (G) HeLa cells were co-transfected with p65/p66 siRNA-#1 and a SMAD3-expression plasmid, followed by the cell migration assay. Representative images of migratory cells stained with crystal violet were shown on the top. Scale bar is 360 μm. Quantification of migratory cells as mean ± SD was shown on the left bottom. Western blot analysis was shown on the right bottom. * p < 0.05, ** p < 0.01, *** p < 0.001, NS p > 0.05.
![Ijms 23 04363 g007]()


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






