
Enhancing SARS-CoV-2 Surveillance through Regular Genomic Sequencing in Spain: The RELECOV Network

 , , , , , , , , , , , , and add
Show full author list
, , , , , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Results
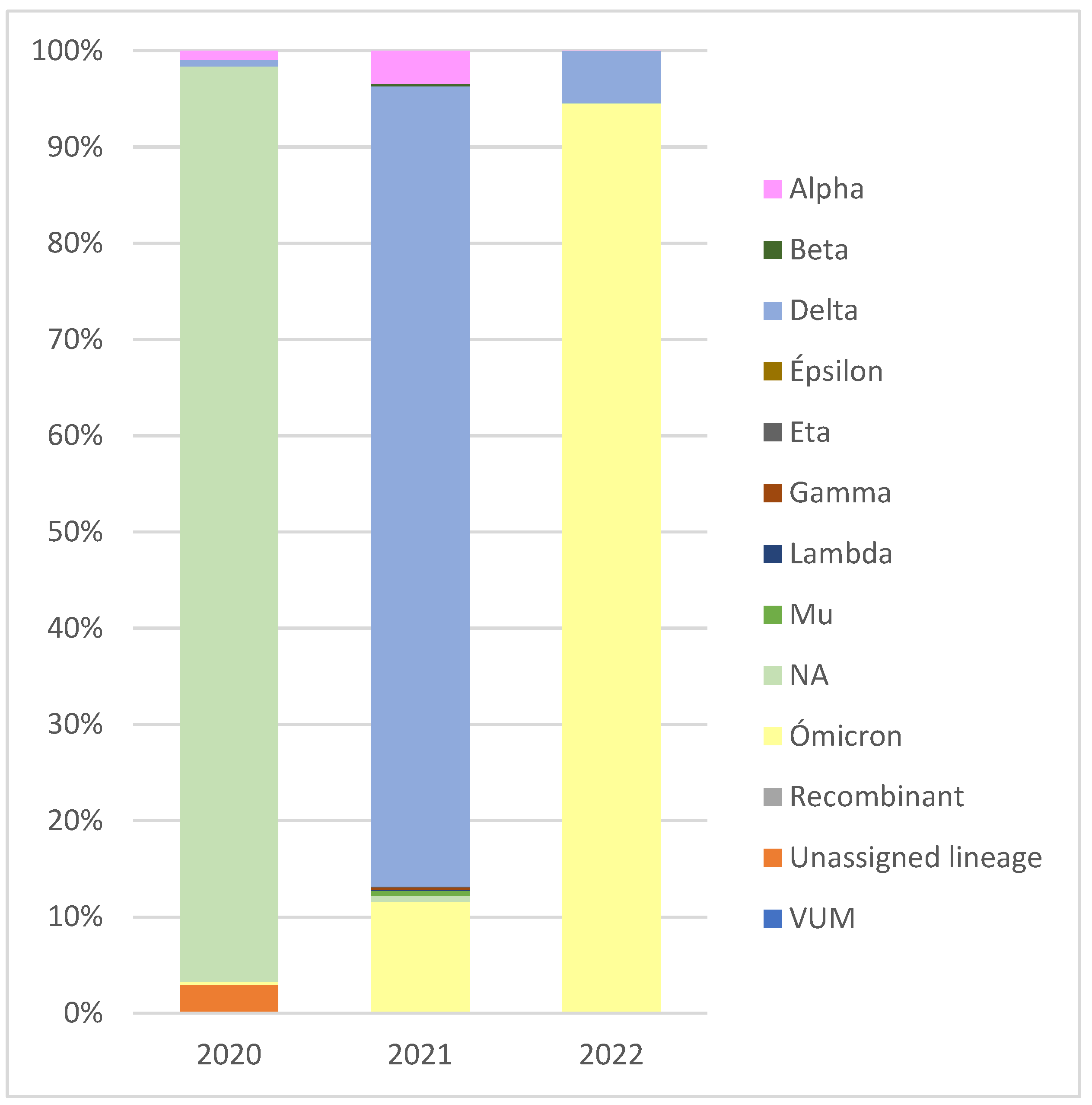
2.1. Surveillance of Virus from Sequences Deposited in GISAID by the RELECOV Network
2.2. Tracking Changes in Delta and Omicron Variants
2.2.1. Delta Variant
2.2.2. Omicron Variant
2.3. Quality Control Assessment for SARS-CoV-2 Sequencing (QCA)
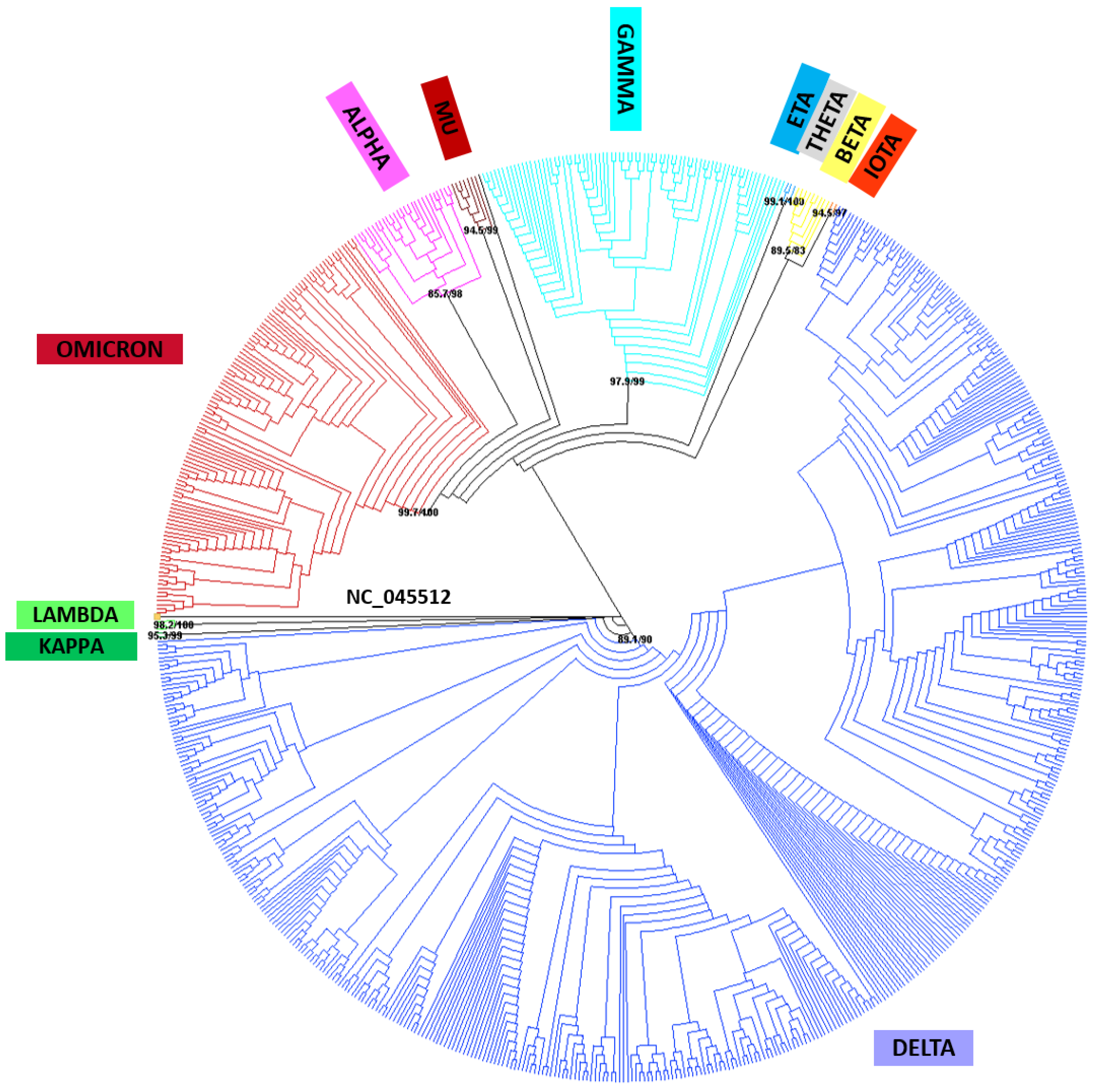
2.4. Reference SARS-CoV-2 Phylogenetic Analysis Implementation
3. Discussion
4. Materials and Methods
4.1. SARS-CoV-2 Viral Sequences Deposited in GISAID by the RELECOV Network Members
4.2. Tracking the Changes in the SARS-CoV-2 Delta and Omicron Variants
4.3. Quality Control Assessment (QCA) of SARS-CoV-2 Viral Genome Sequencing
4.4. Phylogenetic Analysis of SARS-CoV-2 Viral Genome Sequences
- Correspondence to a specific variant described by the WHO.
- First described sequences with complete viral genome, high coverage (according to GISAID those sequences with less than 1% of undefined bases (NNNs) and insertions and/or deletions verified by the submitter), and complete collection date.
- At least 3–6 sequences per lineage and/or sublineage described for each variant.
- Does not present unresolved nucleotide positions.
- Size of the fragment between position 55 and 29,674 (ORF10 end) refers to the reference sequence NC_04552 available in NCBI [43]
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Statement on the Second Meeting of the International Health Regulations (2005) Emergency Committee Regarding the Outbreak of Novel Coronavirus (2019-nCoV). Available online: https://www.who.int/news/item/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov) (accessed on 1 October 2022).
- WHO. Responding to Community Spread of COVID-19. Available online: https://apps.who.int/iris/bitstream/handle/10665/331421/WHO-COVID-19-Community_Transmission-2020.1-eng.pdf (accessed on 1 October 2022).
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, A.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N.; Barakat, A.; Bedford, T.; Bhiman, J.N.; Caly, L.; et al. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Markoulatos, P.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. The Neighborhood of the Spike Gene Is a Hotspot for Modular Intertypic Homologous and Nonhomologous Recombination in Coronavirus Genomes. Mol. Biol. Evol. 2022, 39, msab292. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Papakyriakou, A.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G.; Amoutzias, G.D. Comparative Analysis of SARS-CoV-2 Variants of Concern, Including Omicron, Highlights Their Common and Distinctive Amino Acid Substitution Patterns, Especially at the Spike ORF. Viruses 2022, 14, 707. [Google Scholar] [CrossRef]
- Tay, J.H.; Porter, A.F.; Wirth, W.; Duchene, S. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate. Mol. Biol. Evol. 2022, 39, msac013. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef] [PubMed]
- WHO. SARS-CoV-2 Genomic Sequencing for Public Health Goals: Interim Guidance, 8 January 2021. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-genomic_sequencing-2021.1 (accessed on 1 October 2022).
- ECDC. Guidance for Representative and Targeted Genomic SARS-CoV-2 Monitoring. Available online: https://www.ecdc.europa.eu/en/publications-data/guidance-representative-and-targeted-genomic-sars-cov-2-monitoring (accessed on 1 November 2022).
- European-Commission. Coronavirus: Preparing Europe for the Increased Threat of Variants. 2021. Available online: https://ec.europa.eu/commission/presscorner/detail/en/ip_21_641 (accessed on 1 October 2022).
- Ministerio de Sanidad. Integración de la Secuenciación Genómica en la Vigilancia del SARS-Cov-2. 2021. Available online: https://www.sanidad.gob.es/profesionales/saludPublica/ccayes/alertasActual/nCov/documentos/Integracion_de_la_secuenciacion_genomica-en_la_vigilancia_del_SARS-CoV-2.pdf (accessed on 1 November 2022).
- Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses 2022, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Announces Simple, Easy-to-Say Labels for SARS-CoV-2 Variants of Interest and Concern. Available online: https://www.who.int/news/item/31-05-2021-who-announces-simple-easy-to-say-labels-for-sars-cov-2-variants-of-interest-and-concern (accessed on 1 October 2022).
- Hughes, L.; Gangavarapu, K.; Latif, A.A.; Mullen, J.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.; et al. Outbreak.info genomic reports: Scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- ECDC. ECDC Strategic Framework for the Integration of Molecular and Genomic Typing into European Surveillance and Multi-country Outbreak Investigations. Available online: https://www.ecdc.europa.eu/en/publications-data/ecdc-strategic-framework-integration-molecular-and-genomic-typing-european (accessed on 1 October 2022).
- Colijn, C.; Earn, D.J.; Dushoff, J.; Ogden, N.H.; Li, M.; Knox, N.; Van Domselaar, G.; Franklin, K.; Jolly, G.; Otto, S.P. The need for linked genomic surveillance of SARS-CoV-2. Can. Commun. Dis. Rep. 2022, 48, 131–139. [Google Scholar] [CrossRef]
- Leite, J.A.; Vicari, A.; Perez, E.; Siqueira, M.; Resende, P.; Motta, F.C.; Freitas, L.; Fernandez, J.; Parra, B.; Castillo, A.; et al. Implementation of a COVID-19 Genomic Surveillance Regional Network for Latin America and Caribbean region. PLoS ONE 2022, 17, e0252526. [Google Scholar] [CrossRef]
- Schubert, G.; Achi, V.; Ahuka, S.; Belarbi, E.; Bourhaima, O.; Eckmanns, T.; Johnstone, S.; Kabore, F.; Kra, O.; Mendes, A.; et al. The African Network for Improved Diagnostics, Epidemiology and Management of common infectious Agents. BMC Infect. Dis. 2021, 21, 539. [Google Scholar] [CrossRef]
- Smith, D.; Bashton, M. An integrated national scale SARS-CoV-2 genomic surveillance network. Lancet Microbe 2020, 1, e99–e100. [Google Scholar] [CrossRef]
- Lopez, M.G.; Chiner-Oms, A.; Garcia de Viedma, D.; Ruiz-Rodriguez, P.; Bracho, M.A.; Cancino-Munoz, I.; D’Auria, G.; de Marco, G.; Garcia-Gonzalez, N.; Goig, G.A.; et al. The first wave of the COVID-19 epidemic in Spain was associated with early introductions and fast spread of a dominating genetic variant. Nat. Genet. 2021, 53, 1405–1414. [Google Scholar] [CrossRef]
- WHO. Global Genomic Surveillance Strategy for Pathogens with Pandemic and Epidemic Potential, 2022–2032. Available online: https://www.who.int/publications/i/item/9789240046979 (accessed on 1 February 2023).
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S.; et al. Global landscape of SARS-CoV-2 genomic surveillance and data sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. bioRxiv 2021, 10.1101/2021.04.06.438731. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhou, J.; Tian, M.; Huang, M.; Liu, S.; Xie, Y.; Han, P.; Bai, C.; Han, P.; Zheng, A.; et al. Omicron SARS-CoV-2 mutations stabilize spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape. Nat. Commun. 2022, 13, 4958. [Google Scholar] [CrossRef]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target Ther. 2022, 7, 138. [Google Scholar] [CrossRef]
- Cov-lineages.org. Available online: https://cov-lineages.org/lineage_list.html (accessed on 1 October 2022).
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Ze-Ze, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- Bush, R.M. Predicting adaptive evolution. Nat. Rev. Genet. 2001, 2, 387–392. [Google Scholar] [CrossRef]
- Influenza Virus Characterization: Summary Report, Europe, September 2022. Copenhagen and Stockholm: World Health Organization Regional Office for Europe and European Centre for Disease Prevention and Control; 2022. Licence: CC BY 3.0 IGO. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/Influenza-ECDC-WHO-report-Sep-2022.pdf (accessed on 1 February 2023).
- Brito, A.F.; Semenova, E.; Dudas, G.; Hassler, G.W.; Kalinich, C.C.; Kraemer, M.U.G.; Ho, J.; Tegally, H.; Githinji, G.; Agoti, C.N.; et al. Global disparities in SARS-CoV-2 genomic surveillance. Nat. Commun. 2022, 13, 7003. [Google Scholar] [CrossRef] [PubMed]
- Nucleotide, National Library of Medicine. National Center for Biotechnology Information (NCBI). MD, USA. Available online: https://www.ncbi.nlm.nih.gov/nucleotide/ (accessed on 1 July 2022).
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Number of Sequences | Number of Lineages |
|---|---|---|
| Alpha | 1185 | 3 |
| Beta | 87 | 2 |
| Gamma | 96 | 9 |
| Delta | 18,272 | 126 |
| Mu | 171 | 3 |
| Omicron | 8 | 4 |
| Lambda | 40 | 2 |
| 2 VUM | 1 | 1 |
| Eta | 27 | 1 |
| Epsilon | 1 | 1 |
| 1 NA | 483 | 48 |
| Unassigned lineage | 61 | NA |
| Recombinant | 3 | NA |
| Total | 20,435 | 200 |
| Variant | Number of Sequences | Number of Lineages |
|---|---|---|
| Alpha | 72 | 2 |
| Beta | 2 | 2 |
| Delta | 12,733 | 107 |
| Gamma | 4 | 2 |
| Mu | 15 | 3 |
| Omicron | 15,251 | 48 |
| Unassigned lineage | 16 | NA |
| Recombinant | 1 | NA |
| 1 NA | 49 | 8 |
| Total | 28,143 | 172 |
| Sample | Variant | Lineage | Variant | Lineage |
|---|---|---|---|---|
| Number of Correct Results (%) | Number of Correct Results (%) | |||
| #1 | Alpha | B.1.1.7 | 37 (100) | 36 (97.30) |
| #2 | Beta | B.1.351 | 37 (100) | 37 (100) |
| #3 | 1 NA | A.28 | 36 (97.30) | 36 (97.30) |
| #4 | Mu | B.1.621 | 37 (100) | 37 (100) |
| #5 | Gamma | P.1 | 37 (100) | 34 (91.89) |
| #6 | Delta | AY.9.2 | 37 (100) | 33 (89.19) |
| #7 | Delta | AY.43 | 14 (37.83) | 11 (29.73) |
| #8 | Delta | AY.94 | 36 (97.30) | 31 (83.78) |
| #9 | Delta | AY.94 | 37 (100) | 32 (86.49) |
| #10 | Delta | AY.43 | 37 (100) | 37 (100) |
| Sample | Variant | Lineages | Pre-Shipment 2 Cts | Post-Shipment 2 Cts | |
|---|---|---|---|---|---|
| 56 Days | 96 Days | ||||
| #1 | Alpha | B.1.1.7 | 19.1 | 17.8 | 21.0 |
| #2 | Beta | B.1.351 | 17.6 | 16.0 | 19.2 |
| #3 | 1 NA | A.28 | 22.8 | 19.6 | 21.6 |
| #4 | Mu | B.1.621 | 18.7 | 17.0 | 18.5 |
| #5 | Gamma | P.1 | 17.7 | 18.7 | 20.6 |
| #6 | Delta | AY.9.2 | 25.2 | 22.6 | 25.7 |
| #7 | Delta | AY.43 | 21.3 | 21.6 | 24.7 |
| #8 | Delta | AY.94 | 16.2 | 16.6 | 19.4 |
| #9 | Delta | AY.94 | 16.2 | 19.4 | 20.5 |
| #10 | Delta | AY.43 | 21.3 | 20.8 | 21.9 |
| Sample | Average | 1 SD | 2 CI | Average ± CI 2 (95%) |
|---|---|---|---|---|
| #1 | 19.3 | 1.33 | 1.50 | 19.3 ± 1.50 |
| #2 | 17.6 | 1.31 | 1.48 | 17.6 ± 1.48 |
| #3 | 21.3 | 1.33 | 1.50 | 21.3 ± 1.50 |
| #4 | 18.1 | 0.77 | 0.87 | 18.1 ± 0.87 |
| #5 | 19.0 | 1.21 | 1.36 | 19.0 ± 1.36 |
| #6 | 24.5 | 1.34 | 1.52 | 24.5 ± 1.52 |
| #7 | 22.6 | 1.52 | 1.72 | 22.6 ± 1.72 |
| #8 | 17.4 | 1.44 | 1.63 | 17.4 ± 1.63 |
| #9 | 18.7 | 1.83 | 2.07 | 18.7 ± 2.07 |
| #10 | 21.3 | 0.46 | 0.52 | 21.3 ± 0.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Morón, S.; Iglesias-Caballero, M.; Lepe, J.A.; Garcia, F.; Melón, S.; Marimon, J.M.; García de Viedma, D.; Folgueira, M.D.; Galán, J.C.; López-Causapé, C.; et al. Enhancing SARS-CoV-2 Surveillance through Regular Genomic Sequencing in Spain: The RELECOV Network. Int. J. Mol. Sci. 2023, 24, 8573. https://doi.org/10.3390/ijms24108573
Vázquez-Morón S, Iglesias-Caballero M, Lepe JA, Garcia F, Melón S, Marimon JM, García de Viedma D, Folgueira MD, Galán JC, López-Causapé C, et al. Enhancing SARS-CoV-2 Surveillance through Regular Genomic Sequencing in Spain: The RELECOV Network. International Journal of Molecular Sciences. 2023; 24(10):8573. https://doi.org/10.3390/ijms24108573
Chicago/Turabian StyleVázquez-Morón, Sonia, María Iglesias-Caballero, José Antonio Lepe, Federico Garcia, Santiago Melón, José M. Marimon, Darío García de Viedma, Maria Dolores Folgueira, Juan Carlos Galán, Carla López-Causapé, and et al. 2023. "Enhancing SARS-CoV-2 Surveillance through Regular Genomic Sequencing in Spain: The RELECOV Network" International Journal of Molecular Sciences 24, no. 10: 8573. https://doi.org/10.3390/ijms24108573