
Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines


1
Masonic Cancer Center, University of Minnesota, Minneapolis, MN 55455, USA
2
Department of Medicinal Chemistry, College of Pharmacy, University of Minnesota, Minneapolis, MN 55455, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(9), 5109; https://doi.org/10.3390/ijms23095109
Submission received: 29 March 2022
/
Revised: 29 April 2022
/
Accepted: 30 April 2022
/
Published: 4 May 2022
(This article belongs to the Special Issue N-Nitroso Compounds: Biological and Environmental Significance and Defense Mechanisms)
Abstract
:The tobacco-specific N-nitrosamines 4-(N-nitrosomethylamino)-1-(3-pyridyl)-1-butanone (NNK) and N′-nitrosonornicotine (NNN) always occur together and exclusively in tobacco products or in environments contaminated by tobacco smoke. They have been classified as “carcinogenic to humans” by the International Agency for Research on Cancer. In 1998, we published a review of the biochemistry, biology and carcinogenicity of tobacco-specific nitrosamines. Over the past 20 years, considerable progress has been made in our understanding of the mechanisms of metabolism and DNA adduct formation by these two important carcinogens, along with progress on their carcinogenicity and mutagenicity. In this review, we aim to provide an update on the carcinogenicity and mechanisms of the metabolism and DNA interactions of NNK and NNN.
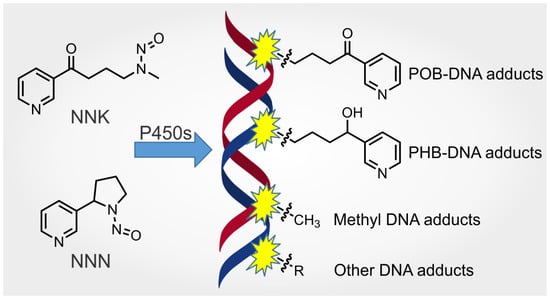
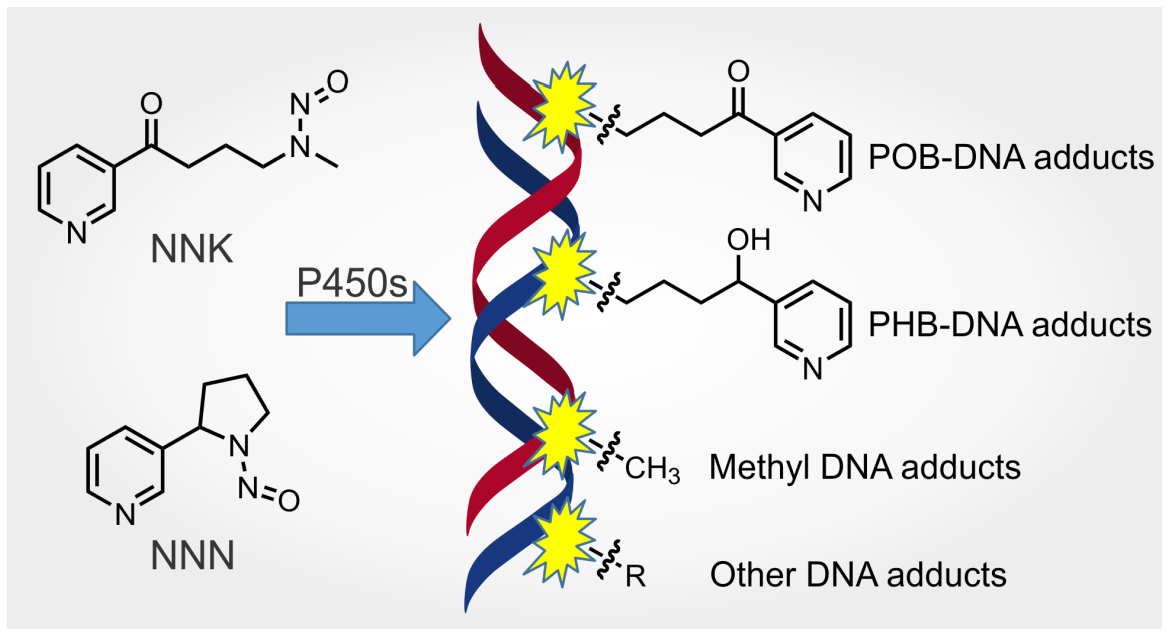
1. Introduction
Tobacco use remains the leading preventable cause of cancer death [1]. Among over 80 carcinogens identified in tobacco and tobacco smoke [2], the tobacco-specific N-nitrosamines (TSNAs) 4-(N-nitrosomethylamino)-1-(3-pyridyl)-1-butanone 5 (NNK, Scheme 1) and N′-nitrosonornicotine 8 (NNN) are particularly noteworthy carcinogens that have been linked to lung, oral cavity, and esophageal cancers in tobacco users. While the levels of commonly occurring nitrosamines, such as N-nitrosodimethylamine (NDMA) and N-nitrosopyrrolidine (NPYR), are generally fairly low in tobacco products and tobacco smoke, NNN and NNK are consistently present in remarkably higher abundance.
The formation of NNK and NNN mainly result from the nitrosation of their precursor amines in tobacco pseudooxy nicotine 4 and nornicotine 7 [3,4]. They can also be formed by nitrosation of nicotine 1, the overwhelmingly abundant tobacco alkaloid, which is not carcinogenic but is highly addictive [5].
Since the comprehensive 1998 review of the biochemistry, biology and carcinogenicity of NNK and NNN [6], significant progress has been made in our understanding the of the metabolism and DNA interactions of these nitrosamines, while several reviews have discussed aspects of the occurrence, metabolism, biomarker and relevant cancer etiology studies of these two carcinogens [7,8,9,10,11], but no comprehensive review of NNK and NNN metabolism and DNA adduct formation has been published. In this paper, we aim to provide an update on the metabolism and DNA interactions of NNK and NNN. Recent carcinogenicity and mutagenicity data are also included.
2. Human Exposure to Carcinogenic Tobacco-Specific N-Nitrosamines
The major source of human exposure to total N-nitrosamines in the U.S. is the consumption of tobacco products, which is estimated to be maximally 25,000 ± 4950 ng per day, in sharp contrast to other sources from food (1900 ± 380 ng/day), alcohol (1000 ± 200 ng/day) and drinking water (120 ± 24 ng/day). The tobacco-specific N-nitrosamines NNN, NNK, N′-nitrosoanatabine (NAT) and N′-nitrosoanabasine (NAB) predominated among the total N-nitrosamines reported in tobacco and tobacco smoke [12].
Since the first identification of NNN in unburned tobacco and tobacco smoke in the 1970s [13,14], multiple studies have evaluated the levels of TSNAs in various tobacco products [2]. In one recent study of 50 U.S. domestic cigarette products, the levels of these nitrosamines in tobacco filler were (mean ± S.D.): NNN, 1900.7 ± 359; NNK, 522.8 ± 157.8; NAB, 72.6 ± 12.4; and NAT, 1386.5 ± 261 ng/g tobacco. In mainstream smoke, they were: NNN, 85 ± 31; NNK, 55 ± 20; NAB, 11.7 ± 3.5; and NAT, 80 ± 28 ng/cigarette. The levels in the mainstream smoke of a 2R4F reference cigarette were: NNN, 133.1 ± 12.5; NNK, 115.6 ± 8.7; NAB, 11.7 ± 3.5; and NAT 119 ± 8.5 ng/cigarette [15]. Other recent studies have compared the levels of TSNAs in smokeless tobacco products consumed in different parts of the world [16,17]. The levels of NNN plus NNK were higher in snus products sold in the U.S. than in those sold in northern Europe [17], while the highest levels of these carcinogens in smokeless tobacco products were found in South-East Asia [9]; for example, the levels of total TSNAs in 22 zarda brands and 4 gul brands ranged 6.3–114 µg/g and 35–56 µg/g tobacco powder, respectively [16]. The high levels of these carcinogens, particularly NNN, in the smokeless tobacco products consumed in South-East Asia are especially notable, since (S)-NNN, the enantiomer that predominates in smokeless tobacco, is a powerful oral-cavity carcinogen in rats. Thus, NNN likely plays an important role in oral cancer induction in smokeless tobacco users. NNK, on the other hand, is a potent inducer of lung tumors in all the animal species tested, independently of the route of administration at low doses. Collectively, the extensive data on the NNN and NNK levels in tobacco products, coupled with the demonstrated carcinogenicity of these products in humans, led to the IARC classification of NNN and NNK, which always occur together in tobacco products, as “carcinogenic to humans” [18].
3. Metabolism and DNA Adduct Formation of NNK and Its Metabolite, NNAL
3.1. Occurrence and Carcinogenicity
NNK is considered to mainly result from the nitrosation of pseudooxy nicotine 4 (Scheme 1), which is present in tobacco in both free and matrix-bound forms [3]. It also results from reactions with nicotine during the curing process. The reaction of nicotine and nitrite in vitro leads to the formation of NNK, with a favorable pH range of 3.4–4.5 [5,19].
Laboratory animal studies clearly demonstrate that NNK is a strong carcinogen, with a preference for inducing tumors of the lung [20]. NNK causes lung tumors in mice, rats and Syrian golden hamsters regardless of the administration pathway, including i.p. injection, s.c. injection, oral dosing in drinking water and oral swabbing [6,21]. NNK also induces tumors in the nasal cavity, liver and pancreas of rats [6]. Rat pancreatic tumors resulted from the metastasis of lung cancer induced by NNK and its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol 11 (NNAL, Scheme 2) [22]. Pancreatic tumorigenesis in NNK-treated rats was enhanced and accelerated by dietary fat [23]. To evaluate the contribution of NNAL enantiomers to the carcinogenicity of NNK, a 90-week rat study with NNK, (S)- or (R)-NNAL was conducted. The three carcinogens were administered in the drinking water at concentrations of 5 ppm. Racemic NNAL was also tested at 10 ppm in the same study. The target organs were the lung and the pancreas. NNK, (S)-NNAL and (R)-NNAL induced lung tumors in nearly all the rats tested. NNK was significantly more carcinogenic than both the NNAL enantiomers (p < 0.001) based on the severity of the lung lesions. The difference between (S)-NNAL and (R)-NNAL was not statistically significant [24].
3.2. Metabolism
3.2.1. Metabolic Activation and Detoxification Pathways
NNK is extensively bioactivated through multiple metabolic pathways (Scheme 2). The major metabolic activation pathways are α-hydroxylation (α-methyl and α-methylene) and carbonyl reduction, followed by α-hydroxylation, while N-oxidation and glucuronidation are considered as detoxification pathways [6].
The carbonyl reduction of NNK to NNAL is catalyzed by several enzymes, including carbonyl reductases, aldo-keto reductases (AKRs) and 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) [25]. This conversion occurs predominately over the other metabolic pathways in nearly all studies in mice, rats, hamsters, patas monkeys and humans [26,27]. In rat pulmonary cells in vitro, carbonyl reduction predominated in all the tested cell types [28]. In A/J mouse lung explants, ~60% of NNK was converted to NNAL after 24 h incubation [29]. In explanted human tissues, NNAL constituted over 90% of the amount of radioactivity from NNK [30]. However, ~10–20% of NNAL is oxidized back to NNK by P450s in A/J mice and rats [31,32,33]. The administration pathways affect the metabolic conversion rates of NNK to NNAL. The conversion appeared to be more efficient via nose-only inhalation compared to i.p. injection and oral gavage [34]. Comparative carcinogenicity studies in A/J mice indicated that NNAL is a strong carcinogen comparable to or slightly weaker than NNK [29,35,36]; similar results were obtained in the carcinogenicity study in rats noted above [24]. Qualitatively and quantitatively similar results were also observed in the levels of DNA and hemoglobin adduct formation in rats treated with NNK and NNAL [31].
The primary bioactivation mechanism of NNK and NNAL is thought to be αhydroxylation. Both NNK and NNAL can be oxidized at the carbons adjacent to the nitroso group, catalyzed primarily by P450 2A13, which is principally expressed in the human lung and nasal cavity [35]. For NNK α-methyl hydroxylation, the intermediate α-hydroxymethylNNK 17 (Scheme 2) is formed and can be detected in a stable form as its O-glucuronide [36]. It generates two intermediates—formaldehyde (23) and the unique diazohydroxide 22. Formaldehyde can be further converted to formic acid and, ultimately, carbon dioxide; diazohydroxide 22 can form the highly reactive species pyridyloxobutyl (POB) diazonium ion 28, which pyridyloxobutylates DNA. For NNK α-methylene hydroxylation, two intermediates, 4-oxo-4-(pyridin-3-yl)butanal 24 (OPB) and methane diazohydroxide 25, are formed. The aldehyde intermediate 24 can be further converted to keto acid 29, which is reduced to hydroxy acid 30. The methane diazohydroxide intermediate 25 spontaneously loses one molecule of H2O and generates the alkylating species methyldiazonium ion 31, which attacks DNA and forms methyl DNA adducts. Similarly, for the α-methyl hydroxylation of NNAL, formaldehyde and the unique pyridylhydroxybutyl (PHB) diazonium ion 32 are formed, with the latter forming PHB DNA adducts that are specific to NNK/NNAL exposure. The α-methylene hydroxylation of NNAL generates the same methyldiazonium ion 31 as is formed from NNK. The other metabolite, 4-hydroxy-4-(pyridine-3-yl)butanal (26), can be further metabolized to hydroxy acid 30. Clearly, α-hydroxylation is the major metabolic pathway to bioactivate NNK/NNAL to exert their carcinogenicity. The highly DNA-reactive electrophiles, including POB diazonium ion 28, PHB diazonium ion 32, methyldiazonium ion 31 and, probably, the aldehydes, such as formaldehyde 23 and OPB 24, are considered responsible for the mutagenicity and carcinogenicity of NNK [6,37].
The N-oxidation of the pyridine ring of NNK/NNAL also occurs in vivo. The enzyme responsible for catalyzing the formation of pyridyl-N-oxides 7 and 12 is not fully characterized. P450 2B1 is the principal responsible enzyme in rat livers but not the lungs; P450 3A4 is suggested to be associated with the formation of the N-oxides of NNK and NNAL in the human liver [6]. NNK-N-oxide is significantly less tumorigenic than NNK and NNAL in mice, and is thus considered a detoxification metabolite [29]. The levels of NNK-N-oxide and NNAL-N-oxide have been quantified in human urine samples. While NNK-N-oxide was not detected in smokers’ urine, NNAL-N-oxide occurred at 0.53 ± 0.36 and 0.41 ± 0.35 pmol/mg creatinine in the urine of smokers and smokeless tobacco users, respectively. This indicates that the pyridine-N-oxidation metabolites of NNK/NNAL only represent a relatively minor detoxification pathway of NNK/NNAL metabolism in humans [38].
The glucuronidation of NNAL represents one of the most important detoxification pathways of NNK/NNAL metabolism. In 1993, we first reported the detection of NNAL glucuronides in the urine of smokers [39]. These metabolites were later widely detected in human samples, such as urine, blood, saliva and amniotic fluid [40,41,42,43,44,45,46,47]. Two glucuronides of NNAL have been identified: 4-(methylnitrosamino)-1-(3-pyridyl)-1-(O-β-D-glucopyranuronosyl)butane 13 (NNAL-O-Gluc) and 4-(methylnitrosamino)-1-(3-pyridyl-N-β-D-glycopyranuronosyl)-1-butanolonium inner salt 14 (NNAL-N-Gluc). NNAL-N-Gluc comprised 50% of the total NNAL-glucuronides in the urine of smokers and 24% in the urine of snuff-dippers [48]. The average level of total NNAL (free NNAL plus NNAL glucuronides) in the urine of 2641 smokers was 1.65 ± 2.13 pmol/mL, of which NNAL-O-Gluc and NNAL-N-Gluc accounted for 48 ± 15% and 22 ± 14% of the total NNAL, respectively [49].
NNK and NNAL are also biotransformed through some minor pathways. Hydroxylation occurring on the pyridine ring of NNK leads to the formation of 4-(methylnitrosamino)-1-(3-(6-hydroxypyridyl))-1-butanone 8 (6-hydroxyNNK), which is a minor metabolite representing approximately 1% of the urinary metabolites in NNK-treated rats and mice [50]. Nicotinamide adenosine dinucleotide phosphate (NADP) analogs of NNK (10, (NNK)ADP+) and NNAL (15, (NNAL)ADP+) were observed in the incubation mixtures with rat liver and pancreatic microsomes. They were suggested to be products of the transglycosylation reactions catalyzed by microsomal NAD+ glycohydrolases [51]. The denitrosation of NNK, a putative detoxification pathway, was observed with the release of nitrite in rat liver microsomal incubations [52]. The decomposition products of NNK denitrosation were proposed to be OPB 24 and myosmine 6 by analogy to the α-radical denitrosation mechanism of NDMA [53]. A new P450-mediated oxidation metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1,4-butanedione (9, CH2-oxo-NNK), was detected in vitro with low conversion rates and catalyzed by P450 2A13 [54]. It can methylate DNA in vitro. No formation of 4-(nitrosoformamido)-1-(3-pyridyl)-1-butanone (CH3-oxo-NNK) was observed in the same incubation mixture, possibly due to its relatively short half-life (6.7 min) compared to that of the CH2-oxo-NNK (35.5 min) [54].
The end metabolites of NNK are primarily keto acid 29 and hydroxy acid 30. In F344 rats, over 99% of gavaged NNK was metabolized in 48 h. The major ultimate metabolite was keto acid (38%), followed by hydroxy acid (14%), NNAL (10%) and NNK-N-oxide (3%) [55]. In male hamsters given s.c. injections of NNK, the major end metabolites in 48-h urine samples were hydroxy acid (16.8%), keto acid (8.4%), NNAL (6.9%). No NNK-N-oxide was detected [56]. In 20 smokers who smoked cigarettes containing [pyridine-d4]NNK for one week, ~86% of NNK was metabolically activated, with [pyridine-d4]keto acid and [pyridine-d4]hydroxy acid as the two principal end metabolites in the urine. The [pyridine-d4]keto acid contributed an average of 28.2% to the sum of these two metabolites [57]. However, in a second study, [pyridine-d4]hydroxy acid was found at an average level of 130 (range: 25–390) fmol/mL, lower than in the first study, in the urine of 87 smokers who smoked cigarettes spiked with [pyridine-d4]NNK for 1 week [58]. The high reduction rate of keto acid to hydroxy acid in humans (85%) was in sharp contrast to that in rats (~1%) [59,60].
3.2.2. Stereochemistry of NNAL and Its Glucuronides
Due to the chiral δ-carbon (carbinol carbon), there are two NNAL enantiomers—(S)-NNAL (11-(S)) and (R)-NNAL (11-(R)) (Scheme 3). The stereochemical aspects of NNAL formation by NNK metabolism have been investigated. In rodent tissues, including microsomes and cytosol from male F344 rat livers and lungs and female A/J mouse livers and lungs, (S)-NNAL predominated at 90–98% of NNAL formed by NNK. In human tissues, including liver microsomes, liver cytosol and red blood cells, (S)-NNAL predominated at 64, 90 and >95% of NNAL, respectively [61]. The preferential formation of (S)-NNAL over its (R)-counterpart is largely due to the high enantioselectivity of NNK reductases [25]. The aldo-keto reductases (AKRs) and carbonyl reductase purified from human liver cytosol produced >90% (S)-NNAL, in contrast to the formation of 35% (R)-NNAL by 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) purified from human liver microsomes. Interestingly, human lung and placenta microsomes catalyzed NNK to NNAL with high selectivity to form (R)-NNAL, with a production of 89% and 87% of total NNAL, respectively [25]. The metabolism of (S)- and (R)-NNAL appears not to be enantioselective in human liver microsomes, even though some selectivity was observed in rat lung microsomes, where (S)-NNAL was preferentially oxidized to its metabolites. Both NNAL enantiomers were metabolized at similar rates by all the oxidative pathways in human liver microsomes [61].
The enantiomeric distribution of NNAL is intriguing. (R)-NNAL predominated in the bile or urine of rats treated with either NNK or NNAL. However, (S)-NNAL was significantly retained in the lungs of rats, comprising an (S)-/(R)-ratio of >20 after 24 h administration of racemic NNAL [62]. (S)-NNAL was also retained in humans, suggesting the existence of an unknown receptor for this enantiomer, possibly in the lungs [63]. (S)-NNAL accounted for 54% of NNAL enantiomers in the urine of human smokers (note that the original assignment of the stereochemistry of the two NNAL enantiomers was reversed. The corrected information was published in a 2000 corrigendum) [64,65].
The diastereomeric distribution of the NNAL glucuronide NNAL-O-Gluc has also been investigated. After the chiral derivatization with Mosher’s reagent, stereochemical configurations of the two NNAL-O-Gluc diastereomers were established [65,66]. (S)-NNAL-O-Gluc predominated in rat urine, while (R)-NNAL-O-Gluc was the major diastereomer in the bile of rats treated with either NNK or NNAL [62,67]. In the urine of patas monkeys and humans, (S)-NNAL-O-Gluc was the predominant diastereomer [26,66,67,68]. For example, it represented 68% of urinary NNAL-O-Gluc in cigarette smokers [64,65].
The stereochemical studies of NNK and NNAL metabolism may provide some explanations for the different carcinogenicities of NNAL enantiomers. In a study in A/J mice, (S)-NNAL showed equal lung tumorigenicity (25.6 ± 7.5 tumors/mouse) to NNK (25.3 ± 9.8 tumors/mouse), and was significantly more tumorigenic than racemic NNAL (12.1 ± 5.6 tumors/mouse) or (R)-NNAL (8.2 ± 3.3 tumors/mouse) [32]. The underlying mechanism was considered to be the preferential pulmonary metabolic activation of (S)-NNAL and its less effective glucuronidation than that of (R)-NNAL. However, an interspecies difference was observed in our chronic drinking water study with F344 rats. In this study, (S)-NNAL was equally potent to (R)-NNAL in causing lung and pancreatic tumors [24].
3.2.3. Stereochemistry of Hydroxy Acid Formation
Similar to NNAL, hydroxy acid is comprised of two enantiomers due to the chiral δ-carbon (Scheme 3). In F344 rat urine, NNK metabolism produces mainly (R)-hydroxy acid (30-(R)), whereas nicotine metabolism generates the predominant (S)-hydroxy acid (30-(S)) [60,65]. This led to the hypothesis that the ratio of (R)-/(S)-hydroxy acid could be a biomarker specific to NNK metabolism. In F344 rats, hydroxy acid accounted for 12% of the NNK dose, in sharp contrast to the 0.1% of the nicotine dose [60]. This difference was considered sufficiently great to distinguish the origin of (R)-hydroxy acid from NNK metabolism versus from a minor 2′-hydroxylation metabolic pathway of nicotine metabolism via the intermediate 4 [69,70]. High stereoselectivity in the formation of hydroxy acid has been suggested due to the highly enantioselective reduction process catalyzed by carbonyl reductase [68].
However, the hypothesis based on the use of the ratio of (R)-/(S)-hydroxy acid as a urinary biomarker does not hold true in humans. The conversion of nicotine to hydroxy acid is a substantial metabolic pathway in humans, accounting for ~5–6% of the estimated nicotine dose [59,71]. Even though the stereochemistry of urinary hydroxy acid is essentially the same—97% of (S)-hydroxy acid in nicotine-treated rats and 98.7% of (S)-hydroxy acid in human nicotine-patch users [59,65]—the substantially high conversion rate of nicotine to hydroxy acid still makes it impossible to distinguish the origin of (R)-hydroxy acid in human urine due to the overwhelming abundance of nicotine (16.2–26.3 mg/g) in cigarette tobacco [72,73].
3.3. DNA Adducts Formed by NNK Metabolism
As shown in Scheme 4, NNK, via α-methyl hydroxylation, generates the POB diazonium ion 28, which is highly reactive and pyridyloxobutylates DNA. The presence of compounds 33 and 39 as solvolysis products provides solid evidence for its formation [74]. The POB diazonium ion also rearranges and forms two secondary products—cyclic oxonium ion 35 and β-carbocation 36—as demonstrated by the solvolysis product 38 and the dehydrogenation product 40, respectively [74]. After the enzymatic reduction of NNK, the major metabolite NNAL generates the PHB diazonium ion 32 in a similar manner, via the α-methyl hydroxylation pathway. This results in the products 41 and 42, which provide the evidence for its formation. The PHB diazonium ion attacks DNA and forms PHB DNA adducts. It also rearranges to the corresponding β-carbocation 37, which has been suggested by the formation of the internal olefin 43 and the 1,3-diol 44 [74]. Formaldehyde 23 is also formed as a byproduct from the α-methyl hydroxylation of NNK and NNAL. Through the α-methylene hydroxylation pathway, both NNK and NNAL generate the methyldiazonium ion 31.
The major DNA adducts formed by NNK metabolism can be generally classified as POB DNA adducts, PHB DNA adducts and methyl DNA adducts. Each class comprises DNA base adducts and phosphate adducts. The quantitation of several DNA adducts has shown their persistence and accumulation in the target tissues of laboratory animals, suggesting their potential as metabolic activation biomarkers of chronic NNK exposure in human tobacco users.
3.3.1. POB DNA Adducts
- (1)
- HPB-releasing DNA Adducts
The POB DNA adducts formed by [3H]NNK in the liver and lung DNA of F344 rats were first demonstrated by the release of [3H]4-hydroxy-1-(3-pyridyl)-1-butanone ([3H]HPB) upon the acid hydrolysis or neutral thermal hydrolysis of the DNA [75]. [3H]HPB accounted for over 50% of the radioactivity in hepatic DNA [75]. The decomposition of the HPB-releasing DNA adducts 45 (Figure 1) in vitro and in vivo was multiphasic, with detectable levels up to 4 weeks post-s.c.-injection in F344 rats [76]. In male F344 rats, after 4 days of consecutive i.p. injections with NNK (15–5000 μg/kg/day), 18–3400 and 58–2180 fmol HPB/mg DNA were released from rat liver and lung DNA, respectively (Table 1) [77]. HPB-releasing adducts were also quantified in the lung DNA of rats chronically treated with NNK or each of the NNAL enantiomers for up to 70 weeks [24]. The levels of HPB-releasing adducts peaked at 9 ± 3 pmol/mg DNA at weeks 10 and 30 in the NNK treatment group and decreased slightly, to 5 ± 2 pmol/mg DNA, at week 70. In the (S)-NNAL-treated rats, the levels of HPB-releasing adducts in the lung DNA peaked at 11 ± 3 pmol/mg DNA at week 50 and decreased to 5 ± 1 pmol/mg DNA at week 70. The total level of HPB-releasing adducts in the lung DNA of rats in the (R)-NNAL treatment group remained low throughout the 70-week study course, ranging from 1 to 2 pmol/mg DNA [24].
Even though HPB can be formed by the reaction of myosmine 6 (Scheme 2), a minor tobacco alkaloid and dietary component, with NaNO2 under acidic conditions in vitro, HPB-releasing DNA adducts were not detected in rats treated with a combination of myosmine plus NaNO2 [78].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Levels of HPB-releasing adducts quantified in laboratory animal and human tissues.
| Animal Data | |||||||
|---|---|---|---|---|---|---|---|
| Animal Species | Administration Pathway | Exposure Amount | Exposure Duration | Target Tissue DNA | HPB-Releasing Adducts a | Ref. | |
| Female A/J mice | Single i.p. injection | 10 μmol/mouse [5-3H]NNK | 12–144 h | Lung | 8.4 pmol/μmol Gua @ 24 h time point | [79] | |
| Male F344 rats | Once daily i.p. injection | 15–5000 μg/kg/day of [5-3H]NNK or [C3H3]NNK | 4 days | Liver | 18–3400 fmol/mg DNA | [77] | |
| Lung | 58–2180 fmol/mg DNA | ||||||
| Single s.c. injection | 2 μmol/rat [5-3H]NNK | 24 h | Liver | 0.67 ± 0.1 pmol/mg DNA | [75] | ||
| Lung | Present | ||||||
| Single s.c. injection | 0.81 mg/kg of [5-3H]NNK | 24 h | Liver | 2.1 ± 0.1 pmol/μmol Gua | [76] | ||
| Lung | 1.6 pmol/μmol Gua | ||||||
| In the drinking water | 5 ppm NNK | 10 weeks | Lung | 9 ± 3 (pmol/mg DNA) | [24] | ||
| 30 weeks | 9 ± 3 | ||||||
| 50 weeks | 6 ± 1 | ||||||
| 70 weeks | 5 ± 2 | ||||||
| 5 ppm (S)-NNAL | 10 weeks | Lung | 5 ± 3 (pmol/mg DNA) | ||||
| 30 weeks | 10 ± 5 | ||||||
| 50 weeks | 11 ± 3 | ||||||
| 70 weeks | 5 ± 1 | ||||||
| 5 ppm (R)-NNAL | 10 weeks | Lung | 1 ± 0 (pmol/mg DNA) | ||||
| 30 weeks | 1 ± 1 | ||||||
| 50 weeks | 2 ± 1 | ||||||
| 70 weeks | 1 ± 1 | ||||||
| Female A/J mice | Single i.p. injection | 10 μmol/mouse [5-3H]NNN | 24 h | Liver | 22.6 (or 25.1) pmol/μmol Gua | [80] | |
| 11 μmol/mouse [5-3H]NNN | Lung | 5.6 pmol/μmol Gua | [79] | ||||
| Male F344 rats | Single s.c. injection | 0.35 μmol/rat [5-3H]NNN | 24 h | Liver | 0.08 ± 0.1 pmol/mg DNA | [75] | |
| Once daily i.p. injection | 0.407 μmol/rat [5-3H]NNN | 3 days | Liver | 0.54 pmol/mg DNA | [81] | ||
| Lung | 0.50 | ||||||
| Nasal mucosa | 0.02 | ||||||
| Esophagus | <0.005 | ||||||
| Kidney | <0.005 | ||||||
| Human Data | |||||||
| Sample source | Smoking status | Subject number | Tissues | HPB-releasing adducts | Note | Ref. | |
| Autopsy samples | Smokers verified by blood nicotine and cotinine, and medical record if necessary | 9 | Tracheobronchus | 16 ± 18 fmol/mg DNA | [80] | ||
| Peripheral lung tissues | 11 ± 16 | ||||||
| Nonsmokers | 8 | Tracheobronchus | 0.9 ± 1.7 | Only 1 subject had significant HPB levels. | |||
| Peripheral lung tissues | 0.9 ± 2.3 | ||||||
| TobPRAC Biorepository | Smokers (>10 cigs/day for at least 1 year) verified by urinary NNN and NNAL. | 30 | Mouthwash oral cells | 12.0 ± 35.1 pmol/mg DNA | [82] | ||
| Buccal cells | 45 ± 57 | ||||||
| Nonsmokers | 15 | Mouthwash oral cells | 0.23 ± 0.43 | Only 3 subjects had significant HPB levels. | |||
| Patients with HNSCC | Smokers (determined by lifetime tobacco use questionnaire) | 30 | Buccal cells | 8.19 ± 17.8 (median 1.51) pmol/mg DNA | [83] | ||
| Patients without HNSCC | 35 | 4.53 ± 14.36 (median 0.23) | |||||
| Patients with lung cancers | Self-reported smokers | 21 | Peripheral lung tissues | 404 ± 258 fmol/mg DNA | [84] | ||
| Self-reported nonsmokers | 11 | 59 ± 56 | |||||
| Tumor-free sudden death victims | Smokers (>15 ng cotinine/mL blood or >100 ng cotinine/mL urine) | 32 | Lung | 92 ± 148 fmol/mg DNA | Primarily road traffic accidents, suicide and sudden cardiac arrest. | [85] | |
| 29 | Mucosa of esophagus | 138 ± 208 | |||||
| 12 | Mucosa of cardia | 93.6 ± 91.9 | |||||
| Nonsmokers verified by blood or urinary cotinine | 56 | Lung | 61 ± 66 | ||||
| 53 | Mucosa of esophagus | 131 ± 130 | |||||
| 18 | Mucosa of cardia | 117 ± 110 | |||||
| Patients with or without upper gastrointestinal disorders | Self-reported smokers | 7 | Mucosal biopsies of the lower esophagus | 4.80 ± 3.57 pmol/mg DNA | One outlier of patient #55 with ulcerative gastritis was excluded due to its exceptionally high level of HPB-releasing adducts (36.98 pmol/mg DNA) | [86] | |
| Self-reported nonsmokers | 7 | 2.86 ± 2.44 | |||||
a 1 pmol/mg DNA = 1.51 pmol/μmol Gua = 0.33 adducts/106 nucleotides. These conversion factors were calculated based on 1 mg DNA containing approximately 3 μmol nucleotides, whereas dGuo accounted for ~22% of total nucleotides [87].
- (2)
- POB DNA Base Adducts
In an effort to characterize the chemical structures of HPB-releasing DNA adducts, Peterson et al. found that Gua adducts predominated in the reaction mixture of 4-((acetoxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone 16 (NNKOAc, Scheme 2) with different polymers and oligomers [88]. O6-(4-(3-Pyridyl)-4-oxobut-1-yl)-2′-deoxyguanosine 56 (O6-POB-dGuo, Figure 1) was first characterized in calf thymus DNA reacted with NNKOAc as the deglycosylated form, O6-POB-Gua 57 [89]. The major HPB-releasing DNA adducts identified in vitro are two thermally unstable POB DNA base adducts, N7-(4-(3-pyridyl)-4-oxobut-1-yl)-2′-deoxyguanosine 58 (N7-POB-dGuo) and O2-(4-(3-pyridyl)-4-oxobut-1-yl)-2′-deoxycytidine 48 (O2-POB-dCyd) [90,91]. The stable form, N7-POB-Gua 59, accounted for 30–35% of the total HPB-releasing adducts in the DNA treated by NNKOAc [90]. Other adducts that are thermally stable but still release HPB after acid hydrolysis have also been identified in vitro. These include N1-(4-(3-pyridyl)-4-oxobut-1-yl)-2′-deoxyinosine 46 (N1-POB-dIno), N6-(4-(3-pyridyl)-4-oxobut-1-yl)-2′-deoxyadenosine 47 (N6-POB-dAdo), N3-(4-(3-pyridyl)-4-oxobut-1-yl)-2′-deoxycytidine 50 (N3-POB-dCyd), N4-(4-(3-pyridyl)-4-oxobut-1-yl)-2′-deoxycytidine 52 (N4-POB-dCyd), N2-(4-(3-pyridyl)-4-oxobut-1-yl)-2′-deoxyguanosine 53 (N2-POB-dGuo), N2-(2-(3-pyridyl)tetrahydrofuran-2-yl)-2′-deoxyguanosine, or its open chain tautomer (Schiff base) 55, O6-POB-dGuo (56), O2-(4-(3-pyridyl)-4-oxobut-1-yl)thymidine 60 (O2-POB-Thd) and O4-(4-(3-pyridyl)-4-oxobut-1-yl)thymidine 61 (O4-POB-Thd) (Figure 1) [90,91,92,93,94]. A unique dGuo adduct formed by NNKOAc has been identified as N2-(4-(3-pyridyl)-4-oxobut-2-yl)-2′-deoxyguanosine 54 (N2-POBb-dGuo). This adduct was hypothesized to be formed by the rearranged carbocation 36, or the corresponding α,β-unsaturated ketone 40 (Scheme 4) [95].
It is important to note that many adducts identified in vitro have not been detected in vivo. For example, N1-POB-dIno 46 was readily formed in NNKOAc-treated calf thymus DNA but was not detected in rat liver or lung DNA [93]. The Schiff base adduct 55 was only detected in relatively small amounts in the reaction of NNKOAc and dGuo in its reduced form [90]. N3-POB-dCyd 50 and N4-POB-dCyd 52 were formed as minor products in vitro. N3-POB-dCyd was found to easily undergo deamination and form N3-(4-(3-pyridyl)-4-oxobut-1-yl)-2′-deoxyuridine 51 (N3-POB-dUrd) under HPLC purification conditions [92]. The unique N2-POBb-dGuo adduct 54 was not detected in the DNA of rats treated with NNK or NNN [81].
The distribution patterns of POB DNA base adducts in vitro differ greatly from those in vivo. In NNKOAc-treated calf thymus DNA, the relative amounts of the POB DNA adducts were: N7-POB-Gua 59 > O6-POB-dGuo 56 > O2-POB-Thd 60 > O2-POB-Cyt 49 [96]. In the lung DNA of mice administered NNK by a single i.p. dose, N7-POB-Gua was the most abundant POB DNA base adduct, followed by O2-POB-Thd, at slightly lower concentrations. O6-POB-dGuo was substantially lower than O2-POB-Thd. The lowest adduct concentration was that of O2-POB-Cyt [97]. In the liver and lung DNA of rats administered 0.025 or 0.1 mmol/kg body weight of NNK by s.c. injection for 4 consecutive days, N7-POB-Gua and O2-POB-Thd predominated among the four quantified adducts (Table S1). The lowest DNA adduct concentration was that of O6-POB-dGuo [96]. In a 20-week NNK drinking-water study in rats, O2-POB-Thd predominated among the POB DNA adducts detected in rat livers, lungs, nasal respiratory mucosa, nasal olfactory mucosa, oral mucosa and pancreas at each time point. O6-POB-dGuo was only detected in the lung DNA at very low levels [98,99]. A similar trend was observed in our chronic carcinogenicity study of NNK in F344 rats. Throughout the study course of up to 70 weeks, O2-POB-Thd was the most abundant POB DNA base adduct at each time point in NNK-treated rat tissues. O6-POB-dGuo was marginally detectable at very low concentrations [24]. It is noteworthy that the persistence of the POB DNA base adducts varied. For example, the O2-POB-Thd peaked at week 30 with a maximum level of 4809 ± 193 fmol/mg rat lung DNA, while the N7-POB-Gua and the O6-POB-dGuo peaked at week 10, with maximum levels of 970 ± 148 and 34 ± 21 fmol/mg rat lung DNA, respectively [24].
In the same chronic rat study, we also investigated the carcinogenicity of (S)-NNAL and (R)-NNAL. The DNA adducts formed by these two NNAL enantiomers were quantified. The POB DNA base adducts were formed by both enantiomers. However, the concentrations of these POB DNA base adducts were strikingly different. (S)-NNAL produced significantly higher levels of O2-POB-Thd, N7-POB-Gua and O6-POB-dGuo than the (R)-NNAL in all the examined rat tissues at each time point (Table S1). The levels of these three POB DNA base adducts were comparable in the (S)-NNAL treatment group and the NNK treatment group [24]. These results were also consistent with the data from our 20-week rat study with (S)- or (R)-NNAL [98,99]. The difference in the formation of POB DNA base adducts by (S)-NNAL and (R)-NNAL has been proposed to result from (1) the preferential persistence of (S)-NNAL and (2) the higher re-oxidation rates of (S)-NNAL to NNK in the target tissues, such as those of the lungs [98]. Through re-oxidation, NNAL is converted back to NNK, which undergoes the α-methyl hydroxylation pathway, and forms the corresponding POB DNA base adducts.
- (3)
- POB DNA Phosphate Adducts
The phosphate groups in the DNA backbone have free hydroxy groups that can be alkylated by NNK metabolites. Due to the chirality of the phosphorus atom, the POB DNA phosphate adduct 62 (3′ → 5′, Figure 1), if formed, will comprise two or four diastereomers, depending on whether the nucleobases connected to the phosphate group are the same or different. There are up to 10 combinations of nucleobases and, thus, up to 32 diastereomers of POB DNA phosphate adducts that can be formed by DNA phosphate pyridyloxobutylation [100].
In 2002, Tornqvist et al. reported the first evidence of DNA phosphate adduct formation by NNK. The total level of POB DNA phosphate adducts accounted for ~22% of the total pyridyl DNA adducts. However, no structural information was provided using their cob(I)alamin transformation method [101]. We later characterized the chemical structures of POB DNA phosphate adducts using high-resolution mass spectrometry. Up to 30 NNK-derived POB DNA phosphate adducts were characterized in the calf thymus DNA treated with NNKOAc. All the 10 combinations of POB DNA phosphate adducts were detected in the liver and lung DNA of rats treated with NNK [102]. The concentrations of POB DNA phosphate adducts were quantified using the same rat tissues from our chronic carcinogenicity study (Table S1) [24]. The total POB phosphate adducts accounted for 5–9% of the total POB DNA adducts, ranging 89–190 and 218–475 fmol/mg DNA in the rat livers and rat lungs, respectively, over the 70-week study course [103].
The POB DNA phosphate adducts were also formed by (S)-NNAL and (R)-NNAL in the tissues of the rats from the same chronic carcinogenicity study [104]. The (S)-NNAL caused significantly higher levels of POB DNA phosphate adducts than the (R)-NNAL in the rat lung DNA at each time point. The total POB DNA phosphate adducts in the (S)-NNAL treatment group were 1180–4650 fmol/mg lung DNA with the maximum level peaking at 10 weeks. In the (R)-NNAL treatment group, they occurred only at 46–175 fmol/mg lung DNA with no significant changes in the first 50 weeks, followed by decreasing levels at week 70 [104]. This was consistent with the POB DNA base adduct formation in both treatment groups. As discussed above, a high conversion rate of (S)-NNAL re-oxidation to NNK is considered to be responsible for this difference.
The distribution patterns of 10 combinations of POB DNA phosphate adducts were slightly different in the rat livers and lungs. However, adducts such as Cp(POB)C 63 (Figure 1), Ap(POB)C 64 or Cp(POB)A 65 and combinations with Thd appeared to be predominant [102]. Due to the chirality of the phosphorus atom, these adducts may comprise two or four diastereomers. The formation and persistence of the diastereomers were also different. For example, one diastereomer of Tp(POB)T (66-(S) or 66-(R)) remained at low concentrations with a slowly decreasing trend throughout the 70-week study course, while the other diastereomer reached its maximum level at week 30 and remained at nearly the same level up to 70 weeks [102]. This is likely the balanced result of adduct formation and enzymatic repair in vivo. However, the stereochemical aspects of the repair mechanisms of POB DNA phosphate adducts are not well understood [105].
3.3.2. PHB DNA Adducts
As illustrated by Scheme 4, NNK can be reduced to NNAL, which is metabolized to the PHB diazonium ion 32 and the rearranged carbocation 37, which can react with DNA, forming the corresponding PHB DNA base and phosphate adducts.
- (1)
- PHB DNA Base Adducts
To characterize the chemical structures of PHB DNA base adducts, a regiochemically activated form of NNAL—4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol 21 (NNALOAc, Scheme 2)—was reacted with dGuo and DNA in vitro. N7-(4-(3-Pyridyl)-4-hydroxybut-1-yl)-2′-deoxyguasosine 72 (N7-PHB-dGuo, Figure 2) and its stable product, N7-(4-(3-pyridyl)-4-hydroxybut-1-yl)guanine 73 (N7-PHB-Gua), together with O6-(4-(3-pyridyl)-4-hydroxybut-1-yl)-2′-deoxyguanosine 71 (O6-PHB-dGuo) and N2-(4-(3-pyridyl)-4-hydroxybut-1-yl)-2′-deoxyguanosine 70 (N2-PHB-dGuo), were unambiguously detected in these reactions [106]. When NNALOAc reacted with dCyd, Thd and DNA, the adducts to the pyrimidine rings were identified as O2-(4-(3-pyridyl)-4-hydroxybut-1-yl)cytidine 69 (O2-PHB-Cyt), the stable form of O2-PHB-dCyd 68 and O2-(4-(3-pyridyl)-4-hydroxybut-1-yl)thymidine 74 (O2-PHB-Thd) [91]. The dAdo-derived PHB adduct formed in vivo by NNK or NNAL was characterized as N6-(4-(3-pyridyl)-4-hydroxybut-1-yl)-2′-deoxyadenosine 67 (N6-PHB-dAdo) [93]. A minor adduct, O4-(4-(3-pyridyl)-4-hydroxybut-1-yl)thymidine 75 (O4-PHB-Thd), was also detected in the cellular genomic DNA exposed to NNALOAc in vitro [107].
In the target tissues of rats treated with 10 ppm NNK or each of the two NNAL enantiomers in the drinking water for 20 weeks, three major PHB DNA base adducts, O2-PHB-Thd 74, N7-PHB-Gua 73 and O6-PHB-dGuo 71, were quantified. As shown in Table S1, O2-PHB-Thd predominated among the three PHB DNA base adducts in all the rat tissues. The highest concentrations of O2-PHB-Thd were observed in the nasal respiratory mucosa of all three treatment groups. Similarly, N7-PHB-Gua and O6-PHB-dGuo were also found in their highest levels in the nasal respiratory mucosa in the NNK treatment group and the (S)-NNAL treatment group. However, the lung tissue contained nearly equal or even higher concentrations of the three PHB DNA base adducts compared with the nasal respiratory mucosa in the rats from the (R)-NNAL treatment group. More strikingly, the total PHB DNA base adducts formed in the (R)-NNAL treatment group exceeded, to a significantly greater extent, those from each of the other two groups [99,108]. This difference in PHB DNA base adduct distributions across the different treatment groups was consistent with that of the POB DNA base adducts formed by NNK or NNAL enantiomers. This was hypothesized to have been due to the different metabolic profile of (R)-NNAL, which has a slower conversion rate to NNK [108]. NNK- and (S)-NNAL-PHB DNA base adducts were also detected in the mitochondrial DNA in the lungs and livers of rats from the same study. The levels of total POB- and PHB-DNA base adducts in the lung mitochondrial DNA (mtDNA) were higher than those in the nuclear DNA (nDNA). No difference was observed in the liver DNA [109].
In the lungs and pancreas (and liver, in some cases) of rats chronically administered NNK or (S)-NNAL or (R)-NNAL in drinking water for up to 70 weeks, the adduct-formation pattern was consistent with the 20-week rat study. As shown in Table S1, O2-PHB-Thd predominated among the three quantified adducts (O2-PHB-Thd, O6-PHB-dGuo and N7-PHB-Gua) in the rat lung DNA in all three treatment groups. It persisted at the highest concentrations of 4009–6581 fmol/mg DNA throughout the 70 weeks in the (R)-NNAL treatment group, while it still occurred at high levels of ~1000 fmol/mg DNA in the NNK or (S)-NNAL treatment group (Figure 3). A similar pattern was observed for N7-PHB-Gua, which persisted in significantly higher concentrations in the (R)-NNAL treatment group than in the other two. However, it did not persist throughout the 70-week study course, decreasing from ~860 to ~380 fmol/mg DNA at week 50. In the pancreatic DNA, both O2-PHB-Thd and N7-PHB-Gua persisted at significantly lower levels, but with a highly similar distribution pattern, as observed in the lung DNA [24]. The levels of N6-PHB-dAdo in the liver and lung DNA of rats treated by (R)-NNAL for 50 weeks (24 ± 2 and 125 ± 34 fmol/mg DNA, respectively) were also significantly higher than those in the same tissues of rats treated by NNK (6 ± 1 and 35 ± 5 fmol/mg DNA, respectively) or (S)-NNAL (5 ± 1 and 36 ± 8 fmol/mg DNA) [93].
- (2)
- PHB DNA Phosphate Adducts
PHB DNA phosphate adducts were also quantified in the lung tissues of rats chronically treated with NNK, (S)-NNAL, or (R)-NNAL from the carcinogenicity study noted above (Table S1) [103,104]. A total of 107 structurally unique PHB DNA phosphate adducts 76 (Figure 2) were characterized in the lung DNA of rats treated with NNK [103]. In (S)- or (R)-NNAL-treated rats, the total PHB DNA phosphate adducts occurred at levels of 3480–4180 or 4530–6920 fmol/mg DNA throughout the 70-week study course. In the lungs of the NNK-treated rats, the total PHB DNA phosphate adducts occurred at levels of 3950–8160 fmol/mg DNA. In contrast to the occurrence of PHB DNA base adducts, the PHB DNA phosphate adducts did not display any significant difference across the three treatment groups. They accounted for 38–55, 30–36 and 45–51% of the total measured DNA adducts in the lungs of the rats in the treatment group of NNK, (S)-NNAL or (R)-NNAL, respectively. The diastereomeric differences were also similar to those observed for the POB DNA phosphate adducts. For example, some combinations, such as Tp(PHB)T 81, Cp(PHB)T 79 or Tp(PHB)C 80, Ap(PHB)C 77 or Cp(PHB)A 78 in the rat livers and Cp(PHB)T or Tp(PHB)C in the rat lungs, appeared to be the predominant adducts among all the PHB DNA phosphate adducts in the NNK-treatment group.
3.3.3. Methyl DNA Adducts
- (1)
- Methyl DNA Base Adducts
O6-Methylguanine 82 (O6-Me-Gua, Figure 4) has been detected in target tissues—the nasal mucosa, lungs and livers—of F344 rats treated with NNK both acutely (4 h after a single i.v. injection of 87 mg/kg NNK) and chronically (daily i.p. injection of 40 mg/kg NNK for 14 days) [110]. Other methyl adducts, such as N7-methylguanine 83 (N7-Me-Gua) and O4-methylthymidine 84 (O4-Me-Thd), were also detected, with accumulation and persistence, in the respiratory tissues of rats repeatedly exposed to NNK via daily i.p. injections of 100 mg/kg for up to 12 days [111].
The levels of methyl DNA base adducts were greater than those of POB DNA base adducts in the livers and lungs of F344 rats administered [5-3H]NNK 88 (Figure 5) or [C3H3]NNK 89 by i.p. injection at 3–5000 μg/kg/day for 4 days. The levels of N7-Me-Gua were 0.22–246 pmol/μmol Gua in the rat liver DNA and 0.23–78.4 pmol/μmol Gua in the rat lung DNA. For comparison, the levels of released HPB from the same rat liver and lung DNA were 0–5.0 and 0–3.2 pmol/μmol Gua, respectively [77]. O6-Me-Gua was detected at its maximal level of 2550 ± 263 fmol/mg DNA at week 5 in the lung DNA of rats treated with 10 ppm NNK in drinking water for 20 weeks. Its levels were substantially higher than those of most of the POB- and PHB-DNA adducts, except for O2-POB-Thd and N7-POB-Gua [112]. We quantified the concentrations of O6-Me-Gua in the lung DNA of the rats from the chronic carcinogenicity study, which were administered 5 ppm NNK or NNAL enantiomers in their drinking water for up to 70 weeks (Table S1) [24]. In the lung DNA of the NNK- and (S)-NNAL-treated rats, O6-Me-Gua peaked at week 10 and decreased dramatically throughout the study course of 70 weeks, occurring at 213 ± 27 and 269 ± 178 fmol/mg DNA at weeks 10 and 34 and 49 ± 22 fmol/mg DNA at week 70. The levels of O6-Me-Gua in the lung DNA of rats from the (R)-NNAL treatment group were much lower, ranging from 17 to 4 fmol/mg DNA in the 70-week study course with no clear trend in changes. No such adduct was detected in the pancreas of rats from the same carcinogenicity study [24]. Considering the well-established miscoding properties of O6-Me-Gua, the discussion of its potential mutagenicity and carcinogenicity is presented in Section 3.5.
- (2)
- Methyl DNA Phosphate Adducts
A methyl DNA phosphate adduct 85 (Figure 4) was also formed in the DNA of rats treated with NNK or each of the two NNAL enantiomers in our chronic carcinogenicity study [113]. It predominated in the lungs of the NNK-treated rats, occurring at 2290 ± 546 fmol/mg DNA at week 10 and increasing moderately to 4510 ± 129 fmol/mg DNA at week 70. The concentrations of the methyl DNA phosphate adducts were slightly lower in the rats treated with (S)-NNAL or (R)-NNAL. In the lungs of the (S)-NNAL-treated rats, the total methyl DNA phosphate adducts occurred at relatively stable levels of 872–1120 fmol/mg DNA across the 70-week study course. In the lungs of the (R)-NNAL-treated rats, these adducts occurred at 874 ± 146 fmol/mg DNA at week 10 and peaked at 1430 ± 140 fmol/mg DNA at week 30 and decreased to 763 ± 90 fmol/mg DNA at week 70 [113].
Among the 32 possible diastereomers of the methyl DNA phosphate adducts, a total of 23, 21 and 22 isomers in all 10 combinations were detected in the rat lung tissues. In the three treatment groups, the most abundant methyl phosphate adducts were Ap(Me)T or Tp(Me)A, Ap(Me)C or Cp(Me)A and Gp(Me)T or Tp(Me)G. Similar to the POB and PHB DNA phosphate adducts, diastereomeric differences in the formation and persistence of methyl DNA phosphate adducts were also clearly observed in the rat lung DNA at various time points [113].
3.3.4. Formaldehyde-Derived DNA Adducts
Since formaldehyde is a metabolite formed by NNK and NNAL α-methyl hydroxylation, formaldehyde-derived DNA adducts were detected in vitro upon the hydrolysis of NNKOAc and NNALOAc [114]. These adducts were also detected in the DNA of rats treated with NNK. Substantial levels of N6-hydroxymethyl-2′-deoxyadenosine 86 (N6-HOCH2-dAdo, Figure 4) were detected in the liver and lung DNA, occurring at 3720 ± 2210 and 615 ± 504 fmol/mg DNA, respectively, when rats were dosed with 0.1 mmol/kg NNK by s.c. injection daily for 4 consecutive days. The levels of the cross-link adduct di-(N6-deoxyadenosyl)methane 87 (N6-dAdo-CH2-N6-dAdo) were significantly lower in the same samples, occurring at 303 ± 290 fmol/mg DNA in the rat livers and 18 fmol/mg DNA in the rat lungs [115].
3.3.5. Summary of DNA Adducts Formed by NNK Metabolism
Three major types of DNA base adduct—POB DNA adducts, PHB DNA adducts and methyl DNA adducts—as well as the minor adducts derived from the NNK metabolite formaldehyde, have been well characterized. The formation and persistence of these DNA adducts in laboratory animals have been extensively studied (Table S1). In rats or mice treated acutely with NNK (from hours to a few days), N7-POB-Gua and O6-Me-dGuo were easily detected in relatively high abundance. However, their high concentrations did not persist for a long time, possibly due to spontaneous depurination (N7-POB-Gua) and rapid O6-alkylguanine-DNA alkyltransferease (AGT)-mediated repair mechanisms (O6-Me-dGuo). In rats treated with NNK or NNAL for up to 20 or 70 weeks, O2-POB-Thd or O2-PHB-Thd clearly predominated among all the quantified DNA adducts. The relatively high levels of O2-POB/PHB-Thd and its long-term persistence in rat tissues suggest that it might be the mostly likely DNA base adduct to be detected in the tissues of people who use tobacco products. Meanwhile, the spontaneous depurination of N7-POB-Gua provides a good chance of detecting this adduct in urine. The specificity of these adducts to tobacco use (as opposed for example to methyl DNA adducts) makes them particularly attractive for mechanistic and biomonitoring studies.
POB and PHB DNA phosphate adducts formed by NNK/NNAL metabolism contribute to a significant portion of the total quantified DNA adducts in rat tissues. The persistence of these phosphate DNA adducts also appears to be longer than many DNA base adducts. However, difficulties in synthesizing the chemical standards of DNA phosphate adducts currently inhibit their accurate quantitation. The lack of apparent mutagenic properties in some POB DNA phosphate adducts (see Section 3.4) also raises some questions regarding their importance in tobacco carcinogenesis.
3.4. Mutagenicity and Genotoxicity of POB and PHB DNA Adducts
A recent review by Peterson [116] provided a summary of the genotoxic properties of NNK-derived DNA damage, including the methyl DNA base adducts O6-Me-dGuo and N7-Me-dGuo and minor methyl DNA base adducts, the POB DNA base adducts O6-POB-dGuo, O2-POB-Thd and basic sites, as well as aldehyde DNA adducts. PHB DNA base adducts are missing from the adduct panel in the review due to the lack of research data. The studies on the mutagenicity and genotoxicity of DNA phosphate adducts are also limited, with some early studies summarized by Farmer et al. in 2010 [100]. Since then, several studies have provided new insights towards the understanding of the mutagenicity and genotoxicity of some POB and PHB DNA base and phosphate adducts (Table 2).
In E. coli cells, O6-POB-dGuo and O6-PHB-dGuo impeded DNA replication to a relatively small or moderate extent, respectively. They both primarily induced G-to-A transitions, but O6-POB-dGuo also induced G-to-T transversions [117]. Similarly, O6-POB-dGuo produced G-to-A transitions and G-to-T transversions and other, more complex, types of mutation in human kidney cells [118]. Computational calculations revealed that both lesions formed stable pairs with C and T, with the latter pair being the least distorted, which agreed well with the experimental data [122]. The NMR data suggested that O6-POB-dGuo adopted wobble base pairing with C. An unusual hydrogen bond occurred between the POB carbonyl oxygen and the partner dCyd’s extra amino proton, forming a triplex and twisting the phosphodiester backbone at the lesion site [123]. O6-PHB-dGuo was suggested to be more mutagenic by stabilizing an intercalated DNA conformation for a deletion mutation [122]. However, no experimental data are currently available to support this computational result. The DNA polymerases induced by SOS were found to play redundant roles in the bypass of two alkylated O6-dGuo lesions [117]. While O6-POB-dGuo has been well documented as a substrate of the AGT enzyme [89,124], O6-PHB-dGuo was also found to be a substrate of this enzyme in one recent study [107].
O2-POB-Thd is a potent mutagen in SOS-induced E. coli and human cells. In SOS-induced wild-type E. coli, O2-POB-Thd induced a mutation frequency of 56%, with the major mutation being T-to-G followed by T-to-A. The T-to-G mutation was reduced in the SOS polymerase-deficient strains [120]. It moderately blocked the replication of DNA and induced almost exclusive T-to-A mutations with a frequency of ~15% in mammalian cells. Multiple translesion synthesis (TLS) DNA polymerases, especially Pol η and Pol ζ, contribute to bypassing O2-POB-Thd [119,121]. Nucleotide excision repair (NER) has been suggested to be involved in repairing this lesion [94,125]. O2-PHB-Thd is also repaired by the NER machinery [107].
O4-POB-Thd was first identified in NNKOAc-treated mammalian cells at levels substantially lower than O2-POB-Thd [94]. Similarly, O4-PHB-Thd also occurred at very low levels in the genomic DNA of mammalian cells exposed to NNALOAc compared to O2-PHB-Thd [107]. Although it is less preferentially formed, O4-POB-Thd has been found to be mutagenic, exclusively inducing T-to-C transversions at ~35% frequency. Similar to O2-POB-Thd, Pol η and Pol ζ are the two polymerases that primarily bypass O4-POB-Thd [119]. The mutagenicity of O4-PHB-Thd remains uninvestigated. However, it is bypassed by the NER machinery. The AGT enzyme seems to be involved in the repair of this lesion [107].
The mutagenicity and genotoxicity of DNA phosphate adducts have not been extensively investigated. In E. coli cells, the Sp configuration of the methyl DNA phosphate adduct was bypassed with higher efficiency than the Rp configuration, which showed suppressed DNA replication. The Sp methyl DNA phosphate adduct induced TT-to-GT and TT-to-GC mutations at the flanking TT dinucleotide site, in an AGT (also termed as Ada)-dependent manner. The replication outcome appeared to be sequence-context-independent of the 5′ dinucleotide adjacent to the lesion. The Rp diastereomer exhibited an error-free replication bypass. SOS-induced TLS polymerases did not appear to be responsible for bypassing this type of DNA lesion [126,127]. The only genotoxicity study of POB DNA phosphate adducts was conducted in E. coli cells by Wu and Wang in 2020 [105]. Surprisingly, no configurations of POB DNA phosphate adducts were found to cause perturbations in DNA replication or mutations in the flanking TT dinucleotides in E. coli cells. They were bypassed independently on SOS-induced DNA polymerases and Ada [105].
3.5. NNK-Derived DNA Adduct Formation in Lung Carcinogenesis
Deuterium isotope effects were clearly observed in the mutagenicity and carcinogenicity of NNK. In a comparative A/J mouse study, [CD3]NNK 90 (Figure 5) induced significantly higher lung tumor multiplicity than NNK, whereas [4,4-D2]NNK 91 was considerably weaker at inducing lung tumorigenesis [128]. The levels of O6-Me-Gua in the pulmonary DNA of mice treated with [4,4-D2]NNK were significantly lower than in those treated with NNK or [CD3]NNK [128,129]. This was in line with the hypothesis that DNA methylation, particularly O6-Me-Gua formation in pulmonary cells, is critical in A/J mouse lung carcinogenesis by NNK [28].
Meanwhile, NNKOAc significantly increased lung tumor multiplicity in A/J mice treated with 0.25 or 0.5 μmol acetoxymethylmethylnitrosamine 94 (AMMN, a model methylating agent, Figure 5) [130]. This enhancement was likely due to the depletion of AGT activity by POB DNA adducts [131,132]. The DNA pyridyloxobutylation by NNK prolonged the persistence time of O6-Me-Gua in mouse lung tissue [79], providing further evidence of the importance of O6-Me-Gua in A/J mouse lung tumorigenesis.
When F344 rats were treated with [14CH3]NNK 92 or [carbonyl-14C]NNK 93 (Figure 5), a higher incorporation of tissue-bound radioactivity in the lungs was observed from [14CH3]NNK than from [carbonyl-14C]NNK [133]. A comparative carcinogenicity study in rats using [CD3]NNK and [4,4-D2]NNK indicated that the pyridyloxobutylation of DNA plays an important role in NNK carcinogenesis, at least in the induction of rat nasal-cavity tumors [129,134]. Methyl DNA adducts, such as O6-Me-Gua and N7-Me-Gua, accumulated and persisted in the DNA of rat target organs (lung, nasal cavity, liver) upon NNK treatment [111]. Two types of DNA adduct—the methyl and POB DNA adducts formed by the α-hydroxylation of NNK—are considered to contribute to NNK mutagenicity and carcinogenicity through a synergistic mechanism.
3.6. Human DNA Adducts Related to NNK Metabolism
The two types of DNA adduct—HPB-releasing DNA adducts and methyl DNA phosphate adducts—discussed below are not specific to NNK metabolism; they may also result from other sources. NNN metabolism also leads to the formation of HPB-releasing DNA adducts, while some unknown sources in tobacco extracts may also produce these DNA adducts (Ma B, Hecht SS, unpublished data). The formation of methyl DNA phosphate adducts in humans may arise endogenously or from other DNA-methylating sources.
3.6.1. HPB-Releasing DNA Adducts in Human Tissues
As summarized in Table 1, the HPB released upon the acid hydrolysis of the peripheral lung and tracheobronchus DNA of cigarette smokers amounted to 11 ± 16 and 16 ± 18 fmol/mg DNA, respectively, in contrast to the low levels found in the DNA of the same tissues from nonsmokers (0.9 ± 2.3 fmol/mg peripheral lung DNA and 0.9 ± 1.7 fmol/mg tracheobronchus DNA) [80]. In the peripheral lung tissues of patients who had adenocarcinoma or squamous cell carcinoma or another diagnosis (1 patient), a significantly higher level of HPB-releasing DNA adducts was observed in self-reported smokers (404 ± 258 fmol/mg DNA) than in self-reported nonsmokers (59 ± 56 fmol/mg DNA) (p < 0.0001) [84]. However, a statistical difference in HPB-releasing adducts between smokers and non-smokers was not observed in the lung DNA of tumor-free sudden-death victims whose smoking status was verified by blood or urinary cotinine levels. The levels of HPB-releasing DNA adducts in the mucosa of the esophagus and cardia from the same two groups did not differ either [85]. In another study with lower esophageal mucosa from patients with or without upper gastrointestinal disorders, there was also no statistical difference in HPB-releasing DNA adduct levels in smokers (4.80 ± 3.57 pmol/mg DNA) versus nonsmokers (2.86 ± 2.44 pmol/mg DNA) after excluding outliers [86].
The levels of HPB released from the oral cell DNA of smokers were 12.0 pmol/mg DNA (mouthwash) and 44.7 pmol/mg DNA (buccal brushings), which were significantly higher than those from samples from nonsmokers (0.23 pmol/mg DNA) [82]. In a larger study with oral samples collected from 65 smokers, 30 of whom had head-and-neck squamous cell carcinoma, the median HPB releasing DNA adduct level was 6.6 times greater in the cancer patients (1.51 pmol/mg DNA) than in the controls (0.23 pmol/mg DNA) [83].
3.6.2. Methyl Phosphate Adducts in Human Lung
In human lung tumor tissues and adjacent normal lung tissues, methyl DNA phosphate adducts were detected in higher levels in the normal lung tissues of 13 current smokers (13 ± 6 adducts/109 nucleotides) than in 17 nonsmokers (8 ± 4 adducts/109 nucleotides). It is important to note that the levels of methyl DNA phosphate adducts remained constant in the lung tissues of untreated rats throughout a 70-week study course, occurring at 5–7 adducts/109 nucleotides. This indicates that the endogenous sources contributing to the formation of methyl DNA phosphate adducts do not accumulate with aging [135].
4. Metabolism and DNA Adduct Formation of NNN
4.1. Occurrence and Carcinogenicity
NNN is a nonvolatile N-nitrosamine present in virtually all tobacco products. During the tobacco curing and aging process, NNN is formed from the nitrosation of nornicotine, a secondary tobacco alkaloid resulting from the demethylation of nicotine via the catalysis of nicotine demethylase [19,136]. NNN can also be formed endogenously through the facile nitrosation of nornicotine, which is present in all tobacco products and is a metabolite of nicotine [137]. NNN, but not NNK, was detected at low levels (~0.5 × 10−3% of nicotine dose) in the urine of rats treated with nicotine and sodium nitrite [138]. It was occasionally detected in very low levels in the urine of some users of oral nicotine replacement therapy products [139,140]. NNN has also been detected in e-cigarette users, with median values of 2.15 pg/mL and 0.1 fmol/mL in the saliva and urine, respectively [141].
The first carcinogenicity study of NNN was reported in 1964, in which mice were administered NNN by i.p. injection once weekly. Pulmonary lesions and adenomas, as well as local invasion of the lungs and bronchii were observed during the first 7 months of treatment [142]. Since then, numerous studies with NNN have demonstrated its strong carcinogenicity in laboratory animals, including mice, rats, Syrian golden hamsters and mink [6,20]. NNN exerts its carcinogenicity on target organs, including the nasal cavity, oral cavity, esophagus, lungs and trachea. The organ specificity of NNN carcinogenicity is species- and administration-pathway-dependent [6]. For example, NNN primarily causes lung adenoma in mice after i.p. injection [19,29,143]. It induces tumors of the esophagus, oral cavity and nasal cavity when administered in drinking water [143,144,145,146,147,148], but predominantly causes nasal cavity tumors after s.c. injection in rats [149,150,151]. NNN together with NNK also induced oral cavity tumors through chronic oral swabbing [21]. Due to the chirality of the 2′-carbon, NNN is comprised of two enantiomers. In our chronic rat study of racemic NNN or NNN enantiomers in drinking water for 17–20 months, (S)-NNN was the stronger carcinogenic NNN enantiomeric component, causing a high incidence of oral and esophageal tumors at 14 ppm, while (R)-NNN was weakly carcinogenic but showed a synergistic enhancement of the carcinogenicity of (S)-NNN [148].
4.2. Metabolism
To exert its carcinogenicity, NNN requires metabolic activation, primarily catalyzed by P450s 2A6 and 3A4 [6]. The identified metabolic pathways of NNN include (1) α-hydroxylation (2′- and 5′-hydroxylation), (2) β-hydroxylation (3′- and 4′-hydroxylation), (3) pyridine-N-glucuronidation, (4) pyridine-N-oxidation and (5) denitrosation (Scheme 5). Among the five metabolic pathways, NNN α-hydroxylation is considered to be the major metabolic activation pathway, due to its potential to generate DNA alkylating agents and to form corresponding adducts with DNA or other biomolecules.
4.2.1. NNN α-Hydroxylation
NNN α-hydroxylation consists of two metabolic pathways—2′-hydroxylation and 5′-hydroxylation—due to the asymmetry of its pyrrolidine ring (Scheme 5). Through the catalysis of, primarily, P450 3A4 and, to some extent, 2A4, 2A5 and 2A13 for only (R)-NNN [149,152], NNN undergoes 2′-hydroxylation and forms the unstable intermediate 2′-hydroxyNNN 105, which spontaneously opens and generates a highly reactive electrophile, the POB diazonium ion 28. 2′-HydroxyNNN also undergoes denitrosation, followed by the loss of one molecule of H2O and forms myosmine 6. The POB diazonium ion 28 forms HPB 33 by solvolysis. The metabolites of HPB are the diol 34 and the keto acid 29.
NNN 5′-hydroxylation is mainly catalyzed by P450 2A6 but also, to a lesser extent, by P450s 2E1 and 2D6 [149,152,153]. After oxidation, the unstable product 5′-hydroxyNNN 106 forms the diazohydroxide 108, which spontaneously loses one molecule of H2O and forms the 4-oxo-1-(pyridine-3-yl)butyl (OPB) diazonium ion 109. The OPB diazonium ion can also rearrange to form the oxonium ion 110. The two ions, 109 and 110, are both highly electrophilic and react with DNA to form adducts. They also undergo solvolysis reactions and form lactol 111 and its ring-opened form 26 in equilibration, both of which are further metabolized to the hydroxy acid 30.
- (1)
- Overall Metabolic Profiles of NNN in Laboratory Animals
The first study of NNN metabolism was conducted by us in 1978 [150]. Using [2′-14C]NNN, 73–85% of the total radioactivity of the dosed compound was detected in the 48-h urine of F344 rats administered NNN by a single s.c. injection. The major metabolites of NNN in rat urine were identified as keto acid 29 and hydroxy acid 30 [150]. The urinary metabolite profiles were qualitatively similar in F344 rats, strain-A mice and Syrian golden hamsters, regardless of the administration pathways. Hydroxy acid 29 (37–53%, respectively, of the dose), keto acid 30 (8–31%), NNN-N-oxide 96 (7–11%), NNN 8 (3–5%) and norcotinine 101 (3–5%) were the principal urinary metabolites [58,154,155]. NNN metabolites formed in patas monkeys were investigated after i.v. injection with [5-3H]NNN [151]. Hydroxy acid 29 was the principal metabolite, accounting for 43.8 ± 4.0% of the total radioactivity eluting from the HPLC. The other metabolites were 3′-hydroxynorcotinine 103 (16.9%), norcotinine-1N-oxide 102 (16.5%), norcotinine 101 (13.1%), 3′-(O-β-D-glucopyranuronosyl)hydroxynorcotinine 104 (5.4%) and keto acid 29 (2.7%). Unchanged NNN accounted for only 0.63 ± 0.15%. Hydroxy acid 29 and norcotinine 101 were the two major metabolites in monkey serum [151]. 3′-Hydroxynorcotinine 103 was quantified in human urine at levels of 393 ± 287 pmol/mL in smokers and 658 ± 491 pmol/mL in current nicotine patch users who were former smokers. This metabolite was not detected in any nonsmoker urine. Considering its relatively high levels in urine samples, it is likely that the detected 3′-hydroxynorcotinine was formed as a minor metabolite of nicotine [156].
- (2)
- Preferential α-Hydroxylation of NNN in Target Tissues
Differential tissue metabolism might provide some insights in determining the organospecific carcinogenicity of N-nitrosamines. Compared to NNK, NNN administered in drinking water was carcinogenic to the oral mucosa and esophagus of rats [143,144,145,146,147,148]. This was partially explained by the higher activity of the esophagus in metabolizing NNN than NNK [157]. The organospecificity of NNN, causing esophageal tumors compared to hepatic tumors, may also be due to the presence of a high-affinity microsomal enzyme in the rat esophagus, which can efficiently catalyze the α-hydroxylation of N-nitrosamines, including NNN [154,155,158,159]. The apparent KM for the total α-hydroxylation of NNN by rat esophageal microsomes was much lower (49 ± 6.5 μM) than that by rat liver microsomes (1.2 ± 0.25 mM) [159].
- (3)
- Selectivity of α-Hydroxylation of NNN in Target Tissues
Keto acid 29 and hydroxy acid 30 are the end metabolites resulting from 2′- and 5′-hydroxylation of NNN in F344 rats, respectively. They were not formed from norcotinine 101, and no significant interconversion (1%) between those two metabolites was observed based on analysis of rat urine [60,160]. Thus, keto acid and hydroxy acid can serve as reliable biomarkers for assessing the two α-hydroxylation pathways of NNN metabolism in laboratory animal models. By quantifying the concentrations of 29 and 30 in in vitro incubation mixtures and in rat urine, catalytic rates of α-hydroxylation of NNN at 2′- and 5′-carbons can be established [161]. However, this does not hold true in humans. A high conversion rate (85%) of keto acid to hydroxy acid was observed based on analysis of human urine [59].
Preferential hydroxylation of NNN at the 2′-carbon has been consistently observed in rat esophagus. In cultured rat esophagus, the ratio of 2′-/5′-hydroxyation metabolites reached 3.4 in a 24 h incubation [157,162]; in rat esophageal microsomes, the ratio was 3.2 ± 0.5 at various NNN concentrations (from 1 μM to 2 mM) [159]. Similarly, 2′-hydroxylation predominated in rat nasal mucosa and rat oral tissues [157,163]. However, it is interesting that 5′-hydroxylation is preferentially observed in rat liver. While both hydroxylation pathways by rat liver microsomes have a similar KM of ~2 mM, the Vmax of 5′-hydroxylation (1.05 nmol/min/mg protein) is nearly 2-fold higher than that of 2′-hydroxyaltion (0.53 nmol/min/mg protein) [161]. The ratio of 2′-/5′-hydroxyation metabolites in rat liver microsomes was 0.71–0.23 depending on NNN concentration [159]. 5′-Hydroxylation also predominated in A/J mouse lung (ratio of 2′-/5′-hydroxyation metabolites: 0.6) and hamster esophagus and trachea (ratio of 2′-/5′-hydroxyation metabolites: 0.3 and 0.7, respectively) [29,164] and in some human tissues including buccal mucosa, trachea, esophagus, lung and bladder [30]. In patas monkeys, hydroxy acid significantly exceeded keto acid in serum concentrations, indicating preferential metabolism by the 5′-hydroxylation pathway in this species [151].
The interspecies and interindividual differences in the selectivity of NNN α-hydroxylation are remarkable. Liver microsomes from Aroclor-pretreated F344 rats showed a 20-fold induction of 2′-hydroxylation of NNN but only a 1.9-fold induction of NNN 5′-hydroxylation. The Vmax’s for NNN 2′- and 5′-hydroxylation by the treated rat liver microsomes were 0.53 and 1.05 nmol/min/mg protein, respectively [161]. The ability to catalyze NNN 2′- and 5′-hydroxylation by human liver microsomes was lower, with mean rates of 0.04 and 0.05 nmol/min/mg protein respectively [165]. A high interindividual variation of the ability of human tissues to metabolize NNN has been observed. Keto acid 29 was not detectable in most individuals but high in one case, similar to a 75-fold variation of benzo[α]pyrene metabolism in cultured human bronchi [30,166].
4.2.2. NNN β-Hydroxylation
NNN β-hydroxylation generates two products—3′-hydroxyNNN 97 and 4′-hydroxyNNN 98 (Scheme 5). They have been detected in the urine of rats administered NNN, however, only at trace levels. The rates of formation of both metabolites by rat liver microsomes were less than 0.01 nmol/min/mg protein [167].
4.2.3. NNN Detoxification Pathways
Pyridine-N-oxidation and glucuronidation of NNN are considered to be the two major detoxification pathways of NNN metabolism. Denitrosation products have also been observed in vivo to a moderate extent.
The metabolite formed by pyridine-N-oxidation is N′-nitrosonornicotine-1N-oxide 96 (NNN-N-oxide, Scheme 5). It accounted for 6.7–9.4% or 10.8% of the dose of NNN in the urine of rats treated by s.c. injection or by gavage, respectively [167,168]. Comparative studies in F344 rats and Syrian golden hamsters demonstrated that NNN-N-oxide was less carcinogenic than NNN, indicating that pyridine-N-oxidation is a detoxification pathway of NNN metabolism [143]. This metabolite has been observed in incubation mixtures of human liver microsomes and cultured human tissues [30,156,169,170,171].
NNN glucuronidation, likely catalyzed mainly by UGT1A4 [172], generates NNN pyridine-N-glucuronide 95 (NNN-N-Gluc, Scheme 5). It has been identified in human urine, accounting for 59.1 ± 26.0% and 61.8 ± 7.7% of the total of free plus glucuronidated NNN in smokers and smokeless tobacco users, respectively [173]. Urinary NNN-N-Gluc together with urinary free NNN comprise “total NNN”, which has successfully served as a biomarker for assessing esophageal cancer risk in smokers [169].
Through denitrosation and sequential oxidation reactions, NNN forms norcotinine 101 and its downstream metabolites likely via the intermediate iso-myosmine 100 [151]. However, mechanisms proceeding via nornicotine or 5′-hydroxyNNN 106 could not be excluded [6]. Norcotinine 101 has been detected in vitro in an incubation mixture of NNN with cultured mouse lung [29] and in vivo in rat urine [160,174]. It accounted for 3–5% of total urinary metabolites of NNN in rats, mice and hamsters [58,154,155] and 29.6% (plus its N-oxidation metabolite) of total NNN metabolites in the serum of patas monkeys [151].
4.2.4. Other Possible Metabolic Pathways
4.2.5. Stereochemistry of NNN Metabolism
(S)-NNN and (R)-NNN are the two enantiomers of NNN existing in tobacco and tobacco products, with (S)-NNN predominating in tobacco [170,175]. Our chronic carcinogenicity study has clearly shown that (S)-NNN is a strong oral and esophageal carcinogen in rats whereas (R)-NNN was less carcinogenic but a potent co-carcinogen to (S)-NNN [148]. The potential difference of metabolic activation of NNN enantiomers in target tissues may be responsible for the observed differences in carcinogenicity.
For NNN α-hydroxylation, enantiomeric differences have been observed in vitro with P450 2As [152]. The kinetic parameter KM for (R)-NNN 5′-hydroxylation is 22 ± 6 μM, which is significantly higher than that of (S)-NNN (2.3 ± 0.6 μM). For 2′-hydroxylation, all P450 2As except 2A6 metabolized (R)-NNN but none were active for (S)-NNN [152]. In cultured rat esophagus, (S)-NNN was preferentially metabolized to 2′-hydroxylation products, whereas (R)-NNN was predominantly metabolized by 5′-hydroxylation. This observation also held true in vivo in rats treated with (R)- or (S)- or racemic NNN by gavage [171].
Due to the potential difference of in vivo metabolism of the two NNN enantiomers, the enantiomeric composition of NNN in human urine may be different from that in tobacco products. In 2000, we analyzed the enantiomers of NNN in commercial cigarette tobacco and smokeless tobacco products—moist snuff and chewing tobacco—in the U.S. market. (S)-NNN comprised 75 ± 8.8% of total NNN in 12 products [175]. An updated analysis of 37 products in 2012 suggested that (S)-NNN was still the predominant enantiomer, representing for 62.9 ± 6.3% of total NNN [170]. Using a chiral stationary phase mass spectrometry method, we quantified (S)- and (R)-NNN in the urine of smokers and smokeless tobacco users; (S)-NNN comprised 67 ± 5% (N = 20) and 56 ± 3% (N = 10) of total NNN respectively [176]. Considering the remarkable similarity of the enantiomeric compositions of NNN in tobacco products and human urine, the overall metabolic biotransformation of (S)-NNN and (R)-NNN probably does not differ much in humans.
4.3. DNA Adducts Formed by NNN Metabolism
As depicted in Scheme 5, intermediates formed by NNN 2′-hydroxylation such as the POB diazonium ion 28 and by NNN 5′-hydroxylation such as the OPB diazonium ion 109 and the oxonium ion 110 have the electrophilic power to react with DNA and form the corresponding DNA adducts. While POB DNA adducts formed by NNN 2′-hydroxylation are the same as NNK-derived adducts, adducts formed by NNN 5′-hydroxylation are structurally unique. They represent a group of DNA adducts with the potential to be NNN-specific bioactivation biomarkers to monitor chronic exposure, uptake, and metabolic activation of this very important tobacco-specific carcinogen.
4.3.1. DNA Adducts Formed by NNN 2′-Hydroxylation
Due to the convergence of the formation of the POB diazonium ion 28 by NNN 2′-hydroxylation (as shown in Scheme 5) and NNK α-methyl hydroxylation (as shown in Scheme 2), most of the POB DNA base and phosphate adducts formed by NNN are similar to those formed by NNK in vitro and in vivo.
- (1)
- POB DNA Base Adducts Formed by NNN 2′-Hydroxylation
The formation of HPB-releasing DNA adducts were readily detected in the liver, lung and nasal mucosa DNA of NNN-exposed mice and rats (Table 1) [75,79,80,81]. The failure to detect HPB-releasing DNA adducts in other tissues was probably due to the detection limit of the analytical method used in those studies [75].
Individual DNA adducts caused by NNN were first characterized and quantified by us in 2007 [177,178]. Three major POB DNA base adducts O2-POB-Thd 60, N7-POB-Gua 59 and O2-POB-Cyt 49 (Figure 1) were detected in the target tissues (esophagus, nasal cavity, oral cavity) and livers and lungs of rats treated with 10 ppm (S)-NNN or (R)-NNN in the drinking water for up to 20 weeks. O6-POB-dGuo 56 was only occasionally detectable in rat nasal mucosa and oral mucosa, mostly in the (R)-NNN treatment group at early time points (weeks 1, 2, 5). At various time intervals, O2-POB-Thd predominated among all the 4 POB DNA adducts, by exceeding N7-POB-Gua in nearly all the examined tissues except for esophagus from both treatment groups. O2-POB-Cyt was formed at lower levels than O2-POB-Thd and N7-POB-Gua in all tissues except for esophagus from the (S)-NNN treatment group at some time points [177,178]. The enantiomeric distribution pattern of those POB DNA adducts formed by NNN is intriguing (Table S2). In the esophagus, oral mucosa and liver, (S)-NNN caused higher concentrations of all the POB DNA base adducts than its (R)-counterpart; however, the trend was inverted in most cases in the nasal olfactory and respiratory mucosa. This was considered to be due to the hepatic first-pass effect of preferential metabolism of (S)-NNN in the liver thus allowing more (R)-NNN to circulate to the nasal cavity and form DNA adducts. NNN-derived POB DNA adducts formed in the highest concentrations in the nasal respiratory mucosa, especially after the treatment with (R)-NNN [177,178].
To further understand the persistence and accumulation of NNN-derived POB DNA adducts in rats, we conducted a chronic carcinogenicity study in F344 rats treated with racemic NNN or its enantiomers in the drinking water for up to 17 months [148,179]. Tissues from rats sacrificed at weeks 10, 30, 50 and 70 were collected and analyzed for the levels of N7-POB-Gua and O2-POB-Thd, the two most abundant POB adducts formed by NNN (Figure 6 and Table S2). As shown in Figure 6, N7-POB-Gua and O2-POB-Thd clearly accumulated to the highest levels in the nasal respiratory mucosa of rats in both the (S)-NNN and (R)-NNN treatment groups. They also persisted in considerably high levels in the nasal olfactory mucosa of rats treated with (R)-NNN. O2-POB-Thd predominated in all the nasal mucosa DNA. However, the nasal mucosa was not the target tissue for carcinogenicity even though the high levels of DNA adducts were formed at this site. This contradiction was likely due to a more deleterious effect on rat survival of the other tumors observed in this study including esophageal and oral tumors than nasal tumors. The relative levels of N7-POB-Gua and O2-POB-Thd in the oral mucosa and esophagus, the two major target tissues for carcinogenicity, were fully consistent with the stronger carcinogenicity of (S)-NNN than (R)-NNN. The formation of O2-POB-dCyd (detected as O2-POB-Cyt) was not observed in the rat tissues from this study [179].
To provide a possible explanation for (R)-NNN as a synergistic co-carcinogen to (S)-NNN, levels of O6-POB-dGuo were quantified in esophageal mucosa DNA of rats treated with NNN enantiomers or racemate (Table S2) [180]. The adduct levels were synergistically increased, in a modest but statistically significant extent, in the racemic NNN treatment group compared to the sum of the amounts formed in the (S)-NNN group or the (R)-NNN group. No synergy in the formation of this adduct was observed in oral cavity or nasal cavity. The formation of other POB DNA base adducts of NNN similarly did not elicit any synergistic enhancement by the treatment of (R)-NNN in the racemic NNN group [180].
- (2)
- POB DNA Phosphate Adducts Formed by NNN 2′-Hydroxylation
POB DNA phosphate adducts formed by NNN 2′-hydroxylation were also quantified in the DNA of rats treated with (S)-NNN or (R)-NNN from our carcinogenicity study (Table S2) [87,148]. The highest amounts of total POB DNA phosphate adducts were observed in the esophageal DNA of rats treated with (S)-NNN, peaking at week 50. Levels of POB DNA phosphate adducts caused by (S)-NNN in esophagus, oral mucosa and liver were higher than those from (R)-NNN treatment. The reverse result was observed in nasal mucosa DNA, which was consistent with our previous results, likely reflecting the hepatic first-pass effect. Some combinations such as Ap(POB)C 64 or Cp(POB)A 65 (Figure 1) persisted throughout the study course of 70 weeks, suggesting their potential as biomarkers for monitoring chronic exposure to NNN and NNK in humans [87].
4.3.2. DNA Adducts Formed by NNN 5′-Hydroxylation
The 5′-hydroxylation pathway generates two unique electrophilic intermediates 109 and 110 (Scheme 5) that are considered to be NNN-specific. Thus, DNA adducts formed by NNN 5′-hydroxylation have the potential to be NNN-specific activation biomarkers for tobacco-related cancer etiology studies.
- (1)
- DNA Base Adducts Formed by NNN 5′-Hydroxylation
The regiochemically activated precursor of 5′-hydroxyNNN, 5′-acetoxy-N′-nitrosonornicotine 107 (5′-acetoxyNNN, Figure 7), was used to investigate DNA adducts possibly formed by the 5′-hydroxylation pathway of NNN metabolism. In the reaction of 5′-acetoxyNNN with dGuo, adducts resulting from NNN 5′-hydroxylation were identified as N2-[2-(3-pyridyl)-N-pyrrolidinyl-5-hydroxy]-2′-deoxyinosine (113, N2-Py-Py(OH)-dI) and N2-[5-(3-pyridyl)tetrahydrofuran-2-yl]-2′-deoxyguanosine (119, N2-Py-THF-dGuo). Their corresponding reduced forms 2-[2-(3-pyridyl)-N-pyrrolidinyl]-2′-deoxyinosine (114, Py-Py-dI) and N2-PHB-dGuo 70 were verified by each authentic synthesized standard. The two reduced adducts 114 and 70 were also detected in the reaction of 5′-acetoxyNNN with calf thymus DNA [181]. Py-Py-dI was also formed in the DNA of rats treated with racemic NNN for 3 weeks [182]. As shown in Table S2, Py-Py-dI preferentially formed in rat lung and nasal mucosa DNA in a dose-dependent manner upon NNN exposure. For example, in the 500 ppm NNN treatment group, Py-Py-dI occurred at 2018 ± 200, 2458 ± 190 and 2015 ± 80 fmol/μmol dGuo in rat lung, nasal olfactory mucosa and nasal respiratory mucosa, respectively. The levels of Py-Py-dI in the other tissues (liver, esophagus and oral mucosa) were substantially lower [182]. The distribution pattern of Py-Py-dI in NNN-treated rats reflected the expression profile of CYP2A3, the primary P450 enzyme responsible for NNN 5′-hydroxylation in rats [152]. It is also interesting to note that the formation of Py-Py-dI is stereospecific. (S)-NNN yielded exclusively the (R)-enantiomer of Py-Py-dI (114-(R), Figure 7), whereas (R)-NNN yielded nearly equal amounts of the two enantiomers of Py-Py-dI. This probably results from racemization of the OPB diazonium ion 109 formed by (R)-NNN [182].
In the reaction of 5′-acetoxyNNN with dAdo, N6-[5-(3-pyridyl)tetrahydrofuran-2-yl)-2′-deoxyadenosine (120, N6-Py-THF-dAdo) and its reduced form upon NaBH3CN treatment—N6-PHB-dAdo 67—were observed [183]. They were also detected in calf thymus DNA treated with 5′-acetoxyNNN, but not in rat liver and lung DNA upon treatment with NNN [184]. N6-[2-(3-Pyridyl)-N-pyrrolidinyl-5-hydroxy]-2′-deoxynebularine (117, N6-Py-Py(OH)-dN), on the other hand, has been detected both in vitro and in vivo. It was readily observed in liver and lung DNA of rats treated with 500 ppm racemic NNN in the drinking water for 3 weeks. Its reduced form 6-[2-(3-pyridyl)-N-pyrrolidinyl]-2′-deoxynebularine (118, Py-Py-dN) was observed only in NaBH3CN-reduced rat DNA but not in NaBH4-reduced samples. This is likely due to the equilibrium between N6-Py-Py(OH)-dN 117 and N6-[4-oxo-1-(pyridine-3-yl)butyl]-2′-deoxyadenosine (115, N6-OPB-dAdo), and the competing reduction of N6-OPB-dAdo 115 to N6-[4-hydroxy-1-(pyridine-3-yl)butyl]-2′-deoxyadenosine (116, N6-HPB-dAdo) [184,185]. N6-HPB-dAdo 116, structurally specific to NNN metabolism, has been quantified in the liver and lung DNA of rats treated with NNN. Its levels, however, were substantially lower than Py-Py-dI 114 in the same tissues (Table S2) [185].
The reduced adduct O2-PHB-Thd 74 was also identified in a hydrolysate of 5′-acetoxyNNN-treated calf thymus DNA after NaBH3CN reduction. This product was hypothesized to arise from the reduction of the unstable intermediate O2-[5-(3-pyridyl)tetrahydrofuran-2-yl]thymidine (121, O2-Py-THF-Thd) [183].
- (2)
- DNA Phosphate Adducts Formed by NNN 5′-Hydroxylation
By analogy to our previous studies with POB DNA phosphate adducts formed by NNN 2′-hydroxyation, we hypothesized that DNA phosphate adducts resulting from the OPB diazonium ion 109 and/or the oxonium ion 110 could also be formed in the reaction of 5′-acetoxyNNN with calf thymus DNA or synthesized dinucleotides. Upon reduction, the simplest DNA phosphate adducts were proposed to be B1p(HPB)B2 122 (Figure 7). However, no evidence of the formation of such adducts was observed (Li Y. and Hecht S.S. unpublished data).
4.4. Mutagenicity and Genotoxicity of NNN-Specific DNA Adducts
Most of the reported mutagenicity and genotoxicity studies reported to date have focused on POB DNA base adducts which can be formed from either NNN (by 2′-hydroxylation) or NNK (see the discussion in Section 3.4). There have been no reports on the newly identified NNN-specific DNA base adducts arising from the 5′-hydroxyaltion pathway. However, considering the structural similarity of the DNA adducts formed by NNN 5′-hydroxylation, for example, Py-Py-dI 114 and Py-Py-dN 118, to other reported mutagenic DNA adducts such as N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine 123 (N6,N6-DHB-dAdo, Figure 8), they are reasonably anticipated to be mutagenic and/or genotoxic [186]. Further studies are warranted to elucidate their roles in the carcinogenesis by NNN.
5. Concluding Remarks
N-nitrosamines comprise a large group of approximately 200 compounds that have been documented to be carcinogenic in more than 30 animal species [187]. Human exposure to these compounds is low in general with some exceptions such as NDMA exposure from certain types of foods or its possible endogenous formation in the stomach [188]. However, exposure to some N-nitrosamines such as NNN and NNK among tobacco users occur in far greater amounts. Considering their common occurrence in tobacco products and very strong carcinogenicity in laboratory animals, NNN and NNK are the 2 N-nitrosamines that have received the widest attention with regard to their metabolism and DNA interactions as related to tobacco carcinogenesis.
The potent pulmonary carcinogenicity and tobacco-specificity of NNK and its metabolism to NNAL which persists in human urine weeks after NNK exposure ceases [189,190], led to studies on NNAL in the urine of non-smokers exposed to secondhand tobacco smoke. These studies contributed significantly to the scientific foundation for the relationship between secondhand tobacco smoke exposure and lung cancer leading to the current nearly universal smoke free indoor environments which were uncommon when this research began in 1992 [47,191,192,193]. Urinary NNAL measurements in the Population Assessment of Tobacco and Health (PATH) study, involving approximately 46,000 adults in the U.S., demonstrated the highest levels of NNAL in smokeless tobacco users, followed by cigarette smokers, both amounts being far greater than in non-users of any tobacco product [194]. Similar results were obtained in the U.S. National Health and Nutrition Examination Survey (NHANES) [195]. Urinary NNAL levels in cigarette smokers were significantly related to lung cancer risk in the prospective Shanghai Cohort Study and Singapore Chinese Health Study, both of which collected the samples years before lung cancer developed [196,197]. These data collectively provide enhanced regulatory rationales related to tobacco use.
The metabolic pathways and DNA adduct structures presented here provide the basis for new insights on mechanisms of N-nitrosamine carcinogenesis. These data can lead to the development of biomarkers of carcinogen exposure and cancer risk. This is particularly critical in exposure scenarios such as smokeless tobacco use in South-East Asia where the relatively high levels of NNN in some products are plausible causes for the commonly occurring oral cancers in that region of the world.
DNA adduct formation caused by N-nitrosamines is considered the necessary step leading to tumor formation [9]. It is important to investigate DNA adducts that are structurally specific to the exposed carcinogen, and quantify the levels of such DNA lesions in human tissues so that predictive or diagnostic information can be obtained relating to cancer incidence in the affected population. While great progress has been made over the past decades, many unknowns remain. For example, the organospecificity of NNK to lung and of NNN to oral, nasal cavities and esophagus has been very well documented by numerous laboratory and epidemiology studies. However, no NNN/NNK-specific DNA adduct (such as POB- and PHB-DNA adducts discussed above) has been detected in the tissues of human tobacco users except for the structurally uncharacterized HPB-releasing DNA adduct. Difficulties in obtaining relevant tissue samples, the timing with respect to exposure, and the low amounts of these carcinogens (a few micrograms per day) taken up even in regular cigarette smokers, are among the principal reasons. Further studies are required on this critical topic.
In summary, much progress has been made in understanding the metabolism and DNA interactions of the tobacco-specific carcinogenic N-nitrosamines NNN and NNK over the past 2–3 decades. We herein provide a comprehensive and updated review on these topics hoping to facilitate research relevant to cancer etiology and prevention.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23095109/s1.
Author Contributions
Conceptualization, Y.L. and S.S.H.; writing—original draft preparation, Y.L.; writing—review and editing, Y.L. and S.S.H. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support for the tobacco-specific N-nitrosamine studies in our laboratory was provided by the U.S. National Cancer Institute through grant CA-81301. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support grant CA-77598.
Acknowledgments
The editorial assistance from Robert (Bob) Carlson is greatly appreciated.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 5′-AcetoxyNNN | 5′-acetoxy-N′-nitrosonornicotine |
| AGT | O6-alkylguanine-DNA alkyltransferease |
| AKRs | aldo-keto reductases |
| AMMN | acetoxymethylmethylnitrosamine |
| dIno | 2′-deoxyinosine |
| dUrd | 2′-deoxyuridine |
| E. Coli | Escherichia coli |
| HPB | 4-hydroxy-1-(3-pyridyl)-1-butanone |
| 11β-HSD1 | 11β-hydroxysteroid dehydrogenase type 1 |
| IARC | International Agency for Research on Cancer |
| i.p. | intraperitoneal |
| i.v. | intravenous |
| N2-POBb-dGuo | N2-(4-(3-pyridyl)-4-oxobut-2-yl)-2′-deoxyguanosine |
| Py-Py-dI | 2-[2-(3-pyridyl)-N-pyrrolidinyl]-2′-deoxyinosine |
| Py-Py-dN | 6-[2-(3-pyridyl)-N-pyrrolidinyl]-2′-deoxynebularine |
| N2-Py-Py(OH)-dI | N2-[2-(3-pyridyl)-N-pyrrolidinyl-5-hydroxy]-2′-deoxyinosine |
| N6-Py-Py(OH)-dN | N6-[2-(3-Pyridyl)-N-pyrrolidinyl-5-hydroxy]-2′-deoxynebularine |
| N2-Py-THF-dGuo | N2-[5-(3-pyridyl)tetrahydrofuran-2-yl]-2′-deoxyguanosine |
| N6-HPB-dAdo | N6-[4-hydroxy-1-(pyridine-3-yl)butyl]-2′-deoxyadenosine |
| N6-OPB-dAdo | N6-[4-oxo-1-(pyridine-3-yl)butyl]-2′-deoxyadenosine |
| N6,N6-DHB-dAdo | N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine |
| NAB | N′-nitrosoanabasine |
| NADP | nicotinamide adenosine dinucleotide phosphate |
| NAT | N′-nitrosoanatabine |
| NER | nucleotide excision repair |
| NHANES | National Health and Nutrition Examination Survey |
| NNAL | 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol |
| NNALOAc | 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol |
| NNAL-N-Gluc | 4-(methylnitrosamino)-1-(3-pyridyl-N-β-D-glycopyranuronosyl)-1-butanolonium inner salt |
| NNAL-O-Gluc | 4-(methylnitrosamino)-1-(3-pyridyl)-1-(O-β-D-glucopyranuronosyl)butane |
| NNC | nitrosamide N′-nitrosonorcotinine |
| NNK | 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone |
| NNKOAc | 4-((acetoxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone |
| NNN | N′-nitrosonornicotine |
| OPB | 4-oxo-4-(pyridin-3-yl)butanal |
| OPB diazonium ion | 4-oxo-1-(pyridine-3-yl)butyl diazonium ion |
| PATH study | Population Assessment of Tobacco and Health study |
| PHB | pyridylhydroxybutyl |
| POB | pyridyloxobutyl |
| pol κ | polymerase κ |
| ppm | parts per million |
| s.c. | subcutaneous |
| S. typhinurium | Salmonella typhinurium |
| TLS | translesion synthesis |
| TSNAs | tobacco-specific N-nitrosamines |
References
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hecht, S.S. Carcinogenic components of tobacco and tobacco smoke: A 2022 update. Food Chem. Toxicol. 2022. manuscript submitted. [Google Scholar]
- Lang, G.; Vuarnoz, A. Matrix-bound 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in tobacco: Quantification and evidence for an origin from lignin-incorporated alkaloids. J. Nat. Prod. 2015, 78, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Ji, H.; Fannin, F.F.; Bush, L.P. Contribution of Nicotine and Nornicotine toward the Production of N′-Nitrosonornicotine in Air-Cured Tobacco (Nicotiana tabacum). J. Nat. Prod. 2016, 79, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Chen, C.B.; Ornaf, R.M.; Jacobs, E.; Adams, J.D.; Hoffmann, D. Reaction of nicotine and sodium nitrite: Formation of nitrosamines and fragmentation of the pyrrolidine ring. J. Org. Chem. 1978, 43, 72–76. [Google Scholar] [CrossRef]
- Hecht, S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef]
- Hecht, S.S.; Stepanov, I.; Carmella, S.G. Exposure and metabolic activation biomarkers of carcinogenic tobacco-specific nitrosamines. Acc. Chem. Res. 2016, 49, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Konstantinou, E.; Fotopoulou, F.; Drosos, A.; Dimakopoulou, N.; Zagoriti, Z.; Niarchos, A.; Makrynioti, D.; Kouretas, D.; Farsalinos, K.; Lagoumintzis, G.; et al. Tobacco-specific nitrosamines: A literature review. Food Chem. Toxicol. 2018, 118, 198–203. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hatsukami, D.K. Smokeless tobacco and cigarette smoking: Chemical mechanisms and cancer prevention. Nat. Rev. Cancer 2022, 22, 143–155. [Google Scholar] [CrossRef]
- Shah, K.A.; Karnes, H.T. A review of the analysis of tobacco-specific nitrosamines in biological matrices. Crit. Rev. Toxicol. 2010, 40, 305–327. [Google Scholar] [CrossRef]
- Ma, B.; Stepanov, I.; Hecht, S.S. Recent studies on DNA adducts resulting from human exposure to tobacco smoke. Toxics 2019, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Gushgari, A.J.; Halden, R.U. Critical review of major sources of human exposure to N-nitrosamines. Chemosphere 2018, 210, 1124–1136. [Google Scholar] [CrossRef]
- Klus, H.; Kuhn, H. Determination of nornicotine nitrosamine in the smoke condensate of nornicotine-rich cigarettes. Fachl. Mitt. Oesterreichischen 1973, 14, 251–257. [Google Scholar]
- Hoffmann, D.; Hecht, S.S.; Ornaf, R.M.; Wynder, E.L. N′-nitrosonornicotine in tobacco. Science 1974, 186, 265–267. [Google Scholar] [CrossRef]
- Edwards, S.H.; Rossiter, L.M.; Taylor, K.M.; Holman, M.R.; Zhang, L.; Ding, Y.S.; Watson, C.H. Tobacco-specific nitrosamines in the tobacco and mainstream smoke of U.S. commercial cigarettes. Chem. Res. Toxicol. 2017, 30, 540–551. [Google Scholar] [CrossRef]
- Nasrin, S.; Chen, G.; Watson, C.J.W.; Lazarus, P. Comparison of tobacco-specific nitrosamine levels in smokeless tobacco products: High levels in products from Bangladesh. PLoS ONE 2020, 15, e0233111. [Google Scholar] [CrossRef]
- Lawler, T.S.; Stanfill, S.B.; Tran, H.T.; Lee, G.E.; Chen, P.X.; Kimbrell, J.B.; Lisko, J.G.; Fernandez, C.; Caudill, S.P.; de Castro, B.R.; et al. Chemical analysis of snus products from the United States and northern Europe. PLoS ONE 2020, 15, e0227837. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Smokeless Tobacco and Some Tobacco-specific N-Nitrosamines. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2007; Volume 89. [Google Scholar]
- Hecht, S.S.; Chen, C.B.; Hirota, N.; Ornaf, R.M.; Tso, T.C.; Hoffmann, D. Tobacco-specific nitrosamines: Formation from nicotine in vitro and during tobacco curing and carcinogenicity in strain A mice. J. Natl. Cancer Inst. 1978, 60, 819–824. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Personal Habits and Indoor Combustions: N′-Nitrosonornicotine and 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2012; Volume 100E, pp. 319–331. [Google Scholar]
- Hecht, S.S.; Rivenson, A.; Braley, J.; DiBello, J.; Adams, J.D.; Hoffmann, D. Induction of oral cavity tumors in F344 rats by tobacco-specific nitrosamines and snuff. Cancer Res. 1986, 46, 4162–4166. [Google Scholar]
- Kovi, R.C.; Johnson, C.S.; Balbo, S.; Hecht, S.S.; O’Sullivan, M.G. Metastasis to the F344 rat pancreas from lung cancer induced by 4-(methylnitrosamino)- 1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)- 1-butanol, constituents of tobacco products. Toxicol. Pathol. 2018, 46, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.; Rivenson, A.; Abbi, R.; Wynder, E.L. A study of tobacco carcinogenesis: Effect of the fat content of the diet on the carcinogenic activity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 1993, 53, 2758–2761. [Google Scholar] [PubMed]
- Balbo, S.; Johnson, C.S.; Kovi, R.C.; James-Yi, S.A.; O’Sullivan, M.G.; Wang, M.; Le, C.T.; Khariwala, S.S.; Upadhyaya, P.; Hecht, S.S. Carcinogenicity and DNA adduct formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F-344 rats. Carcinogenesis 2014, 35, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Breyer-Pfaff, U.; Martin, H.J.; Ernst, M.; Maser, E. Enantioselectivity of carbonyl reduction of 4-methylnitrosamino-1-(3-pyridyl)-1-butanone by tissue fractions from human and rat and by enzymes isolated from human liver. Drug Metab. Dispos. 2004, 32, 915–922. [Google Scholar] [PubMed]
- Hecht, S.S.; Trushin, N.; Reid-Quinn, C.A.; Burak, E.S.; Jones, A.B.; Southers, J.L.; Gombar, C.T.; Carmella, S.G.; Anderson, L.M.; Rice, J.M. Metabolism of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the patas monkey: Pharmacokinetics and characterization of glucuronide metabolites. Carcinogenesis 1993, 14, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.; Engl, J.; Friesenegger, S.; Tricker, A.R. Biotransformation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in lung tissue from mouse, rat, hamster, and man. Chem. Res. Toxicol. 2009, 22, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A.; White, C.M.; Trushin, N.; Hecht, S.S. Cell specificity for the pulmonary metabolism of tobacco-specific nitrosamines in the Fischer rat. Carcinogenesis 1989, 10, 2269–2274. [Google Scholar] [CrossRef]
- Castonguay, A.; Lin, D.; Stoner, G.D.; Radok, P.; Furuya, K.; Hecht, S.S.; Schut, H.A.J.; Klaunig, J.E. A study of chemical carcinogenesis. 45. Comparative carcinogenicity in A/J mice and metabolism by cultured mouse peripheral lung of N′-nitrosonornicotine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, and their analogs. Cancer Res. 1983, 43, 1223–1229. [Google Scholar]
- Castonguay, A.; Stoner, G.D.; Schut, H.A.; Hecht, S.S. Metabolism of tobacco-specific N-nitrosamines by cultured human tissues. Proc. Natl. Acad. Sci. USA 1983, 80, 6694–6697. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S.; Trushin, N. DNA and hemoglobin alkylation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its major metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F344 rats. Carcinogenesis 1988, 9, 1665–1668. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Kenney, P.M.; Hochalter, J.B.; Wang, M.; Hecht, S.S. Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers and metabolites in the A/J mouse. Carcinogenesis 1999, 20, 1577–1582. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.D.; LaVoie, E.J.; Hoffmann, D. On the pharmacokinetics of tobacco-specific N-nitrosamines in Fischer rats. Carcinogenesis 1985, 6, 509–511. [Google Scholar] [CrossRef]
- Hu, S.C.; Bryant, M.S.; Sepehr, E.; Kang, H.K.; Trbojevich, R.; Lagaud, G.; Mehta, D.; Ding, W.; Mittelstaedt, R.A.; Pearce, M.G.; et al. Toxicokinetic and genotoxicity study of NNK in male Sprague Dawley rats following nose-only inhalation exposure, intraperitoneal injection, and oral gavage. Toxicol. Sci. 2021, 182, 10–28. [Google Scholar] [CrossRef]
- Jalas, J.R.; Hecht, S.S.; Murphy, S.E. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem. Res. Toxicol. 2005, 18, 95–110. [Google Scholar] [CrossRef]
- Murphy, S.E.; Spina, D.A.; Nunes, M.G.; Pullo, D.A. Glucuronidation of 4-((hydroxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone, a metabolically activated form of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, by phenobarbital-treated rats. Chem. Res. Toxicol. 1995, 8, 772–779. [Google Scholar] [CrossRef]
- Peterson, L.A.; Urban, A.M.; Vu, C.C.; Cummings, M.E.; Brown, L.C.; Warmka, J.K.; Li, L.; Wattenberg, E.V.; Patel, Y.; Stram, D.O.; et al. Role of aldehydes in the toxic and mutagenic effects of nitrosamines. Chem. Res. Toxicol. 2013, 26, 1464–1473. [Google Scholar] [CrossRef] [Green Version]
- Carmella, S.G.; Borukhova, A.; Akerkar, S.A.; Hecht, S.S. Analysis of human urine for pyridine-N-oxide metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific lung carcinogen. Cancer Epidemiol. Biomark. Prev. 1997, 6, 113–120. [Google Scholar]
- Carmella, S.G.; Akerkar, S.; Hecht, S.S. Metabolites of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers’ urine. Cancer Res. 1993, 53, 721–724. [Google Scholar]
- Carmella, S.G.; Han, S.; Villalta, P.W.; Hecht, S.S. Analysis of total 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in smokers’ blood. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2669–2672. [Google Scholar] [CrossRef] [Green Version]
- Church, T.R.; Anderson, K.E.; Caporaso, N.E.; Geisser, M.S.; Le, C.T.; Zhang, Y.; Benoit, A.R.; Carmella, S.G.; Hecht, S.S. A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol. Biomark. Prev. 2009, 18, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.M.; Koh, W.P.; Murphy, S.E.; Fan, Y.; Wang, R.; Carmella, S.G.; Han, S.; Wickham, K.; Gao, Y.T.; Yu, M.C.; et al. Urinary levels of tobacco-specific nitrosamine metabolites in relation to lung cancer development in two prospective cohorts of cigarette smokers. Cancer Res. 2009, 69, 2990–2995. [Google Scholar] [CrossRef] [Green Version]
- Milunsky, A.; Carmella, S.G.; Ye, M.; Hecht, S.S. A tobacco-specific carcinogen in the fetus. Prenat. Diagn. 2000, 20, 307–310. [Google Scholar] [CrossRef]
- Idris, A.M.; Nair, J.; Friesen, M.; Ohshima, H.; Brouet, I.; Faustman, E.M.; Bartsch, H. Carcinogenic tobacco-specific nitrosamines are present at unusually high levels in the saliva of oral snuff users in Sudan. Carcinogenesis 1992, 13, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Cartanyà-Hueso, À.; Lidón-Moyano, C.; Fu, M.; Perez-Ortuño, R.; Ballbè, M.; Matilla-Santander, N.; Martín-Sánchez, J.C.; Pascual, J.A.; Fernández, E.; Martínez-Sánchez, J.M. Comparison of TSNAs concentration in saliva according to type of tobacco smoked. Environ. Res. 2019, 172, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ortuño, R.; Martínez-Sánchez, J.M.; Fu, M.; Ballbè, M.; Quirós, N.; Fernández, E.; Pascual, J.A. Assessment of tobacco specific nitrosamines (TSNAs) in oral fluid as biomarkers of cancer risk: A population-based study. Environ. Res. 2016, 151, 635–641. [Google Scholar] [CrossRef]
- Hecht, S.S.; Carmella, S.G.; Murphy, S.E.; Akerkar, S.; Brunnemann, K.D.; Hoffmann, D. A tobacco-specific lung carcinogen in the urine of men exposed to cigarette smoke. N. Engl. J. Med. 1993, 329, 1543–1546. [Google Scholar] [CrossRef]
- Carmella, S.G.; Le Ka, K.A.; Upadhyaya, P.; Hecht, S.S. Analysis of N- and O-glucuronides of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human urine. Chem. Res. Toxicol. 2002, 15, 545–550. [Google Scholar] [CrossRef]
- Carmella, S.G.; Ming, X.; Olvera, N.; Brookmeyer, C.; Yoder, A.; Hecht, S.S. High throughput liquid and gas chromatography-tandem mass spectrometry assays for tobacco-specific nitrosamine and polycyclic aromatic hydrocarbon metabolites associated with lung cancer in smokers. Chem. Res. Toxicol. 2013, 26, 1209–1217. [Google Scholar] [CrossRef]
- Desai, D.; Kagan, S.S.; Amin, S.; Carmella, S.G.; Hecht, S.S. Identification of 4-(methylnitrosamino)-1-[3-(6-hydroxypyridyl)]-1-butanone as a urinary metabolite of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in rodents. Chem. Res. Toxicol. 1993, 6, 794–799. [Google Scholar] [CrossRef]
- Peterson, L.A.; Ng, D.K.; Stearns, R.A.; Hecht, S.S. Formation of NADP(H) analogs of tobacco-specific nitrosamines in rat liver and pancreatic microsomes. Chem. Res. Toxicol. 1994, 7, 599–608. [Google Scholar] [CrossRef]
- Castonguay, A.; Pepin, P.; Briere, N. Modulation of 4-(methylnitrosamino)-1-(3-pyridyl)-1 butanone demethylation and denitrosation by rat liver microsomes. Cancer Lett. 1991, 59, 67–74. [Google Scholar] [CrossRef]
- Lee, V.M.; Keefer, L.K.; Archer, M.C. An evaluation of the roles of metabolic denitrosation and alpha-hydroxylation in the hepatotoxicity of N-Nitrosodimethylamine. Chem. Res. Toxicol. 1996, 9, 1319–1324. [Google Scholar] [CrossRef]
- Carlson, E.S.; Upadhyaya, P.; Hecht, S.S. Evaluation of nitrosamide formation in the cytochrome P450-mediated metabolism of tobacco-specific nitrosamines. Chem. Res. Toxicol. 2016, 29, 2194–2205. [Google Scholar] [CrossRef]
- Hecht, S.S.; Young, R.; Chen, C.B. Metabolism in the F344 rat of 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific carcinogen. Cancer Res. 1980, 40, 4144–4150. [Google Scholar]
- Hoffmann, D.; Castonguay, A.; Rivenson, A.; Hecht, S.S. Comparative carcinogenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine in Syrian Golden hamsters. Cancer Res. 1981, 41, 2386–2393. [Google Scholar]
- Stepanov, I.; Upadhyaya, P.; Carmella, S.G.; Feuer, R.; Jensen, J.; Hatsukami, D.K.; Hecht, S.S. Extensive metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1764–1773. [Google Scholar] [CrossRef] [Green Version]
- Jing, M.; Wang, Y.; Upadhyaya, P.; Jain, V.; Yuan, J.M.; Hatsukami, D.K.; Hecht, S.S.; Stepanov, I. Liquid chromatography-electrospray ionization-tandem mass spectrometry quantitation of urinary [pyridine-D4]4-hydroxy-4-(3-pyridyl)butanoic acid, a biomarker of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone metabolic activation in smokers. Chem. Res. Toxicol. 2014, 27, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S.; Hatsukami, D.K.; Bonilla, L.E.; Hochalter, J.B. Quantitation of 4-oxo-4-(3-pyridyl)butanoic acid and enantiomers of 4-hydroxy-4-(3-pyridyl)butanoic acid in human urine: A substantial pathway of nicotine metabolism. Chem. Res. Toxicol. 1999, 12, 172–179. [Google Scholar] [CrossRef]
- Trushin, N.; Hecht, S.S. Stereoselective metabolism of nicotine and tobacco-specific N-nitrosamines to 4-hydroxy-4-(3-pyridyl)butanoic acid in rats. Chem. Res. Toxicol. 1999, 12, 164–171. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Carmella, S.G.; Guengerich, F.P.; Hecht, S.S. Formation and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers in vitro in mouse, rat and human tissues. Carcinogenesis 2000, 21, 1233–1238. [Google Scholar] [CrossRef]
- Wu, Z.; Upadhyaya, P.; Carmella, S.G.; Hecht, S.S.; Zimmerman, C.L. Disposition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in bile duct-cannulated rats: Stereoselective metabolism and tissue distribution. Carcinogenesis 2002, 23, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S.; Carmella, S.G.; Ye, M.; Le, K.-a.; Jensen, J.A.; Zimmerman, C.L.; Hatsukami, D.K. Quantitation of metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone after cessation of smokeless tobacco use. Cancer Res. 2002, 62, 129–134. [Google Scholar] [PubMed]
- Carmella, S.G.; Ye, M.; Upadhyaya, P.; Hecht, S.S. Stereochemistry of metabolites of a tobacco-specific lung carcinogen in smokers’ urine. Cancer Res. 1999, 59, 3602–3605. [Google Scholar] [PubMed]
- Hecht, S.S.; Spratt, T.E.; Trushin, N. Corrigendum: Absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 2000, 21, 850. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Spratt, T.E.; Trushin, N. Absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 1997, 18, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Eklind, K.I.; Toussaint, M.; Amin, S.G.; Chung, F.L. Characterization of a glucuronide metabolite of 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and its dose-dependent excretion in the urine of mice and rats. Carcinogenesis 1990, 11, 1819–1823. [Google Scholar] [CrossRef]
- Dow, J.; Berg, C. Stereoselectivity of the carbonyl reduction of dolasetron in rats, dogs, and humans. Chirality 1995, 7, 342–348. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hochalter, J.B.; Villalta, P.W.; Murphy, S.E. 2′-Hydroxylation of nicotine by cytochrome P450 2A6 and human liver microsomes: Formation of a lung carcinogen precursor. Proc. Natl. Acad. Sci. USA 2000, 97, 12493–12497. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.E. Biochemistry of nicotine metabolism and its relevance to lung cancer. J. Biol. Chem. 2021, 296, 100722. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Fourth Report on Human Exposure to Environmental Chemicals, Updated Tables, (March 2021); U.S. Department of Health and Human Services: Atlanta, GA, USA, 2021.
- Kuiper, N.; Coats, E.M.; Crawford, T.N.; Gammon, D.G.; Loomis, B.; Watson, C.H.; Melstrom, P.C.; Lavinghouze, R.; Rogers, T.; King, B.A. Trends in manufacturer-reported nicotine yields in cigarettes sold in the United States, 2013–2016. Prev. Chronic Dis. 2020, 17, E148. [Google Scholar] [CrossRef]
- Lawler, T.S.; Stanfill, S.B.; de Castro, B.R.; Lisko, J.G.; Duncan, B.W.; Richter, P.; Watson, C.H. Surveillance of nicotine and pH in cigarette and cigar filler. Tob. Regul. Sci. 2017, 3 (Suppl. 1), 101–116. [Google Scholar] [CrossRef] [Green Version]
- Spratt, T.E.; Peterson, L.A.; Confer, W.L.; Hecht, S.S. Solvolysis of model compounds for alpha-hydroxylation of N′-nitrosonornicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: Evidence for a cyclic oxonium ion intermediate in the alkylation of nucleophiles. Chem. Res. Toxicol. 1990, 3, 350–356. [Google Scholar] [CrossRef]
- Hecht, S.S.; Spratt, T.E.; Trushin, N. Evidence for 4-(3-pyridyl)-4-oxobutylation of DNA in F344 rats treated with the tobacco-specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Carcinogenesis 1988, 9, 161–165. [Google Scholar] [CrossRef]
- Peterson, L.A.; Mathew, R.; SE, B.P.M.; Trushin, N.; Hecht, S.S. In vivo and in vitro persistence of pyridyloxobutyl DNA adducts from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 1991, 12, 2069–2072. [Google Scholar] [CrossRef]
- Murphy, S.E.; Palomino, A.; Hecht, S.S.; Hoffmann, D. Dose-response study of DNA and hemoglobin adduct formation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 1990, 50, 5446–5452. [Google Scholar]
- Hecht, S.S.; Han, S.; Kenney, P.M.; Wang, M.; Lindgren, B.; Wang, Y.; Lao, Y.; Hochalter, J.B.; Upadhyaya, P. Investigation of the reaction of myosmine with sodium nitrite in vitro and in rats. Chem. Res. Toxicol. 2007, 20, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.A.; Hecht, S.S. A study of chemical carcinogenesis.141. O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991, 51, 5557–5564. [Google Scholar]
- Foiles, P.G.; Akerkar, S.A.; Carmella, S.G.; Kagan, M.; Stoner, G.D.; Resau, J.H.; Hecht, S.S. Mass spectrometric analysis of tobacco-specific nitrosamine-DNA adducts in smokers and nonsmokers. Chem. Res. Toxicol. 1991, 4, 364–368. [Google Scholar] [CrossRef]
- Spratt, T.E.; Trushin, N.; Lin, D.; Hecht, S.S. Analysis for N2-(pyridyloxobutyl)deoxyguanosine adducts in DNA of tissues exposed to tritium-labeled 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Chem. Res. Toxicol. 1989, 2, 169–173. [Google Scholar] [CrossRef]
- Stepanov, I.; Muzic, J.; Le, C.T.; Sebero, E.; Villalta, P.; Ma, B.; Jensen, J.; Hatsukami, D.; Hecht, S.S. Analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB)-releasing DNA adducts in human exfoliated oral mucosa cells by liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol. 2013, 26, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Ruszczak, C.; Jain, V.; Khariwala, S.S.; Lindgren, B.; Hatsukami, D.K.; Stepanov, I. Optimized liquid chromatography nanoelectrospray-high-resolution tandem mass spectrometry method for the analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in human oral cells. Chem. Res. Toxicol. 2016, 29, 1849–1856. [Google Scholar] [CrossRef] [Green Version]
- Hölzle, D.; Schlöbe, D.; Tricker, A.R.; Richter, E. Mass spectrometric analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in human lung. Toxicology 2007, 232, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Schlöbe, D.; Hölzle, D.; Hatz, D.; von Meyer, L.; Tricker, A.R.; Richter, E. 4-Hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in lung, lower esophagus and cardia of sudden death victims. Toxicology 2008, 245, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Heppel, C.W.; Heling, A.K.; Richter, E. Ultrasensitive method for the determination of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts by gas chromatography-high resolution mass spectrometry in mucosal biopsies of the lower esophagus. Anal. Bioanal. Chem. 2009, 393, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, B.; Cao, Q.; Balbo, S.; Zhao, L.; Upadhyaya, P.; Hecht, S.S. Mass spectrometric quantitation of pyridyloxobutyl DNA phosphate adducts in rats chronically treated with N′-nitrosonornicotine. Chem. Res. Toxicol. 2019, 32, 773–783. [Google Scholar] [CrossRef]
- Liu, X.K.; Spratt, T.E.; Murphy, S.E.; Peterson, L.A. Pyridyloxobutylation of guanine residues by 4-[(acetoxymethyl)nitrosamino]-1-(3-pyridyl)-1-butanone generates substrates of O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 1996, 9, 949–953. [Google Scholar] [CrossRef]
- Wang, L.; Spratt, T.E.; Liu, X.K.; Hecht, S.S.; Pegg, A.E.; Peterson, L.A. Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 1997, 10, 562–567. [Google Scholar] [CrossRef]
- Wang, M.; Cheng, G.; Sturla, S.J.; Shi, Y.; McIntee, E.J.; Villalta, P.W.; Upadhyaya, P.; Hecht, S.S. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem. Res. Toxicol. 2003, 16, 616–626. [Google Scholar] [CrossRef]
- Hecht, S.S.; Villalta, P.W.; Sturla, S.J.; Cheng, G.; Yu, N.; Upadhyaya, P.; Wang, M. Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)- 1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitros- amino)-1-(3-pyridyl)-1-butanol with DNA. Chem. Res. Toxicol. 2004, 17, 588–597. [Google Scholar] [CrossRef]
- Michel, A.K.; Zarth, A.T.; Upadhyaya, P.; Hecht, S.S. Identification of 4-(3-pyridyl)-4-oxobutyl-2′-deoxycytidine adducts formed in the reaction of DNA with 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone: A chemically activated form of tobacco-specific carcinogens. ACS Omega 2017, 2, 1180–1190. [Google Scholar] [CrossRef]
- Carlson, E.S.; Upadhyaya, P.; Villalta, P.W.; Ma, B.; Hecht, S.S. Analysis and identification of 2′-deoxyadenosine-derived adducts in lung and liver DNA of F-344 rats treated with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2018, 31, 358–370. [Google Scholar]
- Leng, J.; Wang, Y. Liquid chromatography-tandem mass spectrometry for the quantification of tobacco-specific nitrosamine-induced DNA adducts in mammalian cells. Anal. Chem. 2017, 89, 9124–9130. [Google Scholar] [CrossRef]
- Hecht, S.S.; Lin, D.; Chuang, J.; Castonguay, A. A study of chemical carcinogenesis.91. Reactions with deoxyguanosine of 4-(carbethoxynitrosamino)-1-(3-pyridyl)-1-butanone, a model-compound for α-hydroxylation of tobacco-specific nitrosamines. J. Am. Chem. Soc. 1986, 108, 1292–1295. [Google Scholar] [CrossRef]
- Lao, Y.; Villalta, P.W.; Sturla, S.J.; Wang, M.; Hecht, S.S. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol. 2006, 19, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Urban, A.M.; Upadhyaya, P.; Cao, Q.; Peterson, L.A. Formation and repair of pyridyloxobutyl DNA adducts and their relationship to tumor yield in A/J mice. Chem. Res. Toxicol. 2012, 25, 2167–2178. [Google Scholar] [CrossRef] [Green Version]
- Lao, Y.; Yu, N.; Kassie, F.; Villalta, P.W.; Hecht, S.S. Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2007, 20, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, M.; Villalta, P.W.; Lindgren, B.R.; Upadhyaya, P.; Lao, Y.; Hecht, S.S. Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2009, 22, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.D.; Le Pla, R.C.; Farmer, P.B. Phosphotriester adducts (PTEs): DNA’s overlooked lesion. Mutagenesis 2010, 25, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Haglund, J.; Henderson, A.P.; Golding, B.T.; Törnqvist, M. Evidence for phosphate adducts in DNA from mice treated with 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Chem. Res. Toxicol. 2002, 15, 773–779. [Google Scholar] [CrossRef]
- Ma, B.; Villalta, P.W.; Zarth, A.T.; Kotandeniya, D.; Upadhyaya, P.; Stepanov, I.; Hecht, S.S. Comprehensive high-resolution mass spectrometric analysis of DNA phosphate adducts formed by the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol. 2015, 28, 2151–2159. [Google Scholar] [CrossRef]
- Ma, B.; Zarth, A.T.; Carlson, E.S.; Villalta, P.W.; Upadhyaya, P.; Stepanov, I.; Hecht, S.S. Identification of more than 100 structurally unique DNA-phosphate adducts formed during rat lung carcinogenesis by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 2018, 39, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Zarth, A.T.; Carlson, E.S.; Villalta, P.W.; Stepanov, I.; Hecht, S.S. Pyridylhydroxybutyl and pyridyloxobutyl DNA phosphate adduct formation in rats treated chronically with enantiomers of the tobacco-specific nitrosamine metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Mutagenesis 2017, 32, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, Y. Replication of pyridyloxobutyl phosphotriester lesions in cells. Chem. Res. Toxicol. 2020, 33, 308–311. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Sturla, S.J.; Tretyakova, N.; Ziegel, R.; Villalta, P.W.; Wang, M.; Hecht, S.S. Identification of adducts produced by the reaction of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with deoxyguanosine and DNA. Chem. Res. Toxicol. 2003, 16, 180–190. [Google Scholar] [CrossRef]
- Guo, S.; Leng, J.; Tan, Y.; Price, N.E.; Wang, Y. Quantification of DNA Lesions induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in mammalian cells. Chem. Res. Toxicol. 2019, 32, 708–717. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Kalscheuer, S.; Hochalter, J.B.; Villalta, P.W.; Hecht, S.S. Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2008, 21, 1468–1476. [Google Scholar] [CrossRef] [Green Version]
- Stepanov, I.; Hecht, S.S. Mitochondrial DNA adducts in the lung and liver of F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and (S)-4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2009, 22, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foiles, P.G.; Trushin, N.; Castonguay, A. Measurement of O6-methyldeoxyguanosine in DNA methylated by the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone using a biotin-avidin enzyme-linked immunosorbent assay. Carcinogenesis 1985, 6, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A.; White, C.M.; Boucheron, J.A.; Richardson, F.C.; Swenberg, J.A.; Anderson, M. Accumulation and persistence of DNA adducts in respiratory tissue of rats following multiple administrations of the tobacco specific carcinogen 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1986, 46, 1280–1284. [Google Scholar] [PubMed]
- Upadhyaya, P.; Lindgren, B.R.; Hecht, S.S. Comparative levels of O6-methylguanine, pyridyloxobutyl-, and pyridylhydroxybutyl-DNA adducts in lung and liver of rats treated chronically with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Drug Metab. Dispos. 2009, 37, 1147–1151. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Zarth, A.T.; Carlson, E.S.; Villalta, P.W.; Upadhyaya, P.; Stepanov, I.; Hecht, S.S. Methyl DNA phosphate adduct formation in rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2018, 31, 48–57. [Google Scholar] [CrossRef]
- Cheng, G.; Wang, M.; Upadhyaya, P.; Villalta, P.W.; Hecht, S.S. Formation of formaldehyde adducts in the reactions of DNA and deoxyribonucleosides with α-acetates of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), and N-nitrosodimethylamine (NDMA). Chem. Res. Toxicol. 2008, 21, 746–751. [Google Scholar] [CrossRef]
- Wang, M.; Cheng, G.; Villalta, P.W.; Hecht, S.S. Development of liquid chromatography electrospray ionization tandem mass spectrometry methods for analysis of DNA adducts of formaldehyde and their application to rats treated with N-nitrosodimethylamine or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol. 2007, 20, 1141–1148. [Google Scholar] [CrossRef]
- Peterson, L.A. Context matters: Contribution of specific DNA adducts to the genotoxic properties of the tobacco-specific nitrosamine NNK. Chem. Res. Toxicol. 2017, 30, 420–433. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Leng, J.; Wang, Y. DNA replication studies of N-nitroso compound-induced O6-alkyl-2′-deoxyguanosine lesions in Escherichia coli. J. Biol. Chem. 2019, 294, 3899–3908. [Google Scholar] [CrossRef]
- Pauly, G.T.; Peterson, L.A.; Moschel, R.C. Mutagenesis by O6-[4-oxo-4-(3-pyridyl)butyl]guanine in Escherichia coli and human cells. Chem. Res. Toxicol. 2002, 15, 165–169. [Google Scholar] [CrossRef]
- Du, H.; Leng, J.; Wang, P.; Li, L.; Wang, Y. Impact of tobacco-specific nitrosamine-derived DNA adducts on the efficiency and fidelity of DNA replication in human cells. J. Biol. Chem. 2018, 293, 11100–11108. [Google Scholar] [CrossRef] [Green Version]
- Jasti, V.P.; Spratt, T.E.; Basu, A.K. Tobacco-specific nitrosamine-derived O2-alkylthymidines are potent mutagenic lesions in SOS-induced Escherichia coli. Chem. Res. Toxicol. 2011, 24, 1833–1835. [Google Scholar] [CrossRef]
- Weerasooriya, S.; Jasti, V.P.; Bose, A.; Spratt, T.E.; Basu, A.K. Roles of translesion synthesis DNA polymerases in the potent mutagenicity of tobacco-specific nitrosamine-derived O2-alkylthymidines in human cells. DNA Repair 2015, 35, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.A.; Garden, J.L.; Wetmore, N.T.; Wetmore, S.D. Computational insights into the mutagenicity of two tobacco-derived carcinogenic DNA lesions. Nucleic Acids Res. 2018, 46, 11858–11868. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.A.; Vu, C.; Hingerty, B.E.; Broyde, S.; Cosman, M. Solution structure of an O6-[4-oxo-4-(3-pyridyl)butyl]guanine adduct in an 11 mer DNA duplex: Evidence for formation of a base triplex. Biochemistry 2003, 42, 13134–13144. [Google Scholar] [CrossRef]
- Pegg, A.E. Repair of O6-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100. [Google Scholar] [CrossRef]
- Li, L.; Perdigao, J.; Pegg, A.E.; Lao, Y.; Hecht, S.S.; Lindgren, B.R.; Reardon, J.T.; Sancar, A.; Wattenberg, E.V.; Peterson, L.A. The influence of repair pathways on the cytotoxicity and mutagenicity induced by the pyridyloxobutylation pathway of tobacco-specific nitrosamines. Chem. Res. Toxicol. 2009, 22, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wang, P.; Wang, Y. Cytotoxic and mutagenic properties of alkyl phosphotriester lesions in Escherichia coli cells. Nucleic Acids Res. 2018, 46, 4013–4021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Yuan, J.; Price, N.E.; Wang, Y. Ada protein- and sequence context-dependent mutagenesis of alkyl phosphotriester lesions in Escherichia coli cells. J. Biol. Chem. 2020, 295, 8775–8783. [Google Scholar] [CrossRef]
- Hecht, S.S.; Jordan, K.G.; Choi, C.I.; Trushin, N. Effects of deuterium substitution on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in A/J mice. Carcinogenesis 1990, 11, 1017–1020. [Google Scholar] [CrossRef]
- Trushin, N.; Rivenson, A.; Hecht, S.S. Evidence supporting the role of DNA pyridyloxobutylation in rat nasal carcinogenesis by tobacco-specific nitrosamines. Cancer Res. 1994, 54, 1205–1211. [Google Scholar]
- Peterson, L.A.; Thomson, N.M.; Crankshaw, D.L.; Donaldson, E.E.; Kenney, P.J. Interactions between methylating and pyridyloxobutylating agents in A/J mouse lungs: Implications for 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis. Cancer Res. 2001, 61, 5757–5763. [Google Scholar]
- Mijal, R.S.; Loktionova, N.A.; Vu, C.C.; Pegg, A.E.; Peterson, L.A. O6-pyridyloxobutylguanine adducts contribute to the mutagenic properties of pyridyloxobutylating agents. Chem. Res. Toxicol. 2005, 18, 1619–1625. [Google Scholar] [CrossRef]
- Peterson, L.A.; Liu, X.K.; Hecht, S.S. Pyridyloxobutyl DNA adducts inhibit the repair of O6-methylguanine. Cancer Res. 1993, 53, 2780–2785. [Google Scholar]
- Castonguay, A.; Tjalve, H.; Hecht, S.S. Tissue distribution of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its metabolites in F344 rats. Cancer Res. 1983, 43, 630–638. [Google Scholar]
- Hecht, S.S.; Lin, D.; Castonguay, A.; Rivenson, A. Effects of alpha-deuterium substitution on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Carcinogenesis 1987, 8, 291–294. [Google Scholar] [CrossRef]
- Ma, B.; Villalta, P.W.; Hochalter, J.B.; Stepanov, I.; Hecht, S.S. Methyl DNA phosphate adduct formation in lung tumor tissue and adjacent normal tissue of lung cancer patients. Carcinogenesis 2019, 40, 1387–1394. [Google Scholar] [CrossRef]
- Mirvish, S.S.; Sams, J.; Hecht, S.S. Kinetics of nornicotine and anabasine nitrosation in relation to N′-nitrosonornicotine occurrence in tobacco and to tobacco-induced cancer. J. Natl. Cancer Inst. 1977, 59, 1211–1213. [Google Scholar] [CrossRef]
- Knezevich, A.; Muzic, J.; Hatsukami, D.K.; Hecht, S.S.; Stepanov, I. Nornicotine nitrosation in saliva and its relation to endogenous synthesis of N′-nitrosonornicotine in humans. Nicotine Tob. Res. 2013, 15, 591–595. [Google Scholar] [CrossRef]
- Carmella, S.G.; Borukhova, A.; Desai, D.; Hecht, S.S. Evidence for endogenous formation of tobacco-specific nitrosamines in rats treated with tobacco alkaloids and sodium nitrite. Carcinogenesis 1997, 18, 587–592. [Google Scholar] [CrossRef] [Green Version]
- Stepanov, I.; Carmella, S.G.; Briggs, A.; Hertsgaard, L.; Lindgren, B.; Hatsukami, D.; Hecht, S.S. Presence of the carcinogen N′-nitrosonornicotine in the urine of some users of oral nicotine replacement therapy products. Cancer Res. 2009, 69, 8236–8240. [Google Scholar] [CrossRef] [Green Version]
- Stepanov, I.; Carmella, S.G.; Han, S.; Pinto, A.; Strasser, A.A.; Lerman, C.; Hecht, S.S. Evidence for endogenous formation of N′-nitrosonornicotine in some long-term nicotine patch users. Nicotine Tob. Res. 2009, 11, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, G.; Ma, B.; Yakovlev, G.; Yershova, K.; Le, C.; Jensen, J.; Hatsukami, D.K.; Stepanov, I. Presence of the carcinogen N′-nitrosonornicotine in saliva of e-cigarette users. Chem. Res. Toxicol. 2018, 31, 731–738. [Google Scholar] [CrossRef]
- Boyland, E.; Roe, F.J.C.; Gorrod, J.W. Induction of pulmonary tumours in mice by nitrosonornicotine, a possible constituent of tobacco smoke. Nature 1964, 202, 1126. [Google Scholar] [CrossRef]
- Hecht, S.S.; Young, R.; Maeura, Y. Comparative carcinogenicity in F344 rats and Syrian golden hamsters of N′-nitrosonornicotine and N′-nitrosonornicotine-1-N-oxide. Cancer Lett. 1983, 20, 333–340. [Google Scholar] [CrossRef]
- Hoffmann, D.; Raineri, R.; Hecht, S.S.; Maronpot, R.; Wynder, E.L. A study of tobacco carcinogenesis. XIV. Effects of N′-nitrosonornicotine and N′-nitrosonanabasine in rats. J. Natl. Cancer Inst. 1975, 55, 977–981. [Google Scholar] [CrossRef]
- Singer, G.M.; Taylor, H.W. Carcinogenicity of N′-nitrosonornicotine in Sprague-Dawley rats. J. Natl. Cancer Inst. 1976, 57, 1275–1276. [Google Scholar] [CrossRef]
- Castonguay, A.; Rivenson, A.; Trushin, N.; Reinhardt, J.; Spathopoulos, S.; Weiss, C.J.; Reiss, B.; Hecht, S.S. Effects of chronic ethanol consumption on the metabolism and carcinogenicity of N′-nitrosonornicotine in F344 rats. Cancer Res. 1984, 44, 2285–2290. [Google Scholar]
- Stoner, G.D.; Adams, C.; Kresty, L.A.; Amin, S.G.; Desai, D.; Hecht, S.S.; Murphy, S.E.; Morse, M.A. Inhibition of N′-nitrosonornicotine-induced esophageal tumorigenesis by 3-phenylpropyl isothiocyanate. Carcinogenesis 1998, 19, 2139–2143. [Google Scholar] [CrossRef] [Green Version]
- Balbo, S.; James-Yi, S.; Johnson, C.S.; O’Sullivan, M.G.; Stepanov, I.; Wang, M.; Bandyopadhyay, D.; Kassie, F.; Carmella, S.; Upadhyaya, P.; et al. (S)-N′-Nitrosonornicotine, a constituent of smokeless tobacco, is a powerful oral cavity carcinogen in rats. Carcinogenesis 2013, 34, 2178–2183. [Google Scholar] [CrossRef] [Green Version]
- Patten, C.J.; Smith, T.J.; Friesen, M.J.; Tynes, R.E.; Yang, C.S.; Murphy, S.E. Evidence for cytochrome P450 2A6 and 3A4 as major catalysts for N′-nitrosonornicotine alpha-hydroxylation by human liver microsomes. Carcinogenesis 1997, 18, 1623–1630. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.B.; Hecht, S.S.; Hoffmann, D. Metabolic α-hydroxylation of the tobacco-specific carcinogen, N′-nitrosonornicotine. Cancer Res. 1978, 38, 3639–3645. [Google Scholar]
- Upadhyaya, P.; Zimmerman, C.L.; Hecht, S.S. Metabolism and pharmacokinetics of N′-nitrosonornicotine in the patas monkey. Drug Metab. Dispos. 2002, 30, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.L.; Murphy, S.E.; Hecht, S.S. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N′-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem. Res. Toxicol. 2005, 18, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Inui, Y.; Yun, C.H.; Guengerich, F.P.; Shimada, T. Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis 1992, 13, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Labuc, G.E.; Archer, M.C. Esophageal and hepatic microsomal metabolism of N-nitrosomethylbenzylamine and N-nitrosodimethylamine in the rat. Cancer Res. 1982, 42, 3181–3186. [Google Scholar] [PubMed]
- Von Hofe, E.; Schmerold, I.; Lijinsky, W.; Jeltsch, W.; Kleihues, P. DNA methylation in rat tissues by a series of homologous aliphatic nitrosamines ranging from N-nitrosodimethylamine to N-nitrosomethyldodecylamine. Carcinogenesis 1987, 8, 1337–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyaya, P.; Hecht, S.S. Quantitative analysis of 3’-hydroxynorcotinine in human urine. Nicotine Tob. Res. 2015, 17, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.E.; Heiblum, R.; Trushin, N. Comparative metabolism of N′-nitrosonornicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by cultured F344 rat oral tissue and esophagus. Cancer Res. 1990, 50, 4685–4691. [Google Scholar]
- Huang, Q.; Stoner, G.; Resau, J.; Nickols, J.; Mirvish, S.S. Metabolism of N-Nitrosomethyl-n-amylamine by microsomes from human and rat esophagus. Cancer Res. 1992, 52, 3547–3551. [Google Scholar]
- Murphy, S.E.; Spina, D.A. Evidence for a high-affinity enzyme in rat esophageal microsomes which α-hydroxylates N′-nitrosonornicotine. Carcinogenesis 1994, 15, 2709–2713. [Google Scholar] [CrossRef]
- Hecht, S.S.; Lin, D.; Chen, C.B. Comprehensive analysis of urinary metabolites of N′-nitrosonornicotine. Carcinogenesis 1981, 2, 833–838. [Google Scholar] [CrossRef]
- Chen, C.H.; Fung, P.T.; Hecht, S.S. Assay for microsomal alpha-hydroxylation of N′-nitrosonornicotine and determination of the deuterium isotope effect for alpha-hydroxylation. Cancer Res. 1979, 39, 5057–5062. [Google Scholar]
- Hecht, S.S.; Reiss, B.; Lin, D.; Williams, G.M. Metabolism of N′-nitrosonornicotine by cultured rat esophagus. Carcinogenesis 1982, 3, 453–456. [Google Scholar] [CrossRef]
- Brittebo, E.B.; Castonguay, A.; Furuya, K.; Hecht, S.S. Metabolism of tobacco-specific nitrosamines by cultured rat nasal mucosa. Cancer Res. 1983, 43, 4343–4348. [Google Scholar]
- McCoy, G.; Katayama, S.; Young, R.; Wyatt, M.; Hecht, S. Influence of Chronic Ethanol Consumption on the Metabolism and Carcinogenicity of Tobacco-related Nitrosamines. In IARC Scientific Publication No. 41: N-Nitroso Compounds: Occurrence and Biological Effects; Bartsch, H., O’Neill, I., Castegnaro, M., Okada, M., Eds.; IARC: Lyon, France, 1982; pp. 635–642. [Google Scholar]
- Hecht, S.S.; Chen, C.H.; McCoy, G.D.; Hoffmann, D.; Domellof, L. α-hydroxylation of N-nitrosopyrrolidine and N′-nitrosonornicotine by human liver microsomes. Cancer Lett. 1979, 8, 35–41. [Google Scholar] [CrossRef]
- Harris, C.C.; Autrup, H.; Connor, R.; Barrett, L.A.; McDowell, E.M.; Trump, B.F. Interindividual variation in binding of benzo[a]pyrene to DNA in cultured human bronchi. Science 1976, 194, 1067–1069. [Google Scholar] [CrossRef]
- Hecht, S.S.; Chen, C.B.; Hoffmann, D. Metabolic ß-hydroxylation and N-oxidation of N′-nitrosonornicotine. J. Med. Chem. 1980, 23, 1175–1178. [Google Scholar] [CrossRef]
- Hecht, S.S.; Young, R. Regiospecificity in the metabolism of the homologous cyclic nitrosamines, N′-nitrosonornicotine and N′-nitrosoanabasine. Carcinogenesis 1982, 3, 1195–1199. [Google Scholar] [CrossRef]
- Yuan, J.M.; Knezevich, A.D.; Wang, R.; Gao, Y.T.; Hecht, S.S.; Stepanov, I. Urinary levels of the tobacco-specific carcinogen N′-nitrosonornicotine and its glucuronide are strongly associated with esophageal cancer risk in smokers. Carcinogenesis 2011, 32, 1366–1371. [Google Scholar] [CrossRef]
- Stepanov, I.; Yershova, K.; Carmella, S.; Upadhyaya, P.; Hecht, S.S. Levels of (S)-N′-nitrosonornicotine in U.S. tobacco products. Nicotine Tob. Res. 2013, 15, 1305–1310. [Google Scholar] [CrossRef] [Green Version]
- McIntee, E.J.; Hecht, S.S. Metabolism of N′-nitrosonornicotine enantiomers by cultured rat esophagus and in vivo in rats. Chem. Res. Toxicol. 2000, 13, 192–199. [Google Scholar] [CrossRef]
- Chen, G.; Dellinger, R.W.; Sun, D.; Spratt, T.E.; Lazarus, P. Glucuronidation of tobacco-specific nitrosamines by UGT2B10. Drug Metab. Dispos. 2008, 36, 824–830. [Google Scholar] [CrossRef] [Green Version]
- Stepanov, I.; Hecht, S.S. Tobacco-specific nitrosamines and their pyridine-N-glucuronides in the urine of smokers and smokeless tobacco users. Cancer Epidemiol. Biomark. Prev. 2005, 14, 885–891. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S.; Chen, C.B.; Hoffmann, D. Evidence for metabolic α hydroxylation of N-nitrosopyrrolidine. Cancer Res. 1978, 38, 215–218. [Google Scholar]
- Carmella, S.G.; McIntee, E.J.; Chen, M.; Hecht, S.S. Enantiomeric composition of N′-nitrosonornicotine and N′-nitrosoanatabine in tobacco. Carcinogenesis 2000, 21, 839–843. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Carmella, S.G.; Hecht, S.S. Analysis of N′-nitrosonornicotine enantiomers in human urine by chiral stationary phase liquid chromatography-nanoelectrospray ionization-high resolution tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1044–1045, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Lao, Y.; Yu, N.; Kassie, F.; Villalta, P.W.; Hecht, S.S. Analysis of pyridyloxobutyl DNA adducts in F344 rats chronically treated with (R)- and (S)-N′-nitrosonornicotine. Chem. Res. Toxicol. 2007, 20, 246–256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, M.; Villalta, P.W.; Lindgren, B.R.; Lao, Y.; Hecht, S.S. Quantitation of pyridyloxobutyl DNA adducts in nasal and oral mucosa of rats treated chronically with enantiomers of N′-nitrosonornicotine. Chem. Res. Toxicol. 2009, 22, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Balbo, S.; Wang, M.; Upadhyaya, P.; Khariwala, S.S.; Villalta, P.W.; Hecht, S.S. Quantitation of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)- or (S)-N′-nitrosonornicotine (NNN) in a carcinogenicity study. Chem. Res. Toxicol. 2013, 26, 1526–1535. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Villalta, P.W.; Upadhyaya, P.; Hecht, S.S. Analysis of O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine and other DNA adducts in rats treated with enantiomeric or racemic N′-ntrosonornicotine. Chem. Res. Toxicol. 2016, 29, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, P.; McIntee, E.J.; Villalta, P.W.; Hecht, S.S. Identification of adducts formed in the reaction of 5′-acetoxy-N′-nitrosonornicotine with deoxyguanosine and DNA. Chem. Res. Toxicol. 2006, 19, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Zarth, A.T.; Upadhyaya, P.; Yang, J.; Hecht, S.S. DNA adduct formation from metabolic 5’-hydroxylation of the tobacco-specific carcinogen N′-nitrosonornicotine in human enzyme systems and in rats. Chem. Res. Toxicol. 2016, 29, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, P.; Hecht, S.S. Identification of adducts formed in the reactions of 5′-acetoxy-N′-nitrosonornicotine with deoxyadenosine, thymidine, and DNA. Chem. Res. Toxicol. 2008, 21, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Carlson, E.S.; Zarth, A.T.; Upadhyaya, P.; Hecht, S.S. Investigation of 2’-deoxyadenosine-derived adducts specifically formed in rat liver and lung DNA by N′-nitrosonornicotine metabolism. Chem. Res. Toxicol. 2021, 34, 1004–1015. [Google Scholar] [CrossRef]
- Li, Y.; Hecht, S.S. Identification of an N′-nitrosonornicotine-specific deoxyadenosine adduct in rat liver and lung DNA. Chem. Res. Toxicol. 2021, 34, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Kotapati, S.; Wickramaratne, S.; Esades, A.; Boldry, E.J.; Quirk Dorr, D.; Pence, M.G.; Guengerich, F.P.; Tretyakova, N.Y. Polymerase bypass of N6-deoxyadenosine adducts derived from epoxide metabolites of 1,3-butadiene. Chem. Res. Toxicol. 2015, 28, 1496–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, S.S. Approaches to cancer prevention based on an understanding of N-nitrosamine carcinogenesis. Proc. Soc. Exp. Biol. Med. 1997, 216, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hecht, S.S. Metabolic activation and DNA interactions of carcinogenic n-nitrosamines to which humans are commonly exposed. Int. J. Mol. Sci. 2022, 23, 4559. [Google Scholar] [CrossRef]
- Hecht, S.S.; Carmella, S.G.; Chen, M.; Koch, J.F.D.; Miller, A.T.; Murphy, S.E.; Jensen, J.A.; Zimmerman, C.L.; Hatsukami, D.K. Quantitation of urinary metabolites of a tobacco-specific lung carcinogen after smoking cessation. Cancer Res. 1999, 59, 590–596. [Google Scholar]
- Goniewicz, M.L.; Havel, C.M.; Peng, M.W.; Jacob, P., III; Dempsey, D.; Yu, L.; Zielinska-Danch, W.; Koszowski, B.; Czogala, J.; Sobczak, A.; et al. Elimination kinetics of the tobacco-specific biomarker and lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3421–3425. [Google Scholar] [CrossRef] [Green Version]
- Parsons, W.D.; Carmella, S.G.; Akerkar, S.; Bonilla, L.E.; Hecht, S.S. A metabolite of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in the urine of hospital workers exposed to environmental tobacco smoke. Cancer Epidemiol. Biomark. Prev. 1998, 7, 257–260. [Google Scholar]
- Anderson, K.E.; Carmella, S.G.; Ye, M.; Bliss, R.; Le, C.; Murphy, L.; Hecht, S.S. Metabolites of a tobacco-specific lung carcinogen in the urine of nonsmoking women exposed to environmental tobacco smoke in their homes. J. Natl. Cancer Inst. 2001, 93, 378–381. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.E.; Kliris, J.; Murphy, L.; Carmella, S.G.; Han, S.; Link, C.; Bliss, R.L.; Murphy, S.E.; Hecht, S.S. Metabolites of a tobacco-specific lung carcinogen in nonsmoking casino patrons. Cancer Epidemiol. Biomark. Prev. 2003, 12, 1544–1546. [Google Scholar]
- Xia, B.; Blount, B.C.; Guillot, T.; Brosius, C.; Li, Y.; Van Bemmel, D.M.; Kimmel, H.L.; Chang, C.M.; Borek, N.; Edwards, K.C.; et al. Tobacco-specific nitrosamines (NNAL, NNN, NAT, and NAB) exposures in the US Population Assessment of Tobacco and Health (PATH) Study Wave 1 (2013–2014). Nicotine Tob. Res. 2021, 23, 573–583. [Google Scholar] [CrossRef]
- Rostron, B.L.; Chang, C.M.; van Bemmel, D.M.; Xia, Y.; Blount, B.C. Nicotine and toxicant exposure among U.S. smokeless tobacco users: Results from 1999 to 2012 National Health and Nutrition Examination Survey data. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1829–1837. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.M.; Nelson, H.H.; Carmella, S.G.; Wang, R.; Kuriger-Laber, J.; Jin, A.; Adams-Haduch, J.; Hecht, S.S.; Koh, W.P.; Murphy, S.E. CYP2A6 genetic polymorphisms and biomarkers of tobacco smoke constituents in relation to risk of lung cancer in the Singapore Chinese Health Study. Carcinogenesis 2017, 38, 411–418. [Google Scholar] [CrossRef]
- Yuan, J.M.; Nelson, H.H.; Butler, L.M.; Carmella, S.G.; Wang, R.; Kuriger-Laber, J.; Adams-Haduch, J.; Hecht, S.S.; Gao, Y.-T.; Murphy, S.E. Genetic determinants of cytochrome P450 2A6 and biomarkers of tobacco smoke exposure in relation to risk of lung cancer development in the Shanghai Cohort Study. Int. J. Cancer 2015, 138, 2161–2171. [Google Scholar] [CrossRef]
Scheme 1.
Mechanisms of formation of tobacco-specific N-nitrosamines.

Scheme 2.
Mechanisms of NNK and NNAL metabolism.

Scheme 3.
Stereochemistry of the formation of NNAL and its glucuronides.

Scheme 4.
DNA-reactive intermediates formed by NNK and NNAL metabolism.

Figure 1.
Structures of POB DNA base and phosphate adducts.

Figure 2.
Structures of PHB DNA base and phosphate adducts.

Figure 3.
Levels of PHB DNA adducts in lung and pancreatic DNA of rats treated with 5 ppm NNK or NNAL enantiomers in the drinking water for 10, 30, 50, or 70 weeks. NNK: filled circle; (S)-NNAL: square; (R)-NNAL: cross. Adapted from ref. [24], with the permission of Oxford University Press.
Figure 3.
Levels of PHB DNA adducts in lung and pancreatic DNA of rats treated with 5 ppm NNK or NNAL enantiomers in the drinking water for 10, 30, 50, or 70 weeks. NNK: filled circle; (S)-NNAL: square; (R)-NNAL: cross. Adapted from ref. [24], with the permission of Oxford University Press.

Figure 4.
Structures of methyl DNA adducts and DNA adducts derived from formaldehyde.

Figure 5.
Structures of isotope-labeled NNKs and AMMN.

Scheme 5.
Pathways of NNN metabolism.

Figure 6.
Levels of N7-POB-Gua (open bars) and O2-POB-Thd (striped bars) in the tissues of rats treated with (S)- or (R)-NNN in the drinking water for up to 70 weeks. Adapted with permission from ref. [179]. Copyright 2013 American Chemical Society.
Figure 6.
Levels of N7-POB-Gua (open bars) and O2-POB-Thd (striped bars) in the tissues of rats treated with (S)- or (R)-NNN in the drinking water for up to 70 weeks. Adapted with permission from ref. [179]. Copyright 2013 American Chemical Society.

Figure 7.
DNA adducts formed by NNN 5′-hydroxylation.

Figure 8.
Structure of N6,N6-DHB-dAdo.

Table 2.
Mutagenicity of some POB and PHB DNA adducts.
| DNA Adduct | Mutation | Cell | Ref. |
|---|---|---|---|
| POB DNA adduct | |||
| O6-POB-dGuo | G > A (~90%) and G > T (~2%) | E. coli | [117] |
| G > A (90%); G > T (~3%); other more complex types | HEK293 | [118] | |
| G > A (~75%) and G > T (~3%) | HEK293T | [119] | |
| O2-POB-Thd | T > G (37%) and T > A (12%) | E. coli | [120] |
| T > A (47%) | HEK293T | [121] | |
| T > A (~15%) | HEK293T | [119] | |
| O4-POB-Thd | T > C (~35%) | HEK293T | [119] |
| B1p(POB)B2 | Not mutagenic | E. coli | [105] |
| PHB DNA adduct | |||
| O6-PHB-dGuo | G > A (~95%) | E. coli | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, Y.; Hecht, S.S. Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines. Int. J. Mol. Sci. 2022, 23, 5109. https://doi.org/10.3390/ijms23095109
AMA Style
Li Y, Hecht SS. Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines. International Journal of Molecular Sciences. 2022; 23(9):5109. https://doi.org/10.3390/ijms23095109
Chicago/Turabian StyleLi, Yupeng, and Stephen S. Hecht. 2022. "Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines" International Journal of Molecular Sciences 23, no. 9: 5109. https://doi.org/10.3390/ijms23095109
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.