
H2O2 Mediates VEGF- and Flow-Induced Dilations of Coronary Arterioles in Early Type 1 Diabetes: Role of Vascular Arginase and PI3K-Linked eNOS Uncoupling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Early Diabetes Impairs Flow- and VEGF-Induced Vasodilations
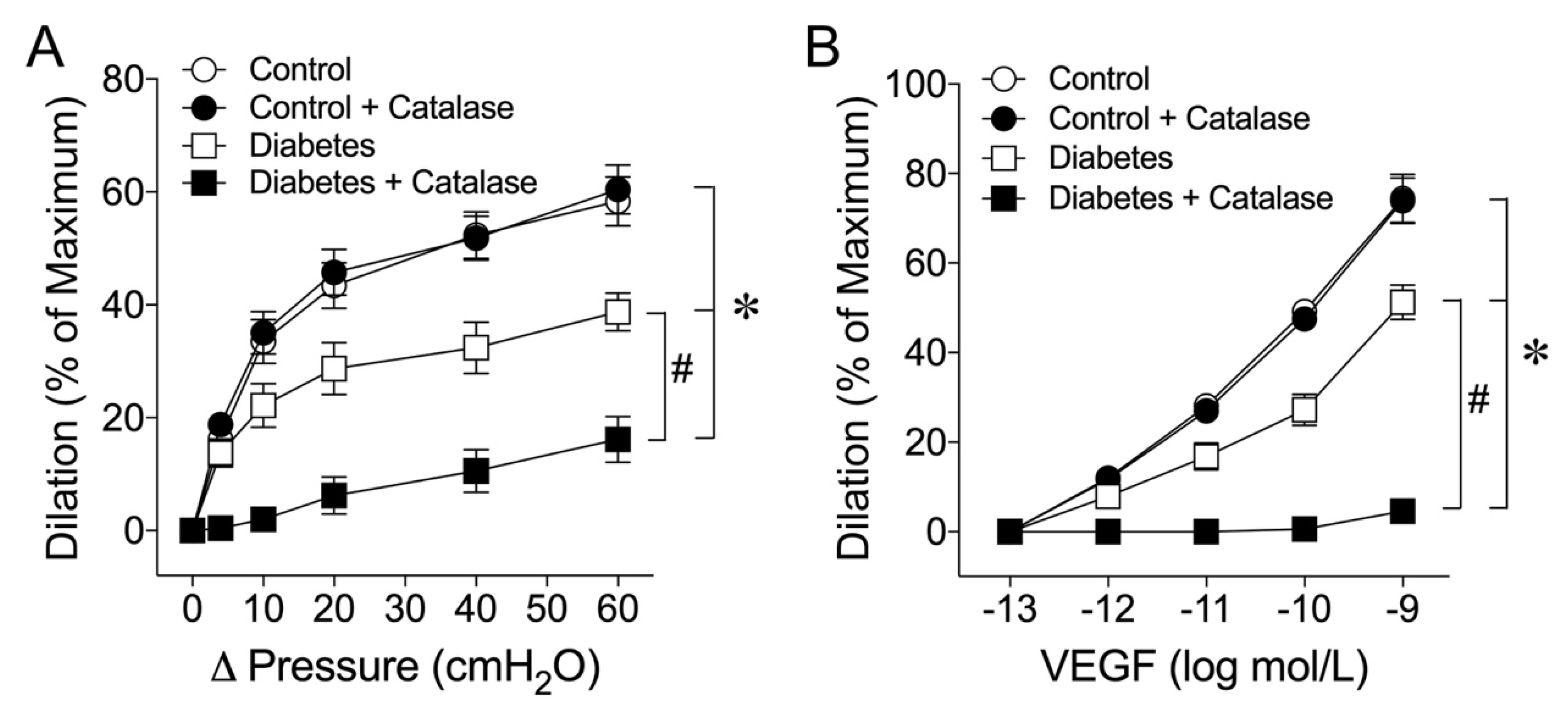
2.2. Role of H2O2 in Flow- and VEGF-Induced Coronary Arteriolar Dilations
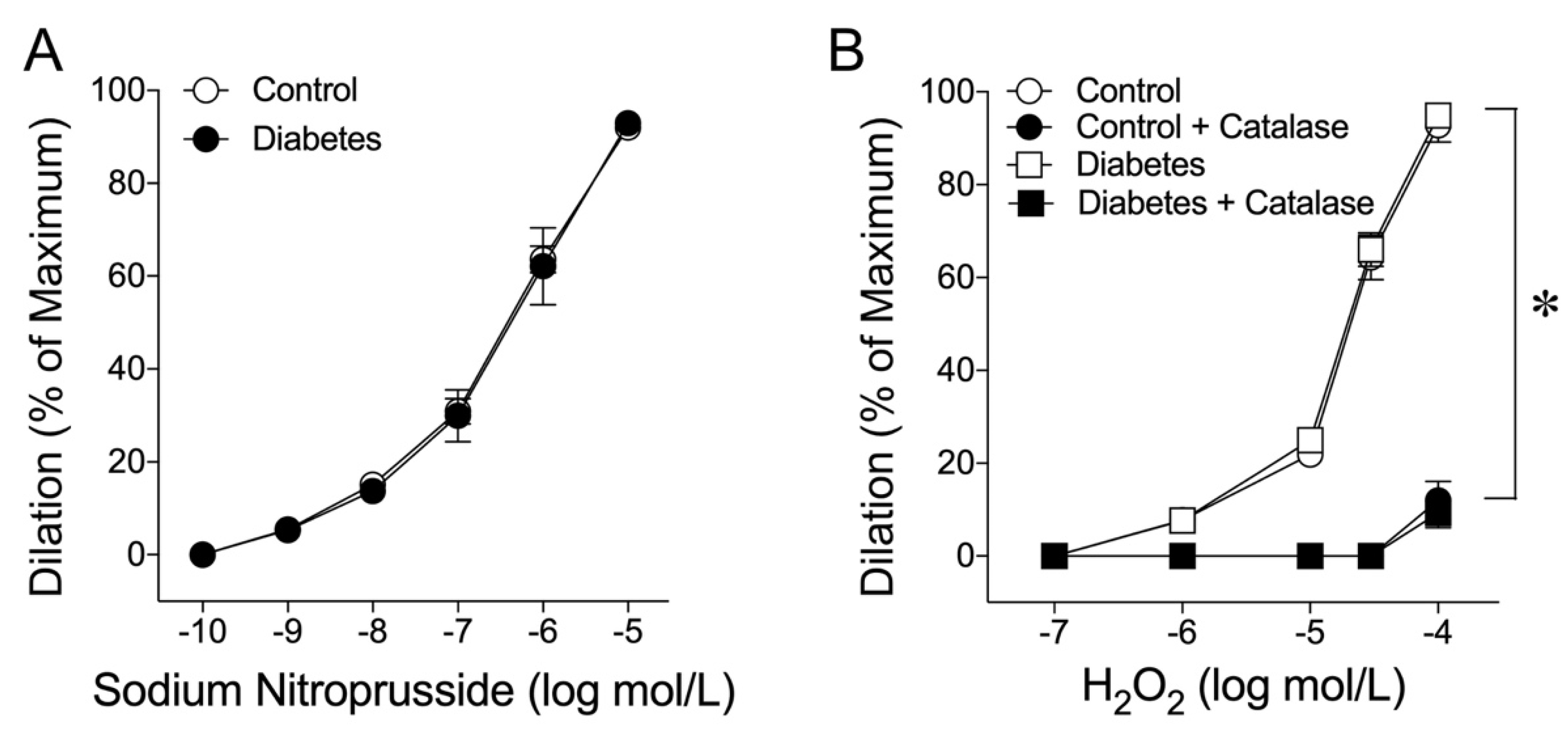
2.3. Coronary Arteriolar Dilations to NO Donor and H2O2
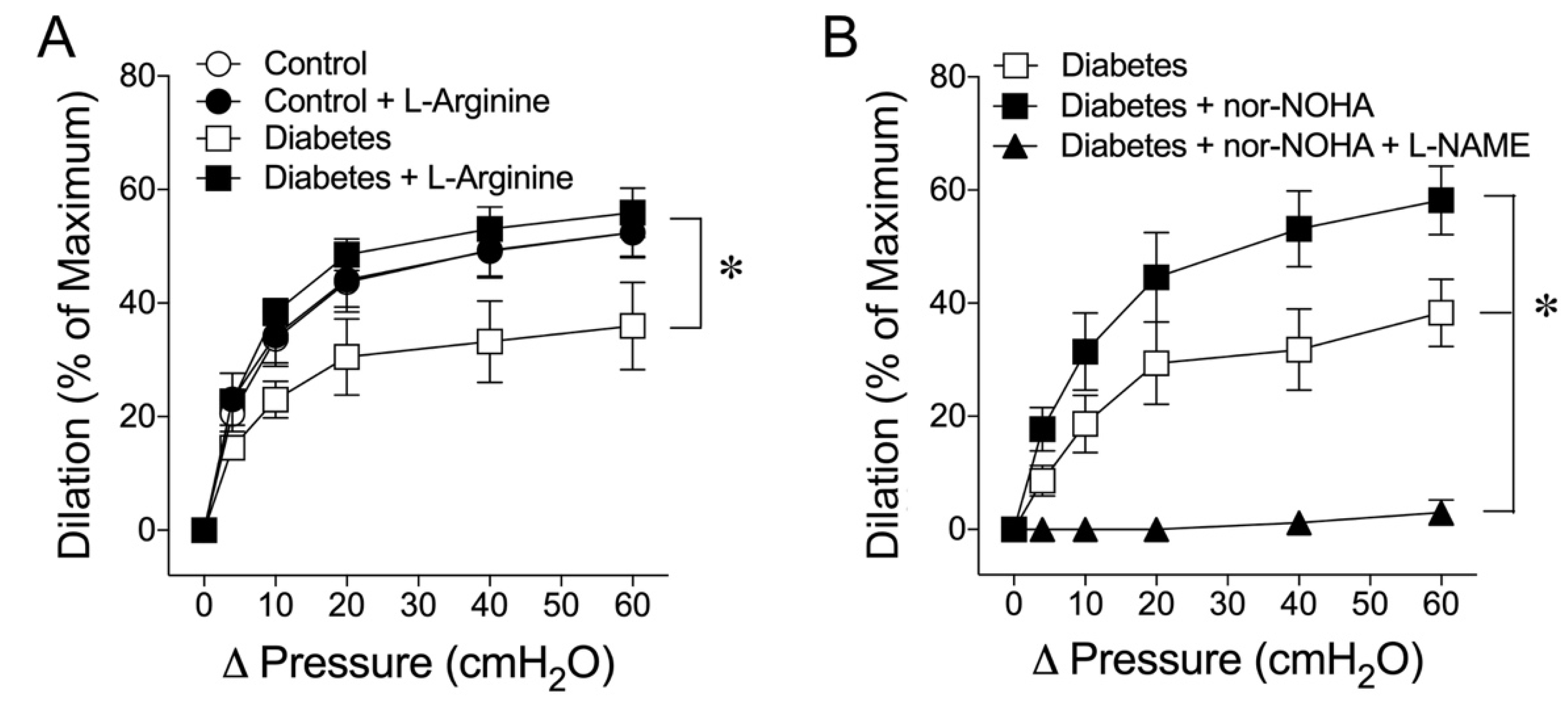
2.4. Vascular L-Arginine Availability and the Role of Arginase in Early Diabetes
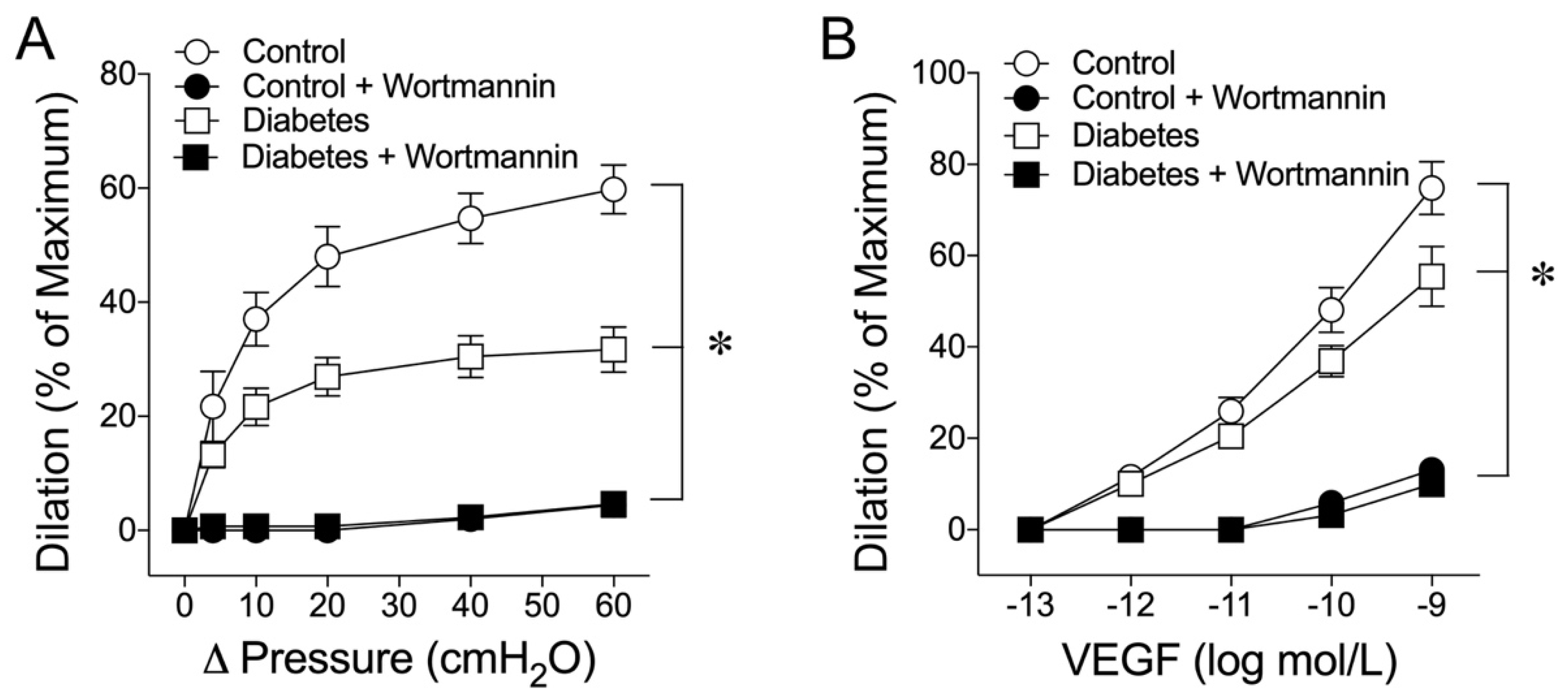
2.5. Role of Phosphoinositide 3-Kinase (PI3K) in Flow- and VEGF-Induced Vasodilations
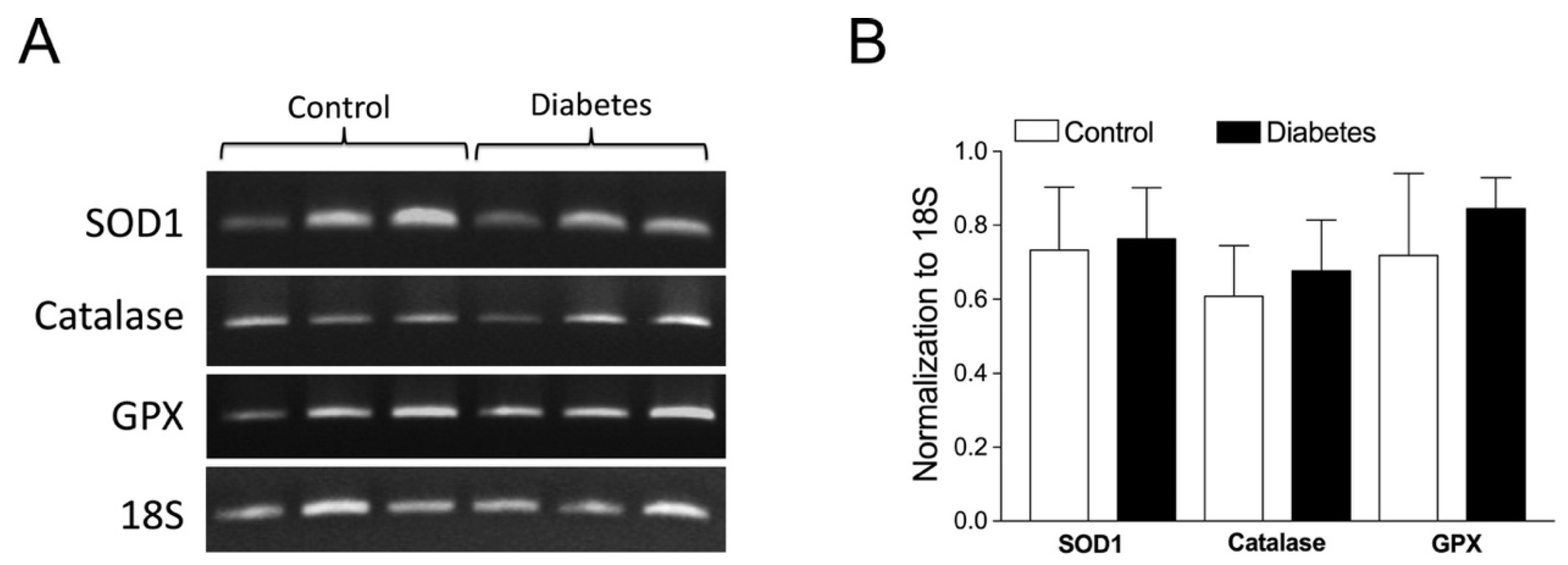
2.6. Role of Vascular Superoxide Dismutase 1 (SOD1), Catalase, and Glutathione Peroxidase (GPX) mRNA Expressions
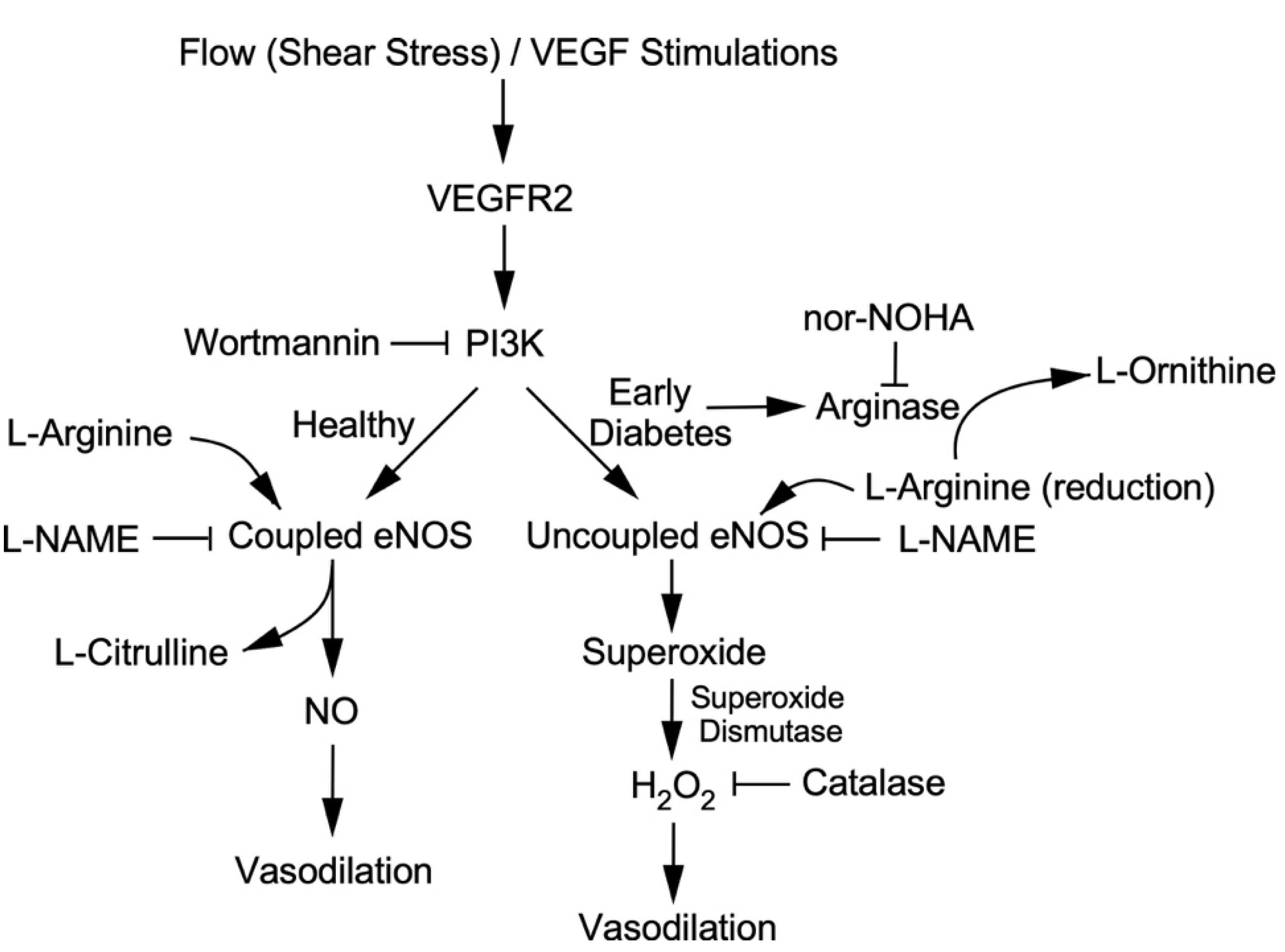
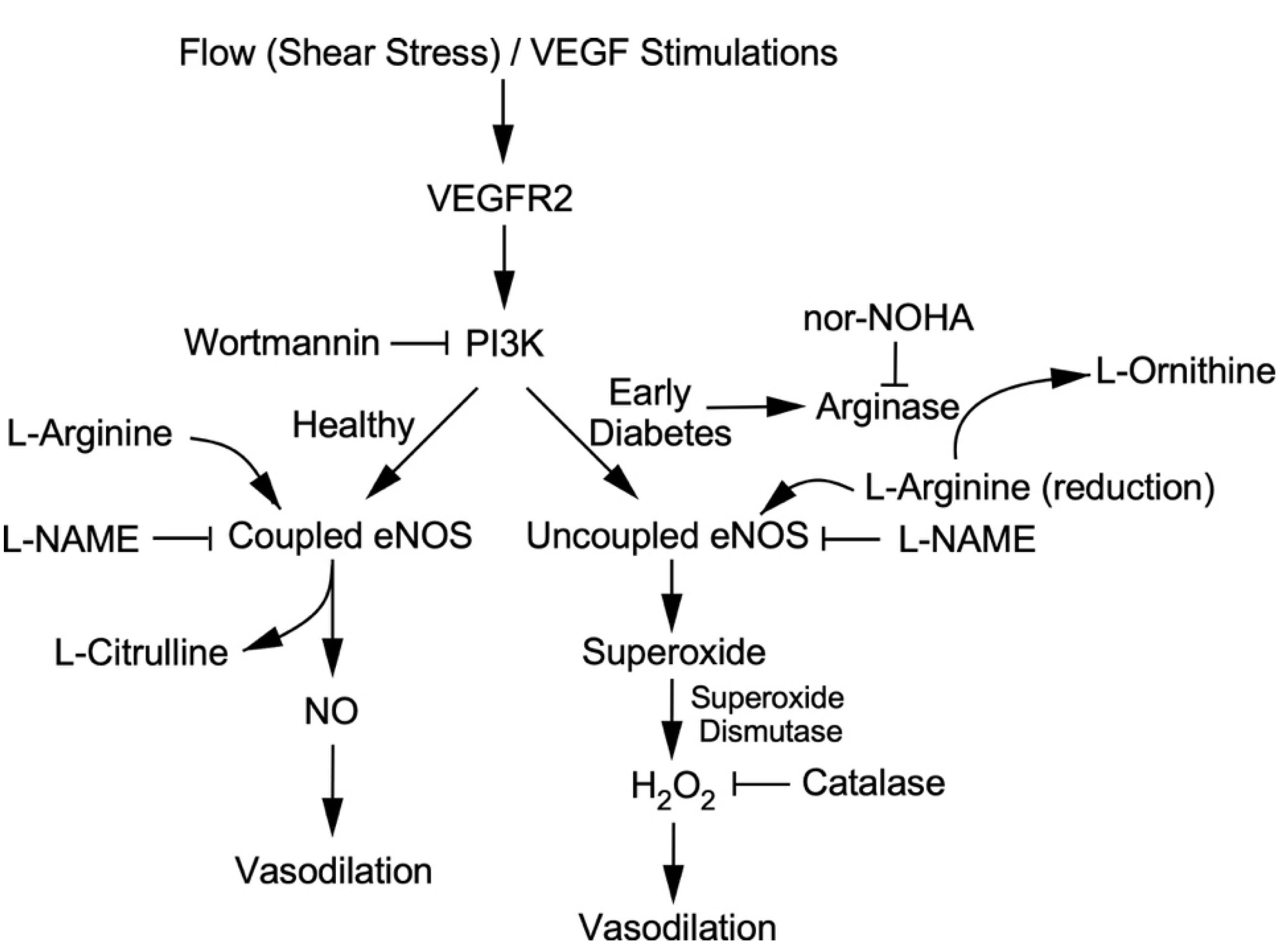
3. Discussion
4. Materials and Methods
4.1. Porcine Diabetes Model
4.2. Isolation and Cannulation of Coronary Arterioles
4.3. Study of Vasomotor Function
4.4. RNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis of Antioxidant Transcripts
4.5. Chemicals
4.6. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momose, M.; Abletshauser, C.; Neverve, J.; Nekolla, S.G.; Schnell, O.; Standl, E.; Schwaiger, M.; Bengel, F.M. Dysregulation of coronary microvascular reactivity in asymptomatic patients with type 2 diabetes mellitus. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 1675–1679. [Google Scholar] [CrossRef] [PubMed]
- Tickerhoof, M.M.; Farrell, P.A.; Korzick, D.H. Alterations in rat coronary vasoreactivity and vascular protein kinase C isoforms in Type 1 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2694–H2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, J.O.; Quinones, M.J.; Hernandez-Pampaloni, M.; Facta, A.D.; Schindler, T.H.; Sayre, J.W.; Hsueh, W.A.; Schelbert, H.R. Coronary circulatory dysfunction in insulin resistance, impaired glucose tolerance, and type 2 diabetes mellitus. Circulation 2005, 111, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, V.L.; Naya, M.; Foster, C.R.; Gaber, M.; Hainer, J.; Klein, J.; Dorbala, S.; Blankstein, R.; Di Carli, M.F. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 2012, 126, 1858–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabe, S.A.; Feng, J.; Sellke, F.W.; Abid, M.R. Mechanisms and clinical implications of endothelium-dependent vasomotor dysfunction in coronary microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H819–H841. [Google Scholar] [CrossRef] [PubMed]
- Koller, A.; Balasko, M.; Bagi, Z. Endothelial regulation of coronary microcirculation in health and cardiometabolic diseases. Intern. Emerg. Med. 2013, 8 (Suppl. S1), 51–54. [Google Scholar] [CrossRef] [Green Version]
- Kibel, A.; Selthofer-Relatic, K.; Drenjancevic, I.; Bacun, T.; Bosnjak, I.; Kibel, D.; Gros, M. Coronary microvascular dysfunction in diabetes mellitus. J. Int. Med. Res. 2017, 45, 1901–1929. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Loffredo, G.; Rinaldi, L.; Catalini, C.; Gjeloshi, K.; Albanese, G.; Di Martino, A.; et al. Coronary microvascular dysfunction in diabetes mellitus: Pathogenetic mechanisms and potential therapeutic options. Biomedicines 2022, 10, 2274. [Google Scholar] [CrossRef]
- Boodhwani, M.; Sodha, N.R.; Mieno, S.; Xu, S.H.; Feng, J.; Ramlawi, B.; Clements, R.T.; Sellke, F.W. Functional, cellular, and molecular characterization of the angiogenic response to chronic myocardial ischemia in diabetes. Circulation 2007, 116, I-31–I-37. [Google Scholar] [CrossRef] [Green Version]
- Hein, T.W.; Xu, X.; Ren, Y.; Xu, W.; Tsai, S.H.; Thengchaisri, N.; Kuo, L. Requisite roles of LOX-1, JNK, and arginase in diabetes-induced endothelial vasodilator dysfunction of porcine coronary arterioles. J. Mol. Cell. Cardiol. 2019, 131, 82–90. [Google Scholar] [CrossRef]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am. J. Physiol. 1990, 259, H1063–H1070. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.C.; Kuo, L. Interaction between adenosine and flow-induced dilation in coronary microvascular network. Am. J. Physiol. 1997, 272, H1571–H1581. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Chilian, W.M.; Davis, M.J. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am. J. Physiol. 1991, 261, H1706–H1715. [Google Scholar] [CrossRef] [PubMed]
- Goodwill, A.G.; Dick, G.M.; Kiel, A.M.; Tune, J.D. Regulation of coronary blood flow. Compr. Physiol. 2017, 7, 321–382. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.T.; Pham, I.; Valensi, P.; Rousseau, H.; Vicaut, E.; Laguillier-Morizot, C.; Nitenberg, A.; Cosson, E. Flow-mediated-paradoxical vasoconstriction is independently associated with asymptomatic myocardial ischemia and coronary artery disease in type 2 diabetic patients. Cardiovasc. Diabetol. 2014, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, C.J.; Agnew, C.E.; McCann, A.; Hamilton, P.K.; Quinn, C.E.; McCall, D.O.; Plumb, R.D.; McClenaghan, V.C.; McGivern, R.C.; Harbinson, M.T.; et al. Impaired flow-mediated dilatation response in uncomplicated Type 1 diabetes mellitus: Influence of shear stress and microvascular reactivity. Clin. Sci. 2011, 121, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, J.; Luo, J.Y.; Tian, X.Y.; Cheang, W.S.; Xu, J.; Lau, C.W.; Wang, L.; Wong, W.T.; Wong, C.M.; et al. Upregulation of angiotensin (1-7)-mediated signaling preserves endothelial function through reducing oxidative stress in diabetes. Antioxid Redox Signal. 2015, 23, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, J.A.; Muller-Delp, J.M.; Delp, M.D.; Mattox, M.L.; Laughlin, M.H.; Parker, J.L. Exercise training enhances vasodilation responses to vascular endothelial growth factor in porcine coronary arterioles exposed to chronic coronary occlusion. Circulation 2004, 109, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Hein, T.W.; Rosa, R.H., Jr.; Ren, Y.; Xu, W.; Kuo, L. VEGF receptor-2-linked PI3K/calpain/SIRT1 activation mediates retinal arteriolar dilations to VEGF and shear stress. Investig. Opthalmol. Vis. Sci. 2015, 56, 5381–5389. [Google Scholar] [CrossRef]
- Mercier, C.; Rousseau, M.; Geraldes, P. Growth factor deregulation and emerging role of phosphatases in diabetic peripheral artery disease. Front. Cardiovasc. Med. 2020, 7, 619612. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Bartoli, M.; Behzadian, M.A.; El-Remessy, A.E.; Al-Shabrawey, M.; Platt, D.H.; Caldwell, R.W. Vascular endothelial growth factor and diabetic retinopathy: Pathophysiological mechanisms and treatment perspectives. Diabetes Metab. Res. Rev. 2003, 19, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.G.; Ueba, H.; Tanimoto, T.; Lungu, A.O.; Frame, M.D.; Berk, B.C. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ. Res. 2003, 93, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanes, M.G.; Oubaha, M.; Rautureau, Y.; Gratton, J.P. Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for Akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells. J. Biol. Chem. 2007, 282, 10660–10669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling endothelial dysfunction by modulating NOS uncoupling: New insights into its pathogenesis and therapeutic possibilities. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E481–E495. [Google Scholar] [CrossRef] [Green Version]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial dysfunction: Is there a hyperglycemia-induced imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Daiber, A.; Ullrich, V.; Mülsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arter. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef]
- van Etten, R.W.; de Koning, E.J.; Verhaar, M.C.; Gaillard, C.A.; Rabelink, T.J. Impaired NO-dependent vasodilation in patients with Type II (non-insulin-dependent) diabetes mellitus is restored by acute administration of folate. Diabetologia 2002, 45, 1004–1010. [Google Scholar] [CrossRef] [Green Version]
- Thengchaisri, N.; Shipley, R.; Ren, Y.; Parker, J.; Kuo, L. Exercise training restores coronary arteriolar dilation to NOS activation distal to coronary artery occlusion: Role of hydrogen peroxide. Arter. Thromb. Vasc. Biol. 2007, 27, 791–798. [Google Scholar] [CrossRef]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thengchaisri, N.; Kuo, L. Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: Role of cyclooxygenase and potassium channels. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2255–H2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, H.; Morikawa, K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J. Mol. Cell. Cardiol. 2005, 39, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.W.; Zhang, C.; Wang, W.; Chang, C.I.; Thengchaisri, N.; Kuo, L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: Counteracting role of arginase. FASEB J. 2003, 17, 2328–2330. [Google Scholar] [CrossRef] [PubMed]
- Thengchaisri, N.; Hein, T.W.; Wang, W.; Xu, X.; Li, Z.; Fossum, T.W.; Kuo, L. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arter. Thromb. Vasc. Biol. 2006, 26, 2035–2042. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hein, T.W.; Wang, W.; Miller, M.W.; Fossum, T.W.; McDonald, M.M.; Humphrey, J.D.; Kuo, L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension 2004, 44, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Johnson, F.K.; Johnson, R.A.; Peyton, K.J.; Shebib, A.R.; Durante, W. Arginase promotes skeletal muscle arteriolar endothelial dysfunction in diabetic rats. Front. Immunol. 2013, 4, 119. [Google Scholar] [CrossRef] [Green Version]
- Tooke, J.E. Microvascular function in human diabetes. A physiological perspective. Diabetes 1995, 44, 721–726. [Google Scholar] [CrossRef]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef]
- Fuller, J.H. Mortality trends and causes of death in diabetic patients. Diabete Metab. 1993, 19, 96–99. [Google Scholar]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Myogenic activity in isolated subepicardial and subendocardial coronary arterioles. Am. J. Physiol. Heart Circ. Physiol. 1988, 255, H1558–H1562. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, A.J.; Dankelman, J.; VanBavel, E.; Spaan, J.A. Balance between myogenic, flow-dependent, and metabolic flow control in coronary arterial tree: A model study. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2224–H2237. [Google Scholar] [CrossRef] [PubMed]
- Quyyumi, A.A.; Dakak, N.; Andrews, N.P.; Gilligan, D.M.; Panza, J.A.; Cannon, R.O., 3rd. Contribution of nitric oxide to metabolic coronary vasodilation in the human heart. Circulation 1995, 92, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Canty, J.M., Jr.; Iyer, V.S. Hydrogen peroxide and metabolic coronary flow regulation. J. Am. Coll. Cardiol. 2007, 50, 1279–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, T.J.; Slorach, C.; Mahmud, F.H.; Dunger, D.B.; Deanfield, J.; Deda, L.; Elia, Y.; Har, R.L.; Hui, W.; Moineddin, R.; et al. Early changes in cardiovascular structure and function in adolescents with type 1 diabetes. Cardiovasc. Diabetol. 2016, 15, 31. [Google Scholar] [CrossRef] [Green Version]
- de, M.M.A.S.; Clemente, E.L.; de Lourdes Guimaraes Rodrigues, M.; Torres Valenca, D.C.; Gomes, M.B. Assessment of microvascular endothelial function in type 1 diabetes using laser speckle contrast imaging. J. Diabetes Complicat. 2017, 31, 753–757. [Google Scholar] [CrossRef]
- Pitkanen, O.P.; Nuutila, P.; Raitakari, O.T.; Ronnemaa, T.; Koskinen, P.J.; Iida, H.; Lehtimaki, T.J.; Laine, H.K.; Takala, T.; Viikari, J.S.; et al. Coronary flow reserve is reduced in young men with IDDM. Diabetes 1998, 47, 248–254. [Google Scholar] [CrossRef]
- Dankowski, R.; Wierzchowiecki, M.; Naskret, D.; Michalski, M.; Kandziora, M.; Biegalski, W.; Poprawski, K. Association between retinopathy, microalbuminuria and coronary perfusion in young patients with type 1 diabetes mellitus. Kardiol. Pol. 2008, 66, 262–268. [Google Scholar]
- Sundell, J.; Laine, H.; Nuutila, P.; Ronnemaa, T.; Luotolahti, M.; Raitakari, O.; Knuuti, J. The effects of insulin and short-term hyperglycaemia on myocardial blood flow in young men with uncomplicated type I diabetes. Diabetologia 2002, 45, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Hein, T.W.; Belardinelli, L.; Kuo, L. Adenosine A2A receptors mediate coronary microvascular dilation to adenosine: Role of nitric oxide and ATP-sensitive potassium channels. J. Pharmacol. Exp. Ther. 1999, 291, 655–664. [Google Scholar]
- Hein, T.W.; Kuo, L. cAMP-independent dilation of coronary arterioles to adenosine: Role of nitric oxide, G proteins, and KATP channels. Circ. Res. 1999, 85, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, R.; Egashira, K.; Arimura, K.; Machida, Y.; Ide, T.; Tsutsui, H.; Shimokawa, H.; Takeshita, A. Increased inactivation of nitric oxide is involved in impaired coronary flow reserve in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2619–H2625. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.J.; Kuo, L.; Davis, M.J.; Chilian, W.M. Distribution and control of coronary microvascular resistance. Adv. Exp. Med. Biol. 1993, 346, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Yada, T.; Shimokawa, H.; Hiramatsu, O.; Shinozaki, Y.; Mori, H.; Goto, M.; Ogasawara, Y.; Kajiya, F. Important role of endogenous hydrogen peroxide in pacing-induced metabolic coronary vasodilation in dogs in vivo. J. Am. Coll. Cardiol. 2007, 50, 1272–1278. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, S.; Zhang, C.; Tune, J.D.; Potter, B.; Kiyooka, T.; Rogers, P.A.; Knudson, J.D.; Dick, G.M.; Swafford, A.; Chilian, W.M. Hydrogen peroxide: A feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2614–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yada, T.; Shimokawa, H.; Hiramatsu, O.; Haruna, Y.; Morita, Y.; Kashihara, N.; Shinozaki, Y.; Mori, H.; Goto, M.; Ogasawara, Y.; et al. Cardioprotective role of endogenous hydrogen peroxide during ischemia-reperfusion injury in canine coronary microcirculation in vivo. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1138–H1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Hein, T.W.; Wang, W.; Chang, C.I.; Kuo, L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J. 2001, 15, 1264–1266. [Google Scholar] [CrossRef] [Green Version]
- Berkowitz, D.E.; White, R.; Li, D.; Minhas, K.M.; Cernetich, A.; Kim, S.; Burke, S.; Shoukas, A.A.; Nyhan, D.; Champion, H.C.; et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 2003, 108, 2000–2006. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.A.; Durante, W.; Craig, T.; Peyton, K.J.; Myers, J.G.; Stewart, R.M.; Johnson, F.K. Vascular arginase contributes to arteriolar endothelial dysfunction in a rat model of hemorrhagic shock. J. Trauma 2010, 69, 384–391. [Google Scholar] [CrossRef]
- Kuo, L.; Hein, T.W. Vasomotor regulation of coronary microcirculation by oxidative stress: Role of arginase. Front. Immunol. 2013, 4, 237. [Google Scholar] [CrossRef] [Green Version]
- Hein, T.W.; Omae, T.; Xu, W.; Yoshida, A.; Kuo, L. Role of arginase in selective impairment of endothelium-dependent nitric oxide synthase-mediated dilation of retinal arterioles during early diabetes. Investig. Opthalmol. Vis. Sci. 2020, 61, 36. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.M.; Maleque, M.A.; Motley, E.D. 3-Morpholinosyndnonimine inhibits 5-hydroxytryptamine-induced phosphorylation of nitric oxide synthase in endothelial cells. Cell. Mol. Biol. 2003, 49, 1385–1389. [Google Scholar] [PubMed]
- Asada, M.; Ebihara, S.; Yamanda, S.; Niu, K.; Okazaki, T.; Sora, I.; Arai, H. Depletion of serotonin and selective inhibition of 2B receptor suppressed tumor angiogenesis by inhibiting endothelial nitric oxide synthase and extracellular signal-regulated kinase 1/2 phosphorylation. Neoplasia 2009, 11, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.B.; Ju, H.; Venema, V.J.; Liang, H.; Zou, R.; Michell, B.J.; Chen, Z.P.; Kemp, B.E.; Venema, R.C. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J. Biol. Chem. 2001, 276, 16587–16591. [Google Scholar] [CrossRef] [Green Version]
- Boo, Y.C.; Hwang, J.; Sykes, M.; Michell, B.J.; Kemp, B.E.; Lum, H.; Jo, H. Shear stress stimulates phosphorylation of eNOS at Ser635 by a protein kinase A-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1819–H1828. [Google Scholar] [CrossRef] [Green Version]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef]
- Dayanir, V.; Meyer, R.D.; Lashkari, K.; Rahimi, N. Identification of tyrosine residues in vascular endothelial growth factor receptor-2/FLK-1 involved in activation of phosphatidylinositol 3-kinase and cell proliferation. J. Biol. Chem. 2001, 276, 17686–17692. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.Y.; Wang, T.; Cai, H. An ezrin/calpain/PI3K/AMPK/eNOSs1179 signaling cascade mediating VEGF-dependent endothelial nitric oxide production. Circ. Res. 2009, 104, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sud, N.; Fonseca, F.V.; Hou, Y.; Black, S.M. Shear stress stimulates nitric oxide signaling in pulmonary arterial endothelial cells via a reduction in catalase activity: Role of protein kinase Cδ. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L105–L116. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Siow, R.C.; Mann, G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: A role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signal. 2011, 14, 469–487. [Google Scholar] [CrossRef]
- Hein, T.W.; Potts, L.B.; Xu, W.; Yuen, J.Z.; Kuo, L. Temporal development of retinal arteriolar endothelial dysfunction in porcine type 1 diabetes. Investig. Opthalmol. Vis. Sci. 2012, 53, 7943–7949. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Xu, W.; Rosa, R.H., Jr.; Kuo, L.; Hein, T.W. Hyperglycemia enhances constriction of retinal venules via activation of the reverse-mode sodium-calcium exchanger. Diabetes 2019, 68, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhao, M.; Abbey, C.A.; Tsai, S.H.; Xie, W.; Pham, D.; Chapman, S.; Bayless, K.J.; Hein, T.W.; Rosa, R.H., Jr.; et al. Newly identified peptide, peptide Lv, promotes pathological angiogenesis. J. Am. Heart Assoc. 2019, 8, e013673. [Google Scholar] [CrossRef] [PubMed]
- Thengchaisri, N.; Hein, T.W.; Ren, Y.; Kuo, L. Activation of coronary arteriolar PKCβ2 impairs endothelial NO-mediated vasodilation: Role of JNK/Rho kinase signaling and xanthine oxidase activation. Int. J. Mol. Sci. 2021, 22, 9763. [Google Scholar] [CrossRef]
- Hein, T.W.; Kuo, L. LDLs impair vasomotor function of the coronary microcirculation: Role of superoxide anions. Circ. Res. 1998, 83, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Thengchaisri, N.; Hein, T.W.; Ren, Y.; Kuo, L. Endothelin-1 impairs coronary arteriolar dilation: Role of p38 kinase-mediated superoxide production from NADPH oxidase. J. Mol. Cell. Cardiol. 2015, 86, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.H.; Lu, G.; Xu, X.; Ren, Y.; Hein, T.W.; Kuo, L. Enhanced endothelin-1/Rho-kinase signalling and coronary microvascular dysfunction in hypertensive myocardial hypertrophy. Cardiovasc. Res. 2017, 113, 1329–1337. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thengchaisri, N.; Kuo, L.; Hein, T.W. H2O2 Mediates VEGF- and Flow-Induced Dilations of Coronary Arterioles in Early Type 1 Diabetes: Role of Vascular Arginase and PI3K-Linked eNOS Uncoupling. Int. J. Mol. Sci. 2023, 24, 489. https://doi.org/10.3390/ijms24010489
Thengchaisri N, Kuo L, Hein TW. H2O2 Mediates VEGF- and Flow-Induced Dilations of Coronary Arterioles in Early Type 1 Diabetes: Role of Vascular Arginase and PI3K-Linked eNOS Uncoupling. International Journal of Molecular Sciences. 2023; 24(1):489. https://doi.org/10.3390/ijms24010489
Chicago/Turabian StyleThengchaisri, Naris, Lih Kuo, and Travis W. Hein. 2023. "H2O2 Mediates VEGF- and Flow-Induced Dilations of Coronary Arterioles in Early Type 1 Diabetes: Role of Vascular Arginase and PI3K-Linked eNOS Uncoupling" International Journal of Molecular Sciences 24, no. 1: 489. https://doi.org/10.3390/ijms24010489