
Porcine Relaxin but Not Serelaxin Shows Residual Bioactivity after In Vitro Simulated Intestinal Digestion—Clues for the Development of New Relaxin Peptide Agonists Suitable for Oral Delivery

 ,
, 
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
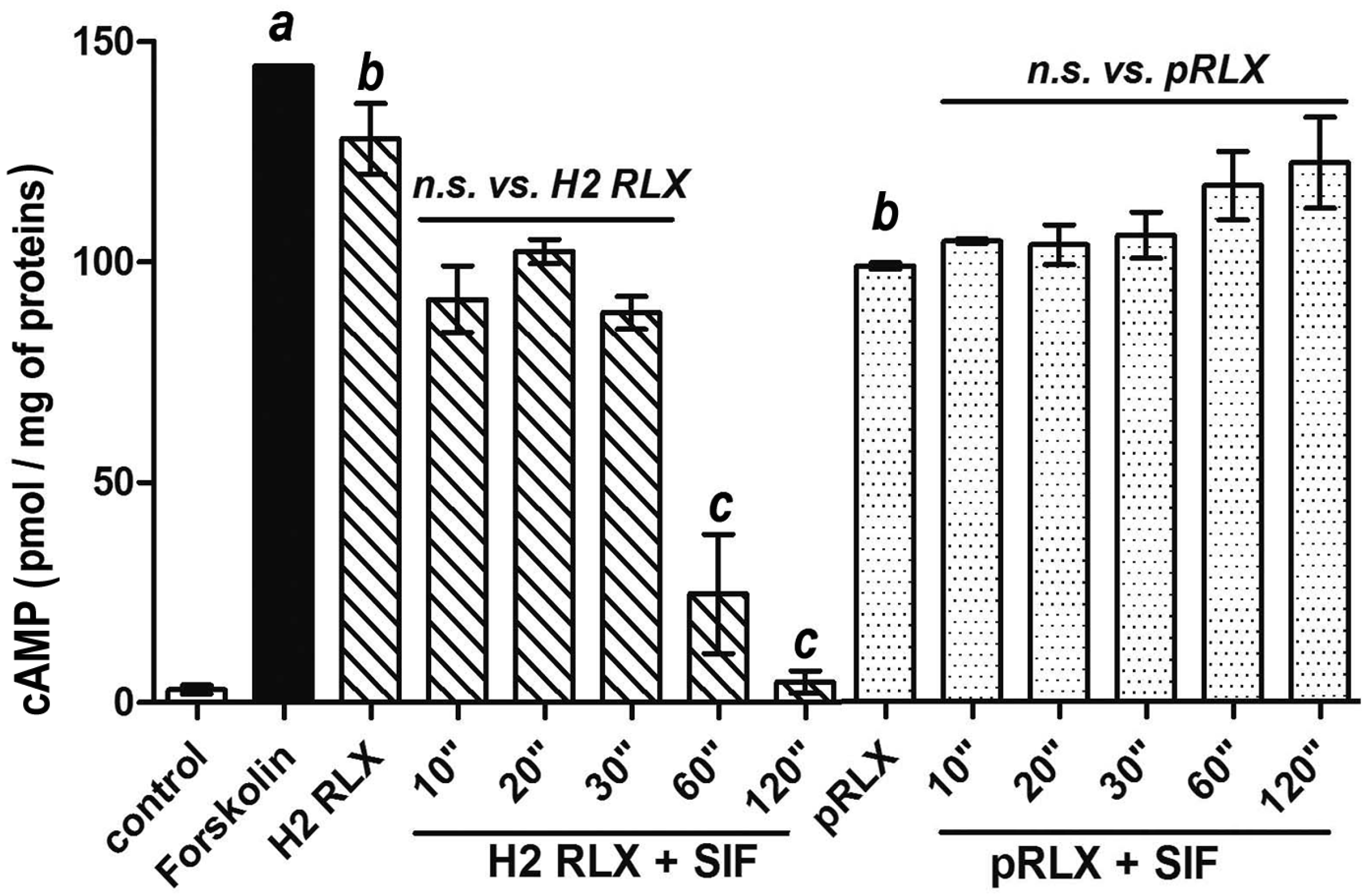
2.1. cAMP Response by THP-1 Cells
2.2. Time Course of pRLX Digestion
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Simulated Intestinal Digestion
4.3. HPLC Analysis
4.4. Cell Culture and cAMP Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nistri, S.; Bigazzi, M.; Bani, D. Relaxin as a cardiovascular hormone: Physiology, pathophysiology and therapeutic promises. Cardiovasc. Hematol. Agents Med. Chem. 2007, 5, 101–108. [Google Scholar] [CrossRef]
- Du, X.J.; Bathgate, R.A.; Samuel, C.S.; Dart, A.M.; Summers, R.J. Cardiovascular effects of relaxin: From basic science to clinical therapy. Nat. Rev. Cardiol. 2010, 7, 48–58. [Google Scholar] [CrossRef]
- Samuel, C.S.; Royce, S.G.; Hewitson, T.D.; Denton, K.M.; Cooney, T.E.; Bennett, R.G. Anti-fibrotic actions of relaxin. Br. J. Pharmacol. 2017, 174, 962–976. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, M.; Du, X.J.; Dschietzig, T.B.; Summers, R.J. The actions of relaxin on the human cardiovascular system. Br. J. Pharmacol. 2017, 174, 933–949. [Google Scholar] [CrossRef] [Green Version]
- Metra, M.; Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Pang, P.S.; Ponikowski, P.; Voors, A.A.; et al. RELAX-AHF-2 Committees Investigators. Effects of serelaxin in patients with acute heart failure. N. Engl. J. Med. 2019, 381, 716–726. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Davison, B.A.; Cotter, G.; Maggioni, A.P.; Sato, N.; Chioncel, O.; Ertl, G.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; et al. Effects of serelaxin in patients admitted for acute heart failure: A meta-analysis. Eur. J. Heart Fail. 2020, 22, 315–329. [Google Scholar] [CrossRef]
- Chen, S.A.; Perlman, A.J.; Spanski, N.; Peterson, C.M.; Sanders, S.W.; Jaffe, R.; Martin, M.; Yalcinkaya, T.; Cefalo, R.C.; Chescheir, N.C.; et al. The pharmacokinetics of recombinant human relaxin in nonpregnant women after intravenous, intravaginal, and intracervical administration. Pharm. Res. 1993, 10, 834–838. [Google Scholar] [CrossRef]
- Hewitson, T.D.; Ho, W.Y.; Samuel, C.S. Antifibrotic properties of relaxin: In vivo mechanism of action in experimental renal tubulointerstitial fibrosis. Endocrinology 2010, 151, 4938–4948. [Google Scholar] [CrossRef] [Green Version]
- Perkins, B.A.; Sherr, J.L.; Mathieu, C. Type 1 diabetes glycemic management: Insulin therapy, glucose monitoring, and automation. Science 2021, 373, 522–527. [Google Scholar] [CrossRef]
- Bratke, H.; Margeirsdottir, H.D.; Assmus, J.; Njølstad, P.R.; Skrivarhaug, T. Does current diabetes technology improve metabolic control? A cross-sectional study on the use of insulin pumps and continuous glucose monitoring devices in a nationwide pediatric population. Diabetes Ther. 2021, 12, 2571–2583. [Google Scholar] [CrossRef]
- Bani, D.; Bigazzi, M. Relaxin as a cardiovascular drug: A promise kept. Curr. Drug Saf. 2011, 6, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Carino, G.P.; Mathiowitz, E. Oral insulin delivery. Adv. Drug Deliv. Rev. 1999, 35, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, L.; Jacques, Y. Oral insulin and buccal insulin: A critical reappraisal. J. Diabetes Sci. Technol. 2009, 3, 568–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathgate, R.A.; Halls, M.L.; van der Westhuizen, E.T.; Callander, G.E.; Kocan, M.; Summers, R.J. Relaxin family peptides and their receptors. Physiol. Rev. 2013, 93, 405–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.A.; Rosengren, K.J.; Samuel, C.S.; Shabanpoor, F.; Chan, L.J.; Bathgate, R.A.; Wade, J.D. The minimal active structure of human relaxin-2. J. Biol. Chem. 2011, 286, 37555–37565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, B.T.; Yang, L.; Sanborn, B.M.; Dessauer, C.W. Phosphoinositide 3-kinase activity is required for biphasic stimulation of cyclic adenosine 3′,5′-monophosphate by relaxin. Mol. Endocrinol. 2003, 17, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Bani, D.; Nistri, S.; Cinci, L.; Giannini, L.; Princivalle, M.; Elliott, L.; Bigazzi, M.; Masini, E. A novel, simple bioactivity assay for relaxin based on inhibition of platelet aggregation. Regul. Pept. 2007, 144, 10–16. [Google Scholar] [CrossRef]
- Halls, M.L.; Bond, C.P.; Sudo, S.; Kumagai, J.; Ferraro, T.; Layfield, S.; Bathgate, R.A.; Summers, R.J. Multiple binding sites revealed by interaction of relaxin family peptides with native and chimeric relaxin family peptide receptors 1 and 2 (LGR7 and LGR8). J. Pharmacol. Exp. Ther. 2005, 313, 677–687. [Google Scholar] [CrossRef]
- Sherwood, C.D.; O’Byrne, E.M. Purification and characterization of porcine relaxin. Arch. Biochem. Biophys. 1974, 160, 185–196. [Google Scholar] [CrossRef]
- Kohsaka, T.; Takahara, H.; Sugawarao, K.; Tagami, S. Endogenous heterogeneity of relaxin and sequence of the major form in pregnant sow ovaries. Biol. Chem. Hoppe-Seyler 1993, 374, 203–210. [Google Scholar] [CrossRef]
- Antunes, F.; Andrade, F.; Ferreira, D.; Nielsen, H.M.; Sarmento, B. Models to predict intestinal absorption of therapeutic peptides and proteins. Curr. Drug Metab. 2013, 14, 4–20. [Google Scholar] [CrossRef]
- Frokjaer, S.; Otzen, D.E. Protein drug stability: A formulation challenge. Nat. Rev. Drug Discov. 2005, 4, 298–306. [Google Scholar] [CrossRef]
- D’Ercole, A.; Nistri, S.; Pacini, L.; Carotenuto, A.; Santoro, F.; Papini, A.M.; Bathgate, R.A.D.; Bani, D.; Rovero, P. Synthetic short-chain peptide analogues of H1 relaxin lack affinity for the RXFP1 receptor and relaxin-like bioactivity. Clues to a better understanding of relaxin agonist design. Front. Pharmacol. 2022, 13, 942178. [Google Scholar] [CrossRef]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Toward oral delivery of biopharmaceuticals: An assessment of the gastrointestinal stability of 17 peptide drugs. Mol. Pharm. 2015, 12, 966–973.175. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Lee, H.Y.; Sherwood, O.D. Porcine and human relaxin bioactivity: Bioactivities of porcine relaxin and human relaxin do not differ in mice and rats. Ann. N. Y. Acad. Sci. 2005, 1041, 126–131. [Google Scholar] [CrossRef]
- Parkes, D.G.; Mace, K.F.; Trautmann, M.E. Discovery and development of exenatide: The first antidiabetic agent to leverage the multiple benefits of the incretin hormone, GLP-1. Expert Opin. Drug. Discov. 2013, 8, 219–244. [Google Scholar] [CrossRef] [PubMed]
- Bani, D.; Yue, S.K.; Bigazzi, M. Clinical profile of relaxin, a possible new drug for human use. Curr. Drug Saf. 2009, 4, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Aungst, B.J. Intestinal permeation enhancers. J. Pharm. Sci. 2000, 89, 429–442. [Google Scholar] [CrossRef]
- Rehmani, S.; Dixon, J.E. Oral delivery of anti-diabetes therapeutics using cell penetrating and transcytosing peptide strategies. Peptides 2018, 100, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Borges, O.; Cordeiro-da-Silva, A.; Romeijn, S.G.; Amidi, M.; de Sousa, A.; Borchard, G.; Junginger, H.E. Uptake studies in rat Peyer’s patches, cytotoxicity and release studies of alginate coated chitosan nanoparticles for mucosal vaccination. J. Control Release 2006, 114, 348–358. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacini, L.; D’Ercole, A.; Papini, A.M.; Bani, D.; Nistri, S.; Rovero, P. Porcine Relaxin but Not Serelaxin Shows Residual Bioactivity after In Vitro Simulated Intestinal Digestion—Clues for the Development of New Relaxin Peptide Agonists Suitable for Oral Delivery. Int. J. Mol. Sci. 2023, 24, 48. https://doi.org/10.3390/ijms24010048
Pacini L, D’Ercole A, Papini AM, Bani D, Nistri S, Rovero P. Porcine Relaxin but Not Serelaxin Shows Residual Bioactivity after In Vitro Simulated Intestinal Digestion—Clues for the Development of New Relaxin Peptide Agonists Suitable for Oral Delivery. International Journal of Molecular Sciences. 2023; 24(1):48. https://doi.org/10.3390/ijms24010048
Chicago/Turabian StylePacini, Lorenzo, Annunziata D’Ercole, Anna Maria Papini, Daniele Bani, Silvia Nistri, and Paolo Rovero. 2023. "Porcine Relaxin but Not Serelaxin Shows Residual Bioactivity after In Vitro Simulated Intestinal Digestion—Clues for the Development of New Relaxin Peptide Agonists Suitable for Oral Delivery" International Journal of Molecular Sciences 24, no. 1: 48. https://doi.org/10.3390/ijms24010048