
Lipocalin-2 Deficiency Diminishes Canonical NLRP3 Inflammasome Formation and IL-1β Production in the Subacute Phase of Spinal Cord Injury

 , , , , , ,
, , , , , ,  , ,
, ,  , and
, and
Abstract
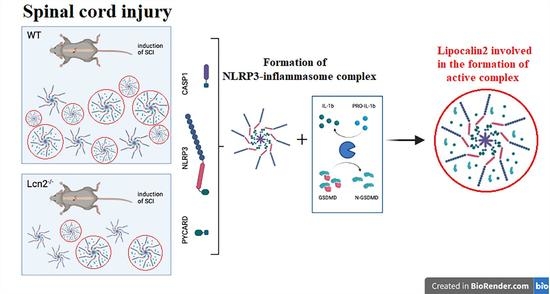
:
1. Introduction
2. Results
2.1. Prediction of the Relationship between LCN2 and NLRP3
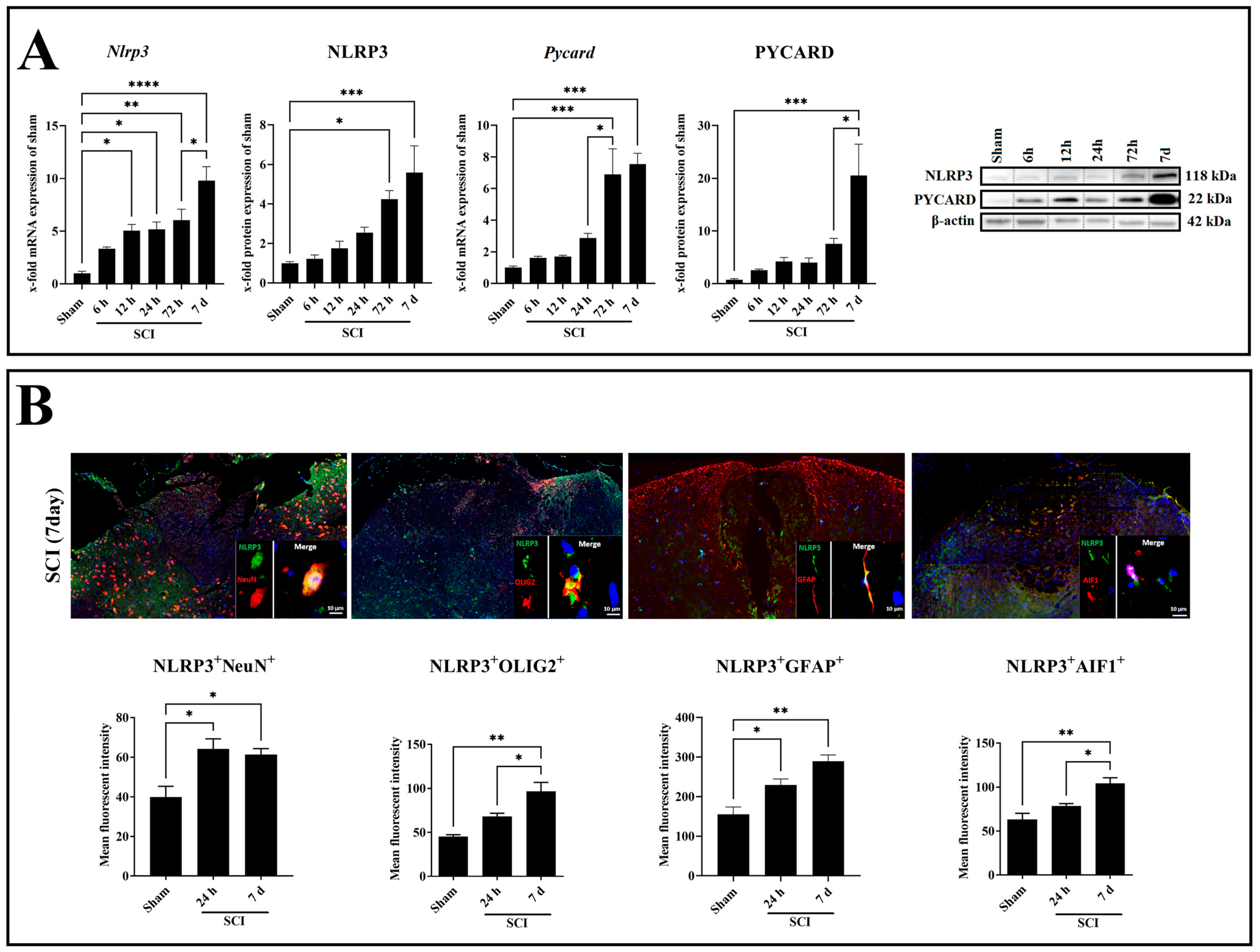
2.2. SCI Leads to Increased Neuroinflammation in a Time-Dependent Manner
2.3. SCI Time-Dependently Increased Inflammasome Activation in Neuron and Glia Cells
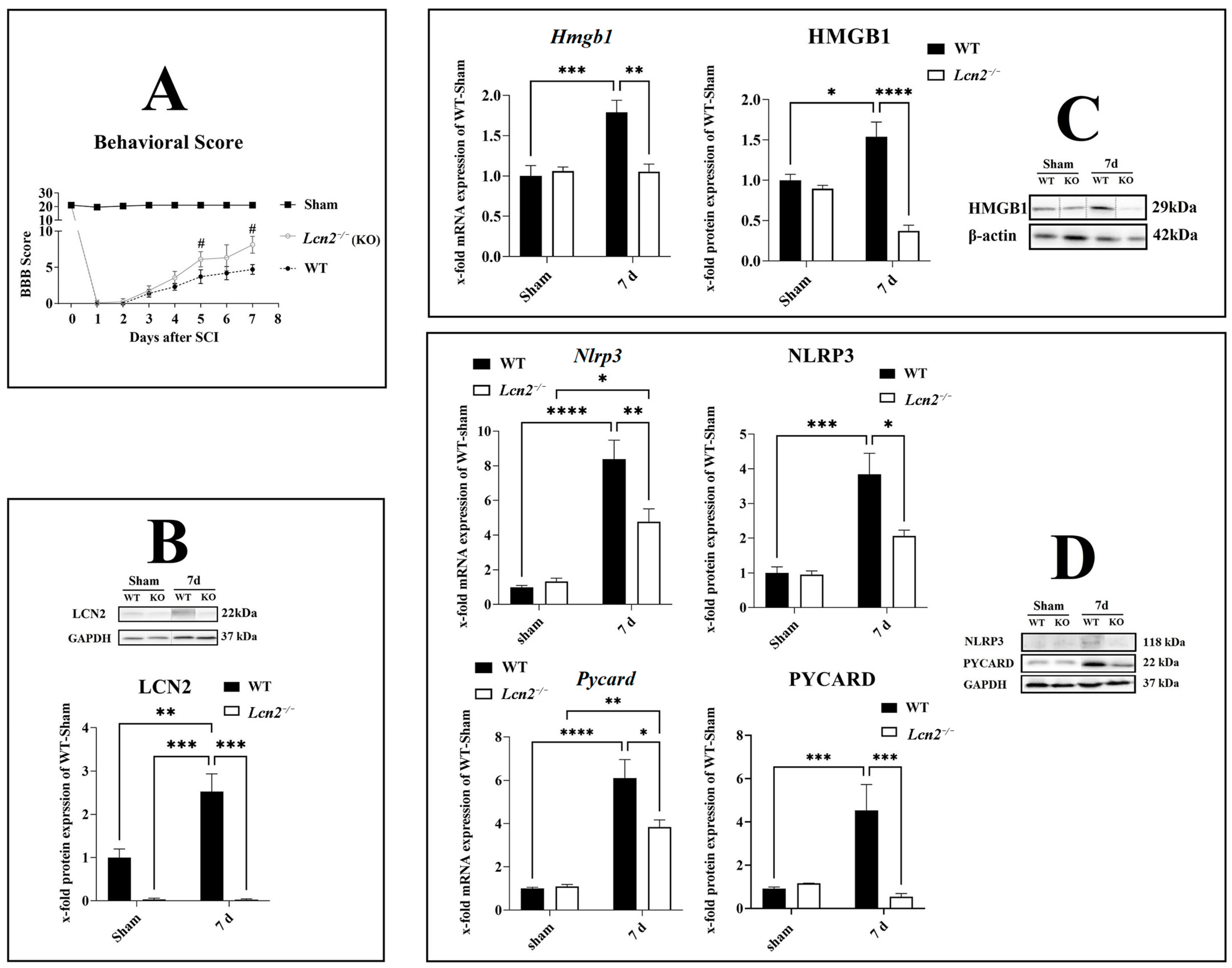
2.4. Lcn2 Deficiency Significantly Improved Locomotor Function after SCI
2.5. Lcn2 Deficiency Significantly Decreased Inflammasome Formation
2.6. Activation of CASP1/GSDMD-NT/IL-1β Axis Reduced in Lcn2−/− Mice
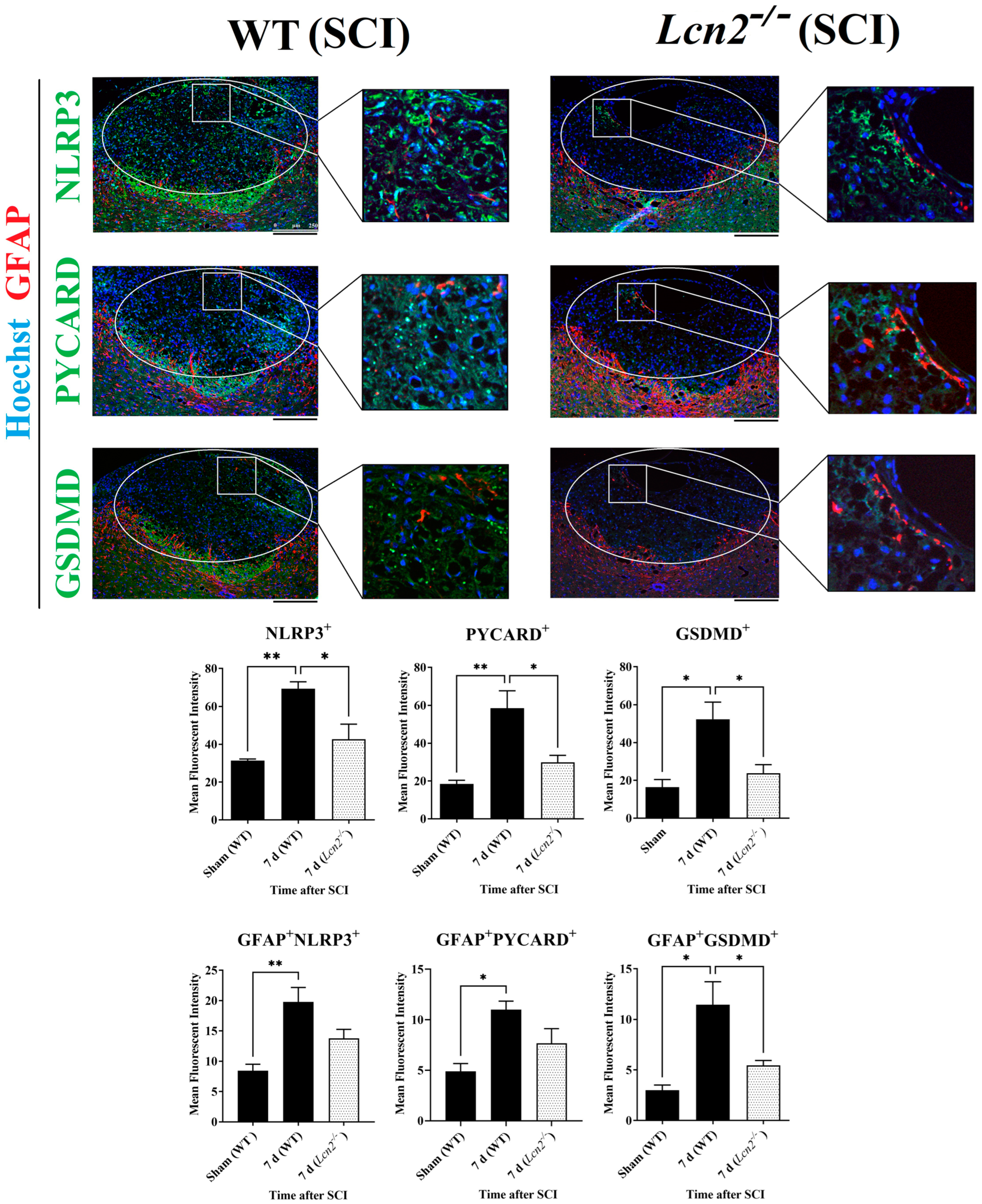
2.7. Lcn2−/− Led to Reduced NLRP3 Activation in Astrocytes and Fewer Signs of Astrogliosis
3. Discussion
4. Material and Methods
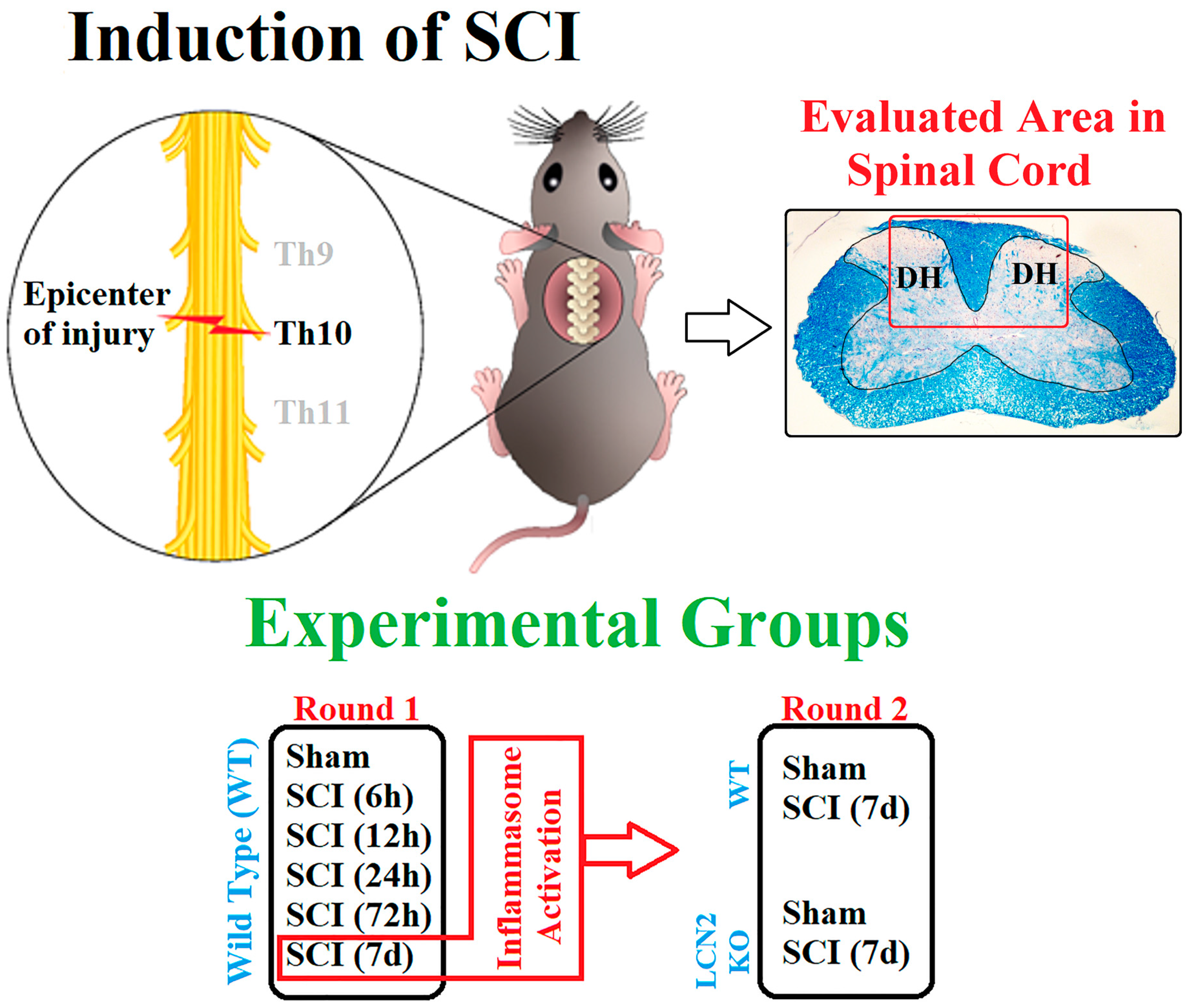
4.1. Animals and Surgery
4.2. Behavioral Assessment
4.3. RNA Extraction and Semiquantitative Real-Time PCR
4.4. Tissue Preparation and Immunofluorescence Staining
4.5. Protein Isolation, SDS-PAGE, and Western Blot
4.6. Bioinformatic Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic spinal cord injury: An overview of pathophysiology, models and acute injury mechanisms. Front. Neurol. 2019, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P.-Y. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef]
- Mortezaee, K.; Khanlarkhani, N.; Beyer, C.; Zendedel, A. Inflammasome: Its role in traumatic brain and spinal cord injury. J. Cell. Physiol. 2017, 233, 5160–5169. [Google Scholar] [CrossRef] [PubMed]
- Zendedel, A.; Johann, S.; Mehrabi, S.; Joghataei, M.T.; Hassanzadeh, G.; Kipp, M.; Beyer, C. Activation and Regulation of NLRP3 Inflammasome by Intrathecal Application of SDF-1a in a Spinal Cord Injury Model. Mol. Neurobiol. 2015, 53, 3063–3075. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Li, C.; Gao, C.; Li, Z.; Yang, J.; Liu, X.; Wang, Y. Effects of hyperbaric oxygen therapy on NACHT domain-leucine-rich-repeat- and pyrin domain-containing protein 3 inflammasome expression in rats following spinal cord injury. Mol. Med. Rep. 2015, 11, 4650–4656. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Yang, J.; Moses, M.A. Lipocalin 2: A multifaceted modulator of human cancer. Cell Cycle 2009, 8, 2347–2352. [Google Scholar] [CrossRef]
- Jin, M.; Kim, J.-H.; Jang, E.; Lee, Y.M.; Han, H.S.; Woo, D.K.; Park, D.H.; Kook, H.; Suk, K. Lipocalin-2 Deficiency Attenuates Neuroinflammation and Brain Injury after Transient Middle Cerebral Artery Occlusion in Mice. J. Cereb. Blood Flow Metab. 2014, 34, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Suk, K. Lipocalin-2 as a therapeutic target for brain injury: An astrocentric perspective. Prog. Neurobiol. 2016, 144, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Edye, M.E.; Lopez-Castejon, G.; Allan, S.M.; Brough, D. Acidosis Drives Damage-associated Molecular Pattern (DAMP)-induced Interleukin-1 Secretion via a Caspase-1-independent Pathway. J. Biol. Chem. 2013, 288, 30485–30494. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Bose, D.; Saha, P.; Sarkar, S.; Seth, R.; Kimono, D.; Albadrani, M.; Nagarkatti, M.; Nagarkatti, P.; Chatterjee, S. Lipocalin 2 induces neuroinflammation and blood-brain barrier dysfunction through liver-brain axis in murine model of nonalcoholic steatohepatitis. J. Neuroinflamm. 2020, 17, 201. [Google Scholar] [CrossRef]
- Al Nimer, F.; Elliott, C.; Bergman, J.; Khademi, M.; Dring, A.M.; Aeinehband, S.; Bergenheim, T.; Christensen, J.R.; Sellebjerg, F.; Svenningsson, A.; et al. Lipocalin-2 is increased in progressive multiple sclerosis and inhibits remyelination. Neurol.-Neuroimmunol. Neuroinflamm. 2016, 3, e191. [Google Scholar] [CrossRef]
- Du, H.; Liang, L.; Li, J.H.; Xiong, Q.; Yu, X.; Yu, H. Lipocalin-2 Alleviates LPS-Induced Inflammation Through Alteration of Macrophage Properties. J. Inflamm. Res. 2021, 14, 4189–4203. [Google Scholar] [CrossRef]
- Behrens, V.; Voelz, C.; Müller, N.; Zhao, W.; Gasterich, N.; Clarner, T.; Beyer, C.; Zendedel, A. Lipocalin 2 as a Putative Modulator of Local Inflammatory Processes in the Spinal Cord and Component of Organ Cross talk After Spinal Cord Injury. Mol. Neurobiol. 2021, 58, 5907–5919. [Google Scholar] [CrossRef]
- Ahn, H.; Lee, G.; Kim, J.; Park, J.; Kang, S.G.; Yoon, S.-I.; Lee, E.; Lee, G.-S. NLRP3 Triggers Attenuate Lipocalin-2 Expression Independent with Inflammasome Activation. Cells 2021, 10, 1660. [Google Scholar] [CrossRef]
- Cruz, D.N.; Gaiao, S.; Maisel, A.; Ronco, C.; Devarajan, P. Neutrophil gelatinase-associated lipocalin as a biomarker of cardiovascular disease: A systematic review. Clin. Chem. Lab. Med. 2012, 50, 1533–1545. [Google Scholar] [CrossRef]
- Chou, W.-H.; Wang, G.; Kumar, V.; Weng, Y.-C. Lipocalin-2 in Stroke. Neuro-Open J. 2015, 2, 38–41. [Google Scholar] [CrossRef]
- Taklimie, F.R.; Gasterich, N.; Scheld, M.; Weiskirchen, R.; Beyer, C.; Clarner, T.; Zendedel, A. Hypoxia Induces Astrocyte-Derived Lipocalin-2 in Ischemic Stroke. Int. J. Mol. Sci. 2019, 20, 1271. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, W.-H.; Lee, M.-S.; Mori, K.; Suk, K. Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J. Neurosci. Res. 2011, 90, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Amo-Aparicio, J.; Martínez-Muriana, A.; Sánchez-Fernández, A.; López-Vales, R. Neuroinflammation Quantification for Spinal Cord Injury. Curr. Protoc. Immunol. 2018, 123, e57. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, A.D.; Fonken, L.K. Glial Cells Shape Pathology and Repair After Spinal Cord Injury. Neurotherapeutics 2018, 15, 554–577. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.B.; Gensel, J.C. Spinal Cord Injury Scarring and Inflammation: Therapies Targeting Glial and Inflammatory Responses. Neurotherapeutics 2018, 15, 541–553. [Google Scholar] [CrossRef]
- Wallisch, J.S.; Simon, D.W.; Bayır, H.; Bell, M.J.; Kochanek, P.M.; Clark, R.S.B. Cerebrospinal Fluid NLRP3 is Increased After Severe Traumatic Brain Injury in Infants and Children. Neurocrit. Care 2017, 27, 44–50. [Google Scholar] [CrossRef]
- Jiang, W.; Li, M.; He, F.; Zhou, S.; Zhu, L. Targeting the NLRP3 inflammasome to attenuate spinal cord injury in mice. J. Neuroinflamm. 2017, 14, 207. [Google Scholar] [CrossRef]
- Sun, X.; Jones, Z.B.; Chen, X.-M.; Zhou, L.; So, K.-F.; Ren, Y. Multiple organ dysfunction and systemic inflammation after spinal cord injury: A complex relationship. J. Neuroinflamm. 2016, 13, 260. [Google Scholar] [CrossRef]
- Gao, S.; Xu, T.; Guo, H.; Deng, Q.; Xun, C.; Liang, W.; Sheng, W. Ameliorative effects of echinacoside against spinal cord injury via inhibiting NLRP3 inflammasome signaling pathway. Life Sci. 2019, 237, 116978. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, H.; Wang, Z.; Gao, F.; Wang, J.; Zhang, W. Wogonoside alleviates inflammation induced by traumatic spinal cord injury by suppressing NF-κB and NLRP3 inflammasome activation. Exp. Ther. Med. 2017, 14, 3304–3308. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 Inflammasome Activity Is Negatively Controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- He, N.; Zheng, X.; He, T.; Shen, G.; Wang, K.; Hu, J.; Zheng, M.; Ding, Y.; Song, X.; Zhong, J.; et al. MCC950 Reduces Neuronal Apoptosis in Spinal Cord Injury in Mice. CNS Neurol. Disord.-Drug Targets 2021, 20, 298–308. [Google Scholar] [CrossRef]
- Jiao, J.; Zhao, G.; Wang, Y.; Ren, P.; Wu, M. MCC950, a Selective Inhibitor of NLRP3 Inflammasome, Reduces the Inflammatory Response and Improves Neurological Outcomes in Mice Model of Spinal Cord Injury. Front. Mol. Biosci. 2020, 7, 37. [Google Scholar] [CrossRef]
- Das, B.; Sarkar, C.; Rawat, V.S.; Kalita, D.; Deka, S.; Agnihotri, A. Promise of the NLRP3 Inflammasome Inhibitors in In Vivo Disease Models. Molecules 2021, 26, 4996. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Zuo, X.; Liang, Z.; Ding, T.; Li, K.; Ma, Y.; Li, P.; Zhu, Z.; Ju, C.; et al. Photobiomodulation inhibits the activation of neurotoxic microglia and astrocytes by inhibiting Lcn2/JAK2-STAT3 crosstalk after spinal cord injury in male rats. J. Neuroinflamm. 2021, 18, 256. [Google Scholar] [CrossRef]
- Song, E.; Jahng, J.W.; Chong, L.P.; Sung, H.K.; Han, M.; Luo, C.; Wu, D.; Boo, S.; Hinz, B.; Cooper, M.A.; et al. Lipocalin-2 induces NLRP3 inflammasome activation via HMGB1 induced TLR4 signaling in heart tissue of mice under pressure overload challenge. Am. J. Transl. Res. 2017, 9, 2723–2735. [Google Scholar]
- Lee, S.; Park, J.-Y.; Lee, W.-H.; Kim, H.; Park, H.-C.; Mori, K.; Suk, K. Lipocalin-2 Is an Autocrine Mediator of Reactive Astrocytosis. J. Neurosci. 2009, 29, 234–249. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Nakamura, K.; Kawakami, T.; Yamamoto, N.; Tomizawa, M.; Fujiwara, T.; Ishii, T.; Harigae, H.; Ogasawara, K. Activation of the NLRP3 inflammasome by cellular labile iron. Exp. Hematol. 2015, 44, 116–124. [Google Scholar] [CrossRef]
- Al Mamun, A.; Wub, Y.; Monalisac, I.; Jiad, C.; Zhoue, K.; Munirf, F.; Xiaoa, J. Role of pyroptosis in spinal cord injury and its therapeutic implications. J. Adv. Res. 2020, 28, 97–109. [Google Scholar] [CrossRef]
- Dubois, H.; Sorgeloos, F.; Sarvestani, S.T.; Martens, L.; Saeys, Y.; Mackenzie, J.; Lamkanfi, M.; Van Loo, G.; Goodfellow, I.; Wullaert, A. Nlrp3 inflammasome activation and Gasdermin D-driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PLOS Pathog. 2019, 15, e1007709. [Google Scholar] [CrossRef]
- Provoost, S.; Maes, T.; Pauwels, N.S.; Berghe, T.V.; Vandenabeele, P.; Lambrecht, B.N.; Joos, G.F.; Tournoy, K.G. NLRP3/Caspase-1–Independent IL-1β Production Mediates Diesel Exhaust Particle-Induced Pulmonary Inflammation. J. Immunol. 2011, 187, 3331–3337. [Google Scholar] [CrossRef]
- Hellenbrand, D.J.; Quinn, C.M.; Piper, Z.J.; Morehouse, C.N.; Fixel, J.A.; Hanna, A.S. Inflammation after spinal cord injury: A review of the critical timeline of signaling cues and cellular infiltration. J. Neuroinflamm. 2021, 18, 284. [Google Scholar] [CrossRef]
- Garcia-Arguello, L.Y.; O’Horo, J.C.; Farrell, A.; Blakney, R.; Sohail, M.R.; Evans, C.T.; Safdar, N. Infections in the spinal cord-injured population: A systematic review. Spinal Cord 2016, 55, 526–534. [Google Scholar] [CrossRef]
- Berger, T.; Togawa, A.; Duncan, G.S.; Elia, A.J.; You-Ten, A.; Wakeham, A.; Fong, H.E.; Cheung, C.C.; Mak, T.W. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 1834–1839. [Google Scholar]
- Basso, D.M.; Beattie, M.S.; Bresnahan, J.C. A Sensitive and Reliable Locomotor Rating Scale for Open Field Testing in Rats. J. Neurotrauma 1995, 12, 1–21. [Google Scholar] [CrossRef]
- Trinh, S.; Kogel, V.; Voelz, C.; Schlösser, A.; Schwenzer, C.; Kabbert, J.; Heussen, N.; Clavel, T.; Herpertz-Dahlmann, B.; Beyer, C.; et al. Gut microbiota and brain alterations in a translational anorexia nervosa rat model. J. Psychiatr. Res. 2020, 133, 156–165. [Google Scholar] [CrossRef]
- Azari, H.; Karimi, E.; Shekari, M.; Tahmasebi, A.; Nikpoor, A.R.; Negahi, A.A.; Sanadgol, N.; Mousavi, P. Construction of a lncRNA–miRNA–mRNA network to determine the key regulators of the Th1/Th2 imbalance in multiple sclerosis. Epigenomics 2021, 13, 1797–1815. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]
- Karimi, E.; Azari, H.; Tahmasebi, A.; Nikpoor, A.R.; Negahi, A.A.; Sanadgol, N.; Shekari, M.; Mousavi, P. LncRNA-miRNA network analysis across the Th17 cell line re-veals biomarker potency of lncRNA NEAT1 and KCNQ1OT1 in multiple sclerosis. J. Cell Mol. Med. 2022, 26, 2351–2362. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.-G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef]
- Ebrahimy, N.; Gasterich, N.; Behrens, V.; Amini, J.; Fragoulis, A.; Beyer, C.; Zhao, W.; Sanadgol, N.; Zendedel, A. Neuroprotective effect of the Nrf2/ARE/miRNA145-5p signaling pathway in the early phase of spinal cord injury. Life Sci. 2022, 304, 120726. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Sense | Antisense | Product Size (bp) | AT (°C) |
|---|---|---|---|---|
| Gsdmd | TACTACTACTCGGCTTTCCCGT | TCGAATCTTGTCCAGGGCATC | 192 | 64 |
| Cyclo A | TTGGGTCCAGGAATGGCAAGA | ACATTGCGAGCAGATGGGGT | 148 | 62 |
| Pycard | CTTGTCAGGGGATGAACTCAAA | GCCATACGACTCCAGATAGTAG | 153 | 60 |
| Casp1 | CCGTGGAGAGAAACAAGGAGT | CCCCTGACAGGATGTCTCCA | 180 | 62 |
| Il-1β | GCCCATCCTCTGTGACTCAT | AGGCCACAGGTATTTTGTCG | 230 | 61 |
| Nlrp3 | CCTGGGGGACTTTGGAATCAG | GATCCTGACAACACGCGGA | 113 | 65 |
| Hmgb1 | GTTACAGAGCGGAGAGAGTG | CCGCAGTTTCCTATCGCTTTG | 130 | 64 |
| Target Protein | Company | Cat. No. | Host | Type | WB | IF |
|---|---|---|---|---|---|---|
| PYCARD | Santa Cruz, Santa Cruz, CA, USA | sc-271054 | mouse | mono | 1:1000 | - |
| CASP1 | Santa Cruz, Santa Cruz, CA, USA | sc56036 | mouse | mono | 1:400 | - |
| NLRP3/cryopyrin | Bioss, Woburn, MA, USA | bs-10021R | rabbit | poly | 1:1000 | 1:300 |
| GSDMD | Abcam, Cambridge, UK | ab219800 | rabbit | poly | 1:1000 | 1:100 |
| HMGB1 | Invitrogen, Braunschweig, Germany | ab-2248274 | rabbit | Poly | 1:1000 | - |
| AIF1 | Millipore, Burlington, MA, USA | MABN92 | mouse | mono | - | 1:500 |
| NeuN | Millipore, Burlington, MA, USA | MAB377 | mouse | mono | - | 1:2000 |
| GFAP | Santa Cruz, Santa Cruz, CA, USA | sc-33673 | mouse | mono | - | 1:1000 |
| OLIG2 | Millipore, Burlington, MA, USA | MABN50 | mouse | mono | - | 1:1000 |
| GAPDH | Santa Cruz, Santa Cruz, CA, USA | sc-25778 | rabbit | poly | 1:5000 | - |
| β-actin | Santa Cruz, Santa Cruz, CA, USA | sc-47778 | mouse | mono | 1:5000 | - |
| IL-1β | Cell Signaling, Danvers, MA, USA | 63124S | rabbit | mono | 1:1000 | |
| LCN2 | Antibodies Online, Philadelphia, PA, USA | ABIN107840 | rabbit | poly | 1:500 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, N.; Scheld, M.; Voelz, C.; Gasterich, N.; Zhao, W.; Behrens, V.; Weiskirchen, R.; Baazm, M.; Clarner, T.; Beyer, C.; et al. Lipocalin-2 Deficiency Diminishes Canonical NLRP3 Inflammasome Formation and IL-1β Production in the Subacute Phase of Spinal Cord Injury. Int. J. Mol. Sci. 2023, 24, 8689. https://doi.org/10.3390/ijms24108689
Müller N, Scheld M, Voelz C, Gasterich N, Zhao W, Behrens V, Weiskirchen R, Baazm M, Clarner T, Beyer C, et al. Lipocalin-2 Deficiency Diminishes Canonical NLRP3 Inflammasome Formation and IL-1β Production in the Subacute Phase of Spinal Cord Injury. International Journal of Molecular Sciences. 2023; 24(10):8689. https://doi.org/10.3390/ijms24108689
Chicago/Turabian StyleMüller, Nina, Miriam Scheld, Clara Voelz, Natalie Gasterich, Weiyi Zhao, Victoria Behrens, Ralf Weiskirchen, Maryam Baazm, Tim Clarner, Cordian Beyer, and et al. 2023. "Lipocalin-2 Deficiency Diminishes Canonical NLRP3 Inflammasome Formation and IL-1β Production in the Subacute Phase of Spinal Cord Injury" International Journal of Molecular Sciences 24, no. 10: 8689. https://doi.org/10.3390/ijms24108689