
Hispolon Methyl Ether, a Hispolon Analog, Suppresses the SRC/STAT3/Survivin Signaling Axis to Induce Cytotoxicity in Human Urinary Bladder Transitional Carcinoma Cell Lines

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. HME Is Cytotoxic to a Panel of Human Urinary Bladder TCC Cell Lines
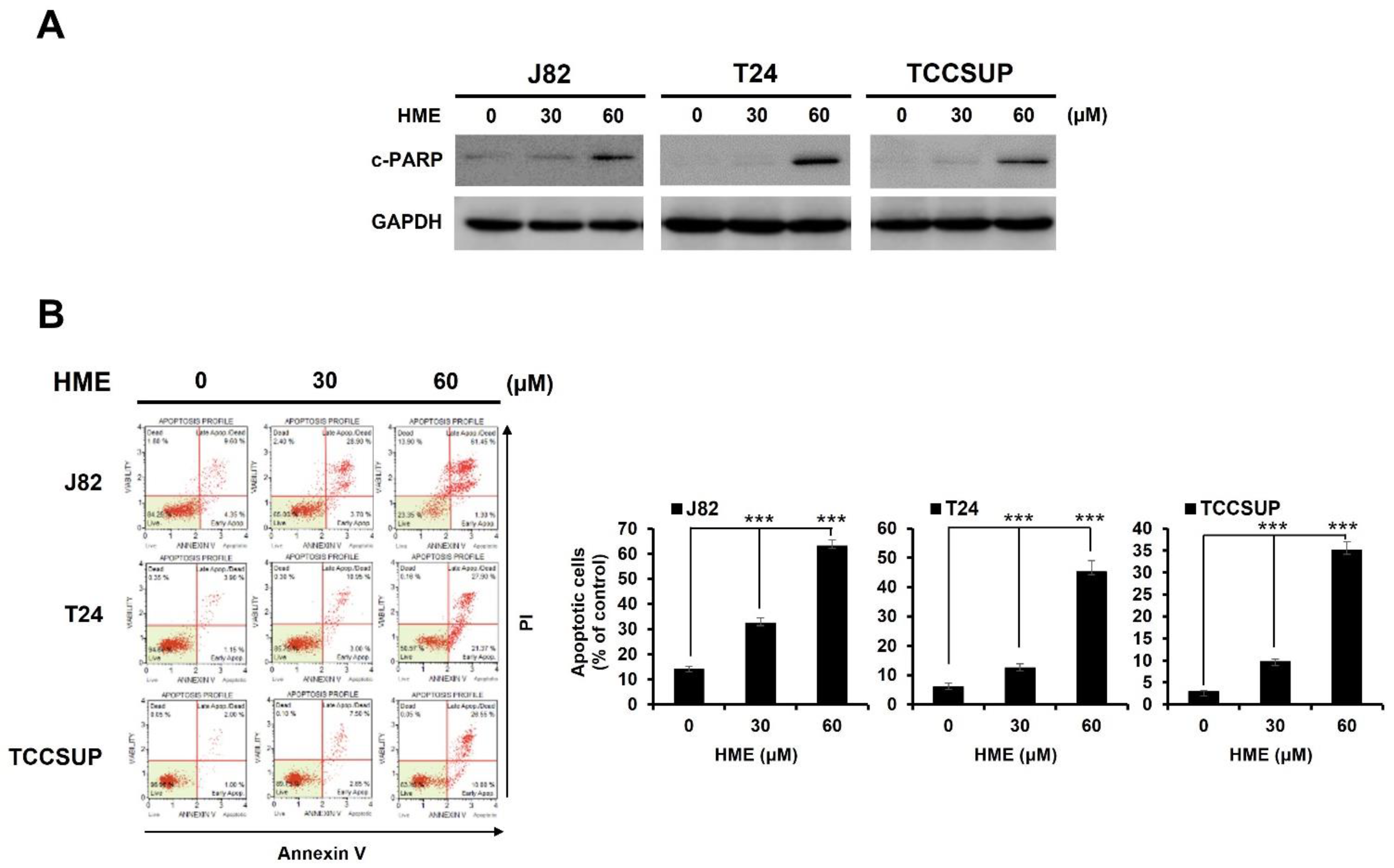
2.2. HME-Induced Bladder Cancer Cytotoxicity Involves Apoptosis Induction
2.3. STAT3 Blockage Is Required for HME-Induced Bladder Cancer Cytotoxicity
2.4. HME Downregulates Survivin, a Canonical STAT3 Downstream Target, to Induce Bladder Cancer Cytotoxicity
2.5. HME Represses SRC to Block STAT3 Activation
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Plasmids
4.3. Cell Culture
4.4. Cytotoxicity Assay
4.5. Apoptosis Assay
4.6. Stable Clone Generation
4.7. Immunoblottinghme
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Khan, R.; Jemal, A. Cancer statistics. In Cancer Rehabilitation: Principles and Practice, 2nd ed.; Stubblefield, M.D., Ed.; Springer Publishing Company: New York, NY, USA, 2019; pp. 10–24. [Google Scholar]
- Lenis, A.T.; Lec, P.M.; Chamie, K.; Mshs, M.D. Bladder cancer: A review. JAMA 2020, 324, 1980–1991. [Google Scholar] [CrossRef]
- Ritch, C.R.; Velasquez, M.C.; Kwon, D.; Becerra, M.F.; Soodana-Prakash, N.; Atluri, V.S.; Almengo, K.; Alameddine, M.; Kineish, O.; Kava, B.R.; et al. Use and validation of the AUA/SUO risk grouping for nonmuscle invasive bladder cancer in a contemporary cohort. J. Urol. 2020, 203, 505–511. [Google Scholar] [CrossRef]
- Von der Maase, H.; Hansen, S.W.; Roberts, J.T.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Bodrogi, I.; Albers, P.; Knuth, A.; Lippert, C.M.; et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: Results of a large, randomized, multinational, multicenter, phase III study. J. Clin. Oncol. 2000, 18, 3068–3077. [Google Scholar] [CrossRef]
- Babjuk, M.; Oosterlinck, W.; Sylvester, R.; Kaasinen, E.; Böhle, A.; Palou-Redorta, J.; Rouprêt, M.; European Association of Urology (EAU). EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder, the 2011 update. Eur. Urol. 2011, 59, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.G.; Oh, W.K.; Galsky, M.D. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J. Clin. 2020, 70, 404–423. [Google Scholar] [CrossRef] [PubMed]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. STAT3 target genes relevant to human cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.Y.; Fan, C.W.; Maa, M.C.; Leu, T.H. Lipopolysaccharide-promoted proliferation of Caco-2 cells is mediated by c-Src induction and ERK activation. BioMedicine 2015, 5, 5. [Google Scholar] [CrossRef]
- Garg, M.; Shanmugam, M.K.; Bhardwaj, V.; Goel, A.; Gupta, R.; Sharma, A.; Baligar, P.; Kumar, A.P.; Goh, B.C.; Wang, L.; et al. The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Med. Res. Rev. 2021, 41, 1291–1336. [Google Scholar] [CrossRef]
- Wang, H.Q.; Man, Q.W.; Huo, F.Y.; Gao, X.; Lin, H.; Li, S.R.; Wang, J.; Su, F.C.; Cai, L.; Shi, Y.; et al. STAT3 pathway in cancers: Past, present, and future. MedComm 2022, 3, e124. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Mahabady, M.K.; Nabavi, N.; Zabolian, A.; Banihashemi, S.M.; Haddadi, A.; Entezari, M.; Hushmandi, K.; Makvandi, P.; et al. Pre-clinical investigation of STAT3 pathway in bladder cancer: Paving the way for clinical translation. Biomed. Pharmacother. 2021, 133, 111077. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Qi, Z.; Pang, Y.; Li, H.; Xie, H.; Wu, J.; Huang, Y.; Zhu, Y.; Shen, Y.; Zhu, Y.; et al. Retinoic acid-related orphan receptor C regulates proliferation, glycolysis, and chemoresistance via the PD-L1/ITGB6/STAT3 signaling axis in bladder cancer. Cancer Res. 2019, 79, 2604–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Chen, X.; Xie, R.; Huang, M.; Dong, W.; Han, J.; Zhang, J.; Zhou, Q.; Li, H.; Huang, J.; et al. DANCR promotes metastasis and proliferation in bladder cancer cells by enhancing IL-11-STAT3 signaling and CCND1 expression. Mol. Ther. 2019, 27, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Hu, X.; Tong, S.; Mo, M.; He, W.; Wang, L.; Li, Y. MEST promotes bladder cancer cell proliferation, migration and invasion via STAT3/Twist-1-mediated EMT. Transl. Cancer Res. 2020, 9, 6178–6188. [Google Scholar] [CrossRef]
- He, H.; Yi, L.; Zhang, B.; Yan, B.; Xiao, M.; Ren, J.; Zi, D.; Zhu, L.; Zhong, Z.; Zhao, X.; et al. USP24-GSDMB complex promotes bladder cancer proliferation via activation of the STAT3 pathway. Int. J. Biol. Sci. 2021, 17, 2417–2429. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, Z.; Chen, H.; Bao, W.; Kuang, X.; Zhou, P.; Gao, Z.; Li, D.; Xie, X.; Yang, C.; et al. SBSN drives bladder cancer metastasis via EGFR/SRC/STAT3 signalling. Br. J. Cancer 2022, 127, 211–222. [Google Scholar] [CrossRef]
- Tsujita, Y.; Horiguchi, A.; Tasaki, S.; Isono, M.; Asano, T.; Ito, K.; Asano, T.; Mayumi, Y.; Kushibiki, T. STAT3 inhibition by WP1066 suppresses the growth and invasiveness of bladder cancer cells. Oncol. Rep. 2017, 38, 2197–2204. [Google Scholar] [CrossRef] [Green Version]
- Korac-Prlic, J.; Degoricija, M.; Vilović, K.; Haupt, B.; Ivanišević, T.; Franković, L.; Grivennikov, S.; Terzić, J. Targeting Stat3 signaling impairs the progression of bladder cancer in a mouse model. Cancer Lett. 2020, 490, 89–99. [Google Scholar] [CrossRef]
- Hindupur, S.V.; Schmid, S.C.; Koch, J.A.; Youssef, A.; Baur, E.M.; Wang, D.; Horn, T.; Slotta-Huspenina, J.; Gschwend, J.E.; Holm, P.S.; et al. STAT3/5 inhibitors suppress proliferation in bladder cancer and enhance oncolytic adenovirus therapy. Int. J. Mol. Sci. 2020, 21, 1106. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Sun, J.; Sadahira, T.; Xu, N.; Wada, K.; Liu, C.; Araki, M.; Xu, A.; Watanabe, M.; Nasu, Y.; et al. Discovery and validation of nitroxoline as a novel STAT3 inhibitor in drug-resistant urothelial bladder cancer. Int. J. Biol. Sci. 2021, 17, 3255–3267. [Google Scholar] [CrossRef]
- Zhu, T.; Kim, S.H.; Chen, C.Y. A medicinal mushroom: Phellinus linteus. Curr. Med. Chem. 2008, 15, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tan, H.; Liu, Q.; Zheng, X.; Zhang, H.; Liu, Y.; Xu, L. A review: The bioactivities and pharmacological applications of Phellinus linteus. Molecules 2019, 24, 1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarfraz, A.; Rasul, A.; Sarfraz, I.; Shah, M.A.; Hussain, G.; Shafiq, N.; Masood, M.; Adem, Ş.; Sarker, S.D.; Li, X. Hispolon: A natural polyphenol and emerging cancer killer by multiple cellular signaling pathways. Environ. Res. 2020, 190, 110017. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Ali, E.S.; Khan, I.N.; Shaw, S.; Uddin, S.J.; Rouf, R.; Dev, S.; Saravi, S.S.S.; Das, N.; Tripathi, S.; et al. Anticancer perspectives on the fungal-derived polyphenolic hispolon. Anticancer Agents Med. Chem. 2020, 20, 1636–1647. [Google Scholar] [CrossRef]
- Ravindran, J.; Subbaraju, G.V.; Ramani, M.V.; Sung, B.; Aggarwal, B.B. Bisdemethylcurcumin and structurally related hispolon analogues of curcumin exhibit enhanced prooxidant, anti-proliferative and anti-inflammatory activities in vitro. Biochem. Pharmacol. 2010, 79, 1658–1666. [Google Scholar] [CrossRef] [Green Version]
- Balaji, N.V.; Ramani, M.V.; Viana, A.G.; Sanglard, L.P.; White, J.; Mulabagal, V.; Lee, C.; Gana, T.J.; Egiebor, N.O.; Subbaraju, G.V.; et al. Design, synthesis and in vitro cell-based evaluation of the anti-cancer activities of hispolon analogs. Bioorg. Med. Chem. 2015, 23, 2148–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.C.; Hsieh, Y.C.; Li, L.H.; Chang, C.C.; Janoušková, K.; Ramani, M.V.; Subbaraju, G.V.; Cheng, K.T.; Chang, C.C. Dehydroxyhispolon methyl ether, a hispolon derivative, inhibits WNT/β-catenin signaling to elicit human colorectal carcinoma cell apoptosis. Int. J. Mol. Sci. 2020, 21, 8839. [Google Scholar] [CrossRef]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of Stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.L.; Huang, G.J.; Lu, T.J.; Wu, J.B.; Wu, C.H.; Yang, T.C.; Iizuka, A.; Chen, Y.F. Hispolon from Phellinus linteus has antiproliferative effects via MDM2-recruited ERK1/2 activity in breast and bladder cancer cells. Food Chem. Toxicol. 2009, 47, 2013–2021. [Google Scholar] [CrossRef]
- Yang, J.; Wang, L.; Guan, X.; Qin, J.J. Inhibiting STAT3 signaling pathway by natural products for cancer prevention and therapy: In vitro and in vivo activity and mechanisms of action. Pharmacol. Res. 2022, 182, 106357. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef]
- Li, F.; Aljahdali, I.; Ling, X. Cancer therapeutics using survivin BIRC5 as a target: What can we do after over two decades of study? J. Exp. Clin. Cancer Res. 2019, 38, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.D.; Wheeler, M.A.; Plescia, J.; Colberg, J.W.; Weiss, R.M.; Altieri, D.C. Urine detection of survivin and diagnosis of bladder cancer. JAMA 2001, 285, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, W.; Chen, N.; Zhang, Y.; Zeng, H.; Chen, X.; He, Y.; Wang, X.; Zhou, Q. Survivin nuclear labeling index: A superior biomarker in superficial urothelial carcinoma of human urinary bladder. Mod. Pathol. 2006, 19, 1487–1497. [Google Scholar] [CrossRef] [Green Version]
- Shariat, S.F.; Ashfaq, R.; Karakiewicz, P.I.; Saeedi, O.; Sagalowsky, A.I.; Lotan, Y. Survivin expression is associated with bladder cancer presence, stage, progression, and mortality. Cancer 2007, 109, 1106–1113. [Google Scholar] [CrossRef]
- Margulis, V.; Lotan, Y.; Shariat, S.F. Survivin: A promising biomarker for detection and prognosis of bladder cancer. World J. Urol. 2008, 26, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Jeon, C.; Kim, M.; Kwak, C.; Kim, H.H.; Ku, J.H. Prognostic role of survivin in bladder cancer: A systematic review and meta-analysis. PLoS ONE 2013, 8, e76719. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Shen, D.; Kong, C.; Zhang, Z.; Zeng, Y.; Lin, X.; Liu, X. NF-κB suppresses apoptosis and promotes bladder cancer cell proliferation by upregulating survivin expression in vitro and in vivo. Sci. Rep. 2017, 7, 40723. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, A.; Finn, R.S. SRC: A century of science brought to the clinic. Neoplasia 2010, 12, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.H.; Yang, H.Y.; Hsu, Y.F.; Chiu, P.T.; Ou, G.; Hsu, M.J. Src contributes to IL6-induced vascular endothelial growth factor-C expression in lymphatic endothelial cells. Angiogenesis 2014, 17, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Qiu, Y. Interaction between Src and a C-terminal proline-rich motif of Akt is required for Akt activation. J. Biol. Chem. 2003, 278, 15789–15793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheskis, B.J.; Greger, J.; Cooch, N.; McNally, C.; Mclarney, S.; Lam, H.S.; Rutledge, S.; Mekonnen, B.; Hauze, D.; Nagpal, S.; et al. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids 2008, 73, 901–905. [Google Scholar] [CrossRef]
- Wu, W.; Sun, Z.; Wu, J.; Peng, X.; Gan, H.; Zhang, C.; Ji, L.; Xie, J.; Zhu, H.; Ren, S.; et al. Trihydrophobin 1 phosphorylation by c-Src regulates MAPK/ERK signaling and cell migration. PLoS ONE 2012, 7, e29920. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, J.; Li, X.; Yang, S.; Yue, S.; Zhang, J.; Yang, X.; Zhu, T.; Jiang, R.; Yang, W. Downregulation of Src enhances the cytotoxic effect of temozolomide through AKT in glioma. Oncol. Rep. 2013, 29, 1395–1398. [Google Scholar] [CrossRef] [Green Version]
- Qi, S.; Feng, Z.; Li, Q.; Qi, Z.; Zhang, Y. Inhibition of ROS-mediated activation Src-MAPK/AKT signaling by orientin alleviates H2O2-induced apoptosis in PC12 cells. Drug Des. Dev. Ther. 2018, 12, 3973–3984. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.P.; Li, S.; Chuang, W.L.; Li, C.H.; Chen, G.J.; Chang, C.C.; Or, C.R.; Lin, P.Y.; Chang, C.C. Blockade of STAT3 signaling contributes to anticancer effect of 5-Acetyloxy-6,7,8,4′-Tetra-Methoxyflavone, a Tangeretin derivative, on human glioblastoma multiforme cells. Int. J. Mol. Sci. 2019, 20, 3366. [Google Scholar] [CrossRef] [Green Version]
- Or, C.R.; Huang, C.W.; Chang, C.C.; Lai, Y.C.; Chen, Y.J.; Chang, C.C. Obatoclax, a pan-bcl-2 inhibitor, downregulates Survivin to induce apoptosis in human colorectal carcinoma cells via suppressing WNT/β-catenin signaling. Int. J. Mol. Sci. 2020, 21, 1773. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, M.-Y.; Yang, W.-T.; Ho, Y.-J.; Chang, G.-M.; Chang, H.-H.; Hsu, C.-Y.; Chang, C.-C.; Chen, Y.-H. Hispolon Methyl Ether, a Hispolon Analog, Suppresses the SRC/STAT3/Survivin Signaling Axis to Induce Cytotoxicity in Human Urinary Bladder Transitional Carcinoma Cell Lines. Int. J. Mol. Sci. 2023, 24, 138. https://doi.org/10.3390/ijms24010138
Kuo M-Y, Yang W-T, Ho Y-J, Chang G-M, Chang H-H, Hsu C-Y, Chang C-C, Chen Y-H. Hispolon Methyl Ether, a Hispolon Analog, Suppresses the SRC/STAT3/Survivin Signaling Axis to Induce Cytotoxicity in Human Urinary Bladder Transitional Carcinoma Cell Lines. International Journal of Molecular Sciences. 2023; 24(1):138. https://doi.org/10.3390/ijms24010138
Chicago/Turabian StyleKuo, Min-Yung, Wei-Ting Yang, Yann-Jen Ho, Ge-Man Chang, Hsiung-Hao Chang, Chao-Yu Hsu, Chia-Che Chang, and Yi-Hsin Chen. 2023. "Hispolon Methyl Ether, a Hispolon Analog, Suppresses the SRC/STAT3/Survivin Signaling Axis to Induce Cytotoxicity in Human Urinary Bladder Transitional Carcinoma Cell Lines" International Journal of Molecular Sciences 24, no. 1: 138. https://doi.org/10.3390/ijms24010138