
Current Drug Development Overview: Targeting Voltage-Gated Calcium Channels for the Treatment of Pain

 , ,
, ,
Abstract
1. Introduction
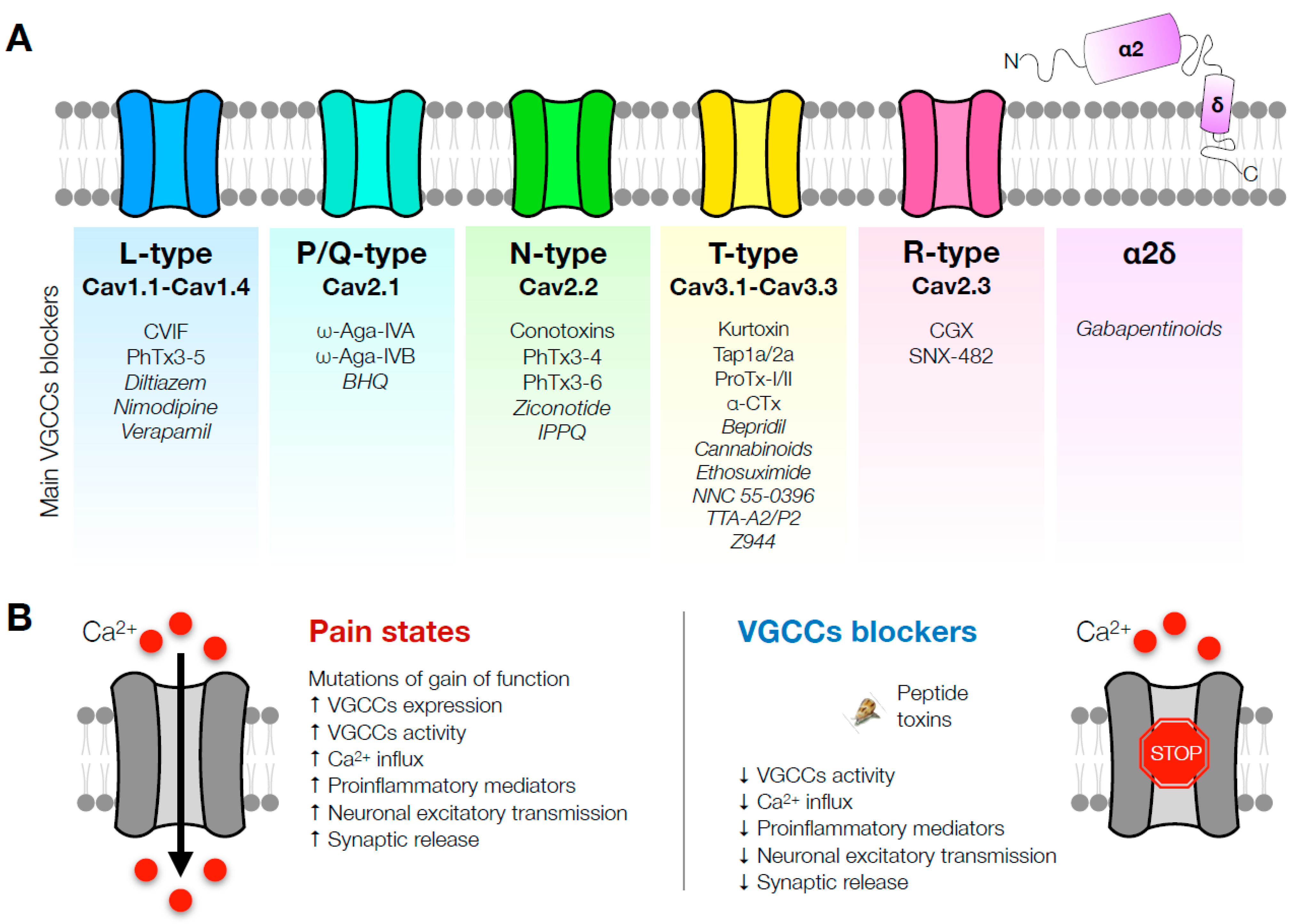
2. L-Type Channels
3. P/Q-Type Channels
4. N-Type Channels
5. Mixed-Target Toxins
6. Mixed-Target Small Molecules
7. T-Type Channels
8. R-Type Channels
9. α2δ Subunit Blockade
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rice, A.S.C.; Smith, B.H.; Blyth, F.M. Pain and the global burden of disease. Pain 2016, 157, 791–796. [Google Scholar] [CrossRef]
- Goldberg, D.S.; McGee, S.J. Pain as a global public health priority. BMC Public Health 2011, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Sessle, B.J. The pain crisis: What it is and what can be done. Pain Res. Treat. 2012, 2012, 703947. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.; Carr, D.B.; Cousins, M. Pain management: A fundamental human right. Anesth. Analg. 2007, 105, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Nalamasu, R.; Nalamachu, S. Evolving Pharmacotherapies for Pain: Drug Development. Phys. Med. Rehabil. Clin. N. Am. 2020, 31, 205–217. [Google Scholar] [CrossRef]
- De Logu, F.; Geppetti, P. Ion Channel Pharmacology for Pain Modulation. Handb. Exp. Pharmacol. 2019, 260, 161–186. [Google Scholar] [CrossRef] [PubMed]
- Mercer Lindsay, N.; Chen, C.; Gilam, G.; Mackey, S.; Scherrer, G. Brain circuits for pain and its treatment. Sci. Transl. Med. 2021, 13, eabj7360. [Google Scholar] [CrossRef]
- Park, J.; Luo, Z.D. Calcium channel functions in pain processing. Channels 2010, 4, 510–517. [Google Scholar] [CrossRef]
- Snutch, T.P. Targeting chronic and neuropathic pain: The N-type calcium channel comes of age. NeuroRx 2005, 2, 662–670. [Google Scholar] [CrossRef]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef]
- Khasar, S.G.; Gold, M.S.; Dastmalchi, S.; Levine, J.D. Selective attenuation of mu-opioid receptor-mediated effects in rat sensory neurons by intrathecal administration of antisense oligodeoxynucleotides. Neurosci. Lett. 1996, 218, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Mirlohi, S.; Bladen, C.; Santiago, M.; Connor, M. Modulation of Recombinant Human T-Type Calcium Channels by Δ9-Tetrahydrocannabinolic Acid In Vitro. Cannabis Cannabinoid Res. 2022, 7, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Alloui, A.; Nargeot, J.; Lory, P.; Eschalier, A.; Bourinet, E.; Chemin, J. T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J. Neurosci. 2009, 29, 13106–13114. [Google Scholar] [CrossRef]
- McArthur, J.R.; Finol-Urdaneta, R.K.; Adams, D.J. Analgesic transient receptor potential vanilloid-1-active compounds inhibit native and recombinant T-type calcium channels. Br. J. Pharmacol. 2019, 176, 2264–2278. [Google Scholar] [CrossRef]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Dobremez, E.; Bouali-Benazzouz, R.; Fossat, P.; Monteils, L.; Dulluc, J.; Nagy, F.; Landry, M. Distribution and regulation of L-type calcium channels in deep dorsal horn neurons after sciatic nerve injury in rats. Eur. J. Neurosci. 2005, 21, 3321–3333. [Google Scholar] [CrossRef]
- Godfraind, T. Discovery and Development of Calcium Channel Blockers. Front. Pharmacol. 2017, 8, 286. [Google Scholar] [CrossRef]
- Radwani, H.; Lopez-Gonzalez, M.J.; Cattaert, D.; Roca-Lapirot, O.; Dobremez, E.; Bouali-Benazzouz, R.; Eiríksdóttir, E.; Langel, Ü.; Favereaux, A.; Errami, M.; et al. Cav1.2 and Cav1.3 L-type calcium channels independently control short- and long-term sensitization to pain. J. Physiol. 2016, 594, 6607–6626. [Google Scholar] [CrossRef]
- Kim, D.S.; Yoon, C.H.; Lee, S.J.; Park, S.Y.; Yoo, H.J.; Cho, H.J. Changes in voltage-gated calcium channel α1 gene expression in rat dorsal root ganglia following peripheral nerve injury. Brain Res. Mol. Brain Res. 2001, 96, 151–156. [Google Scholar] [CrossRef]
- Fossat, P.; Dobremez, E.; Bouali-Benazzouz, R.; Favereaux, A.; Bertrand, S.S.; Kilk, K.; Léger, C.; Cazalets, J.R.; Langel, U.; Landry, M.; et al. Knockdown of L calcium channel subtypes: Differential effects in neuropathic pain. J. Neurosci. 2010, 30, 1073–1085. [Google Scholar] [CrossRef]
- Roca-Lapirot, O.; Radwani, H.; Aby, F.; Nagy, F.; Landry, M.; Fossat, P. Calcium signalling through L-type calcium channels: Role in pathophysiology of spinal nociceptive transmission. Br. J. Pharmacol. 2018, 175, 2362–2374. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, Z.Y.; Lu, J.; Chao, Y.C.; Zhou, X.X.; Huang, Y.; Chen, X.M.; Su, D.S.; Yu, W.F.; Gu, X.Y. Sleep deprivation of rats increases postsurgical expression and activity of L-type calcium channel in the dorsal root ganglion and slows recovery from postsurgical pain. Acta Neuropathol. Commun. 2019, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Wu, W.H.; Yarmush, J.; Zbuzek, V.K. An antinociceptive effect of the intraperitoneal injection of nifedipine in rats, measured by tail-flick test. Life Sci. 1993, 53, PL249–PL253. [Google Scholar] [CrossRef] [PubMed]
- Kawashiri, T.; Egashira, N.; Kurobe, K.; Tsutsumi, K.; Yamashita, Y.; Ushio, S.; Yano, T.; Oishi, R. L type Ca²+ channel blockers prevent oxaliplatin-induced cold hyperalgesia and TRPM8 overexpression in rats. Mol. Pain 2012, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Fukuizumi, T.; Ohkubo, T.; Kitamura, K. Spinally delivered N-, P/Q- and L-type Ca2+-channel blockers potentiate morphine analgesia in mice. Life Sci. 2003, 73, 2873–2881. [Google Scholar] [CrossRef]
- Calcutt, N.A.; Chaplan, S.R. Spinal pharmacology of tactile allodynia in diabetic rats. Br. J. Pharmacol. 1997, 122, 1478–1482. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Pogrel, J.W.; Yaksh, T.L. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J. Pharmacol. Exp. Ther. 1994, 269, 1117–1123. [Google Scholar]
- Kowalska, M.; Prendecki, M.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Molecular factors in migraine. Oncotarget 2016, 7, 50708–50718. [Google Scholar] [CrossRef]
- Choudhuri, R.; Cui, L.; Yong, C.; Bowyer, S.; Klein, R.M.; Welch, K.M.; Berman, N.E. Cortical spreading depression and gene regulation: Relevance to migraine. Ann. Neurol. 2002, 51, 499–506. [Google Scholar] [CrossRef]
- Amrutkar, D.V.; Ploug, K.B.; Olesen, J.; Jansen-Olesen, I. Role for voltage gated calcium channels in calcitonin gene-related peptide release in the rat trigeminovascular system. Neuroscience 2011, 172, 510–517. [Google Scholar] [CrossRef]
- Cekic, E.G.; Soydan, G.; Guler, S.; Babaoglu, M.O.; Tuncer, M. Propranolol-induced relaxation in the rat basilar artery. Vasc. Pharmacol. 2013, 58, 307–312. [Google Scholar] [CrossRef]
- Formisano, R.; Falaschi, P.; Cerbo, R.; Proietti, A.; Catarci, T.; D’Urso, R.; Roberti, C.; Aloise, V.; Chiarotti, F.; Agnoli, A. Nimodipine in migraine: Clinical efficacy and endocrinological effects. Eur. J. Clin. Pharmacol. 1991, 41, 69–71. [Google Scholar] [CrossRef]
- Luo, N.; Di, W.; Zhang, A.; Wang, Y.; Ding, M.; Qi, W.; Zhu, Y.; Massing, M.W.; Fang, Y. A randomized, one-year clinical trial comparing the efficacy of topiramate, flunarizine, and a combination of flunarizine and topiramate in migraine prophylaxis. Pain Med. 2012, 13, 80–86. [Google Scholar] [CrossRef]
- Lacinová, L. Voltage-dependent calcium channels. Gen. Physiol. Biophys. 2005, 24 (Suppl. S1), 1–78. [Google Scholar]
- Striessnig, J.; Pinggera, A.; Kaur, G.; Bock, G.; Tuluc, P. L-type Ca2+ channels in heart and brain. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2014, 3, 15–38. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, K.; Luebke, J.I.; Turner, T.J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995, 18, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Kaneko, M.; Shin, H.S.; Takahashi, T. Presynaptic N-type and P/Q-type Ca2+ channels mediating synaptic transmission at the calyx of Held of mice. J. Physiol. 2005, 568, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.O.; Ren, K.; Sablad, M.; Park, K.T. Medullary N-type and P/Q-type calcium channels contribute to neuropathy-induced allodynia. Neuroreport 2005, 16, 563–566. [Google Scholar] [CrossRef]
- Umeda, M.; Ohkubo, T.; Ono, J.; Fukuizumi, T.; Kitamura, K. Molecular and immunohistochemical studies in expression of voltage-dependent Ca2+ channels in dorsal root ganglia from streptozotocin-induced diabetic mice. Life Sci. 2006, 79, 1995–2000. [Google Scholar] [CrossRef]
- Luvisetto, S.; Marinelli, S.; Panasiti, M.S.; D’Amato, F.R.; Fletcher, C.F.; Pavone, F.; Pietrobon, D. Pain sensitivity in mice lacking the Ca(v)2.1alpha1 subunit of P/Q-type Ca2+ channels. Neuroscience 2006, 142, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, N.; Obama, Y.; Kitamura, N.; Niimi, K.; Takahashi, E.; Itakura, C.; Shibuya, I. Hypoalgesic behaviors of P/Q-type voltage-gated Ca2+ channel mutant mouse, rolling mouse Nagoya. Neuroscience 2009, 160, 165–173. [Google Scholar] [CrossRef]
- Nimmrich, V.; Gross, G. P/Q-type calcium channel modulators. Br. J. Pharmacol. 2012, 167, 741–759. [Google Scholar] [CrossRef] [PubMed]
- Mintz, I.M.; Venema, V.J.; Swiderek, K.M.; Lee, T.D.; Bean, B.P.; Adams, M.E. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature 1992, 355, 827–829. [Google Scholar] [CrossRef]
- Malmberg, A.B.; Yaksh, T.L. Voltage-sensitive calcium channels in spinal nociceptive processing: Blockade of N- and P-type channels inhibits formalin-induced nociception. J. Neurosci. 1994, 14, 4882–4890. [Google Scholar] [CrossRef]
- Diaz, A.; Dickenson, A.H. Blockade of spinal N- and P-type, but not L-type, calcium channels inhibits the excitability of rat dorsal horn neurones produced by subcutaneous formalin inflammation. Pain 1997, 69, 93–100. [Google Scholar] [CrossRef]
- Su, X.; Leon, L.A.; Laping, N.J. Role of spinal Cav2.2 and Cav2.1 ion channels in bladder nociception. J. Urol. 2008, 179, 2464–2469. [Google Scholar] [CrossRef] [PubMed]
- Nebe, J.; Vanegas, H.; Neugebauer, V.; Schaible, H.G. Omega-agatoxin IVA, a P-type calcium channel antagonist, reduces nociceptive processing in spinal cord neurons with input from the inflamed but not from the normal knee joint--an electrophysiological study in the rat in vivo. Eur. J. Neurosci. 1997, 9, 2193–2201. [Google Scholar] [CrossRef]
- Murakami, M.; Nakagawasai, O.; Suzuki, T.; Mobarakeh, I.I.; Sakurada, Y.; Murata, A.; Yamadera, F.; Miyoshi, I.; Yanai, K.; Tan-No, K.; et al. Antinociceptive effect of different types of calcium channel inhibitors and the distribution of various calcium channel alpha 1 subunits in the dorsal horn of spinal cord in mice. Brain Res. 2004, 1024, 122–129. [Google Scholar] [CrossRef]
- Matthews, E.A.; Dickenson, A.H. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain 2001, 92, 235–246. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sakashita, Y. Differential effects of intrathecally administered N- and P-type voltage-sensitive calcium channel blockers upon two models of experimental mononeuropathy in the rat. Brain Res. 1998, 794, 329–332. [Google Scholar] [CrossRef]
- Leão, R.M.; Cruz, J.S.; Diniz, C.R.; Cordeiro, M.N.; Beirão, P.S. Inhibition of neuronal high-voltage activated calcium channels by the omega-phoneutria nigriventer T × 3 − 3 peptide toxin. Neuropharmacology 2000, 39, 1756–1767. [Google Scholar] [CrossRef]
- Dalmolin, G.D.; Silva, C.R.; Rigo, F.K.; Gomes, G.M.; do Nascimento Cordeiro, M.; Richardson, M.; Silva, M.A.R.; Prado, M.A.M.; Gomez, M.V.; Ferreira, J. Antinociceptive effect of Brazilian armed spider venom toxin Tx3-3 in animal models of neuropathic pain. Pain 2011, 152, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Dalmolin, G.D.; Bannister, K.; Gonçalves, L.; Sikandar, S.; Patel, R.; Cordeiro, M.D.N.; Gomez, M.V.; Ferreira, J.; Dickenson, A.H. Effect of the spider toxin Tx3-3 on spinal processing of sensory information in naive and neuropathic rats: An in vivo electrophysiological study. Pain Rep. 2017, 2, e610. [Google Scholar] [CrossRef] [PubMed]
- Pedron, C.; Antunes, F.T.T.; Rebelo, I.N.; Campos, M.M.; Correa, Á.P.; Klein, C.P.; de Oliveira, I.B.; do Nascimento Cordeiro, M.; Gomez, M.V.; de Souza, A.H. Phoneutria nigriventer T × 3 − 3 peptide toxin reduces fibromyalgia symptoms in mice. Neuropeptides 2021, 85, 102094. [Google Scholar] [CrossRef] [PubMed]
- Kors, E.E.; Vanmolkot, K.R.; Haan, J.; Frants, R.R.; van den Maagdenberg, A.M.; Ferrari, M.D. Recent findings in headache genetics. Curr. Opin. Neurol. 2004, 17, 283–288. [Google Scholar] [CrossRef]
- Pietrobon, D. Calcium channels and migraine. Biochim. Biophys. Acta 2013, 1828, 1655–1665. [Google Scholar] [CrossRef]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef]
- Ebersberger, A.; Portz, S.; Meissner, W.; Schaible, H.G.; Richter, F. Effects of N-, P/Q- and L-type calcium channel blockers on nociceptive neurones of the trigeminal nucleus with input from the dura. Cephalalgia 2004, 24, 250–261. [Google Scholar] [CrossRef]
- Tottene, A.; Urbani, A.; Pietrobon, D. Role of different voltage-gated Ca2+ channels in cortical spreading depression: Specific requirement of P/Q-type Ca2+ channels. Channels 2011, 5, 110–114. [Google Scholar] [CrossRef]
- Tottene, A.; Fellin, T.; Pagnutti, S.; Luvisetto, S.; Striessnig, J.; Fletcher, C.; Pietrobon, D. Familial hemiplegic migraine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 13284–13289. [Google Scholar] [CrossRef]
- Inagaki, A.; Frank, C.A.; Usachev, Y.M.; Benveniste, M.; Lee, A. Pharmacological correction of gating defects in the voltage-gated Ca(v)2.1 Ca²⁺ channel due to a familial hemiplegic migraine mutation. Neuron 2014, 81, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Westenbroek, R.E.; Hell, J.W.; Warner, C.; Dubel, S.J.; Snutch, T.P.; Catterall, W.A. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron 1992, 9, 1099–1115. [Google Scholar] [CrossRef]
- Nowycky, M.C.; Fox, A.P.; Tsien, R.W. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 1985, 316, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Westenbroek, R.E.; Hoskins, L.; Catterall, W.A. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J. Neurosci. 1998, 18, 6319–6330. [Google Scholar] [CrossRef]
- Hoppanova, L.; Lacinova, L. Voltage-dependent Ca(V)3.2 and Ca(V)2.2 channels in nociceptive pathways. Pflugers Arch. 2022, 474, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Gonzalez, W.; Fissore, R.A.; Carvacho, I. Conotoxins as Tools to Understand the Physiological Function of Voltage-Gated Calcium (CaV). Mar. Drugs 2017, 15, 313. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, G.; Oliveira, S.M. Animal Venom Peptides Cause Antinociceptive Effects by Voltage-gated Calcium Channels Activity Blockage. Curr. Neuropharmacol. 2022, 20, 1579–1599. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dai, J.; Dai, L.; Deng, M.; Hu, Z.; Hu, W.; Liang, S. Function and solution structure of Huwentoxin-X, a specific blocker of N-type calcium channels, from the Chinese bird spider Ornithoctonus huwena. J. Biol. Chem. 2006, 281, 8628–8635. [Google Scholar] [CrossRef]
- Deng, M.; Luo, X.; Xiao, Y.; Sun, Z.; Jiang, L.; Liu, Z.; Zeng, X.; Chen, H.; Tang, J.; Zeng, W.; et al. Huwentoxin-XVI, an analgesic, highly reversible mammalian N-type calcium channel antagonist from Chinese tarantula Ornithoctonus huwena. Neuropharmacology 2014, 79, 657–667. [Google Scholar] [CrossRef]
- Yousuf, A.; Wu, X.; Bony, A.R.; Sadeghi, M.; Huang, Y.H.; Craik, D.J.; Adams, D.J. αO-Conotoxin GeXIVA isomers modulate N-type calcium (CaV2.2) channels and inwardly-rectifying potassium (GIRK) channels via GABAB receptor activation. J. Neurochem. 2022, 160, 154–171. [Google Scholar] [CrossRef]
- Li, X.; Hu, Y.; Wu, Y.; Huang, Y.; Yu, S.; Ding, Q.; Zhangsun, D.; Luo, S. Anti-hypersensitive effect of intramuscular administration of αO-conotoxin GeXIVA[1,2] and GeXIVA[1,4] in rats of neuropathic pain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 66, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Christie, M.J. α9-nicotinic acetylcholine receptors contribute to the maintenance of chronic mechanical hyperalgesia, but not thermal or mechanical allodynia. Mol. Pain. 2014, 10, 64. [Google Scholar] [CrossRef]
- Romero, H.K.; Christensen, S.B.; Di Cesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1825–E1832. [Google Scholar] [CrossRef] [PubMed]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005, 1059, 149–158. [Google Scholar] [CrossRef]
- Sousa, S.R.; Wingerd, J.S.; Brust, A.; Bladen, C.; Ragnarsson, L.; Herzig, V.; Deuis, J.R.; Dutertre, S.; Vetter, I.; Zamponi, G.W.; et al. Discovery and mode of action of a novel analgesic β-toxin from the African spider Ceratogyrus darlingi. PLoS ONE 2017, 12, e0182848. [Google Scholar] [CrossRef]
- Pope, J.E.; Deer, T.R. Ziconotide: A clinical update and pharmacologic review. Expert. Opin. Pharmacother. 2013, 14, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Duggan, P.J.; Tuck, K.L. Bioactive Mimetics of Conotoxins and other Venom Peptides. Toxins 2015, 7, 4175–4198. [Google Scholar] [CrossRef]
- Sang, C.N.; Barnabe, K.J.; Kern, S.E. Phase IA Clinical Trial Evaluating the Tolerability, Pharmacokinetics, and Analgesic Efficacy of an Intrathecally Administered Neurotensin A Analogue in Central Neuropathic Pain Following Spinal Cord Injury. Clin. Pharmacol. Drug. Dev. 2016, 5, 250–258. [Google Scholar] [CrossRef]
- Bowersox, S.S.; Singh, T.; Nadasdi, L.; Zukowska-Grojec, Z.; Valentino, K.; Hoffman, B.B. Cardiovascular effects of omega-conopeptides in conscious rats: Mechanisms of action. J. Cardiovasc. Pharmacol. 1992, 20, 756–764. [Google Scholar]
- Wright, C.E.; Robertson, A.D.; Whorlow, S.L.; Angus, J.A. Cardiovascular and autonomic effects of omega-conotoxins MVIIA and CVID in conscious rabbits and isolated tissue assays. Br. J. Pharmacol. 2000, 131, 1325–1336. [Google Scholar] [CrossRef]
- Kolosov, A.; Goodchild, C.S.; Cooke, I. CNSB004 (Leconotide) causes antihyperalgesia without side effects when given intravenously: A comparison with ziconotide in a rat model of diabetic neuropathic pain. Pain Med. 2010, 11, 262–273. [Google Scholar] [CrossRef]
- DuBreuil, D.M.; Lopez Soto, E.J.; Daste, S.; Meir, R.; Li, D.; Wainger, B.; Fleischmann, A.; Lipscombe, D. Heat But Not Mechanical Hypersensitivity Depends on Voltage-Gated Ca. J. Neurosci. 2021, 41, 7546–7560. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Starobova, H.; Mueller, A.; Vetter, I.; Lewis, R.J. Subcutaneous ω-Conotoxins Alleviate Mechanical Pain in Rodent Models of Acute Peripheral Neuropathy. Mar. Drugs 2021, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C. Calcium channel diversity: Multiple roles of calcium channel subunits. Curr. Opin. Neurobiol. 2009, 19, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ran, D.; Gomez, K.; Moutal, A.; Patek, M.; Perez-Miller, S.; Khanna, R. Comparison of quinazoline and benzoylpyrazoline chemotypes targeting the CaVα-β interaction as antagonists of the N-type CaV2.2 channel. Channels 2021, 15, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Yu, J.; Yang, X.; Moutal, A.; Chefdeville, A.; Gokhale, V.; Shuja, Z.; Chew, L.A.; Bellampalli, S.S.; Luo, S.; et al. Targeting the CaVα-CaVβ interaction yields an antagonist of the N-type CaV2.2 channel with broad antinociceptive efficacy. Pain 2019, 160, 1644–1661. [Google Scholar] [CrossRef]
- Chen, X.; Liu, D.; Zhou, D.; Si, Y.; Xu, D.; Stamatkin, C.W.; Ghozayel, M.K.; Ripsch, M.S.; Obukhov, A.G.; White, F.A.; et al. Small-molecule CaVα1⋅CaVβ antagonist suppresses neuronal voltage-gated calcium-channel trafficking. Proc. Natl. Acad. Sci. USA 2018, 115, E10566–E10575. [Google Scholar] [CrossRef]
- Gleeson, E.C.; Graham, J.E.; Spiller, S.; Vetter, I.; Lewis, R.J.; Duggan, P.J.; Tuck, K.L. Inhibition of N-type calcium channels by fluorophenoxyanilide derivatives. Mar. Drugs 2015, 13, 2030–2045. [Google Scholar] [CrossRef]
- Sanchez-Campos, N.; Bernaldez-Sarabia, J.; Licea-Navarro, A.F. Conotoxin Patenting Trends in Academia and Industry. Mar. Drugs 2022, 20, 531. [Google Scholar] [CrossRef]
- Peigneur, S.; de Lima, M.E.; Tytgat, J. Phoneutria nigriventer venom: A pharmacological treasure. Toxicon 2018, 151, 96–110. [Google Scholar] [CrossRef]
- Antunes, F.T.T.; Caminski, E.S.; Gomez, M.V.; de Souza, A.H. Phα1β is a Promising Neuroprotective Peptide from the Phoneutria nigriventer ‘Armed’ Spider. Int. J. Pept. Res. Ther. 2022, 28, 70. [Google Scholar] [CrossRef]
- da Silva, J.F.; Binda, N.S.; Pereira, E.M.R.; de Lavor, M.S.L.; Vieira, L.B.; de Souza, A.H.; Rigo, F.K.; Ferrer, H.T.; de Castro, C.J.; Ferreira, J.; et al. Analgesic effects of Phα1β toxin: A review of mechanisms of action involving pain pathways. J. Venom. Anim. Toxins Incl. Trop. Dis. 2021, 27, e20210001. [Google Scholar] [CrossRef]
- Silva, R.B.; Sperotto, N.D.; Andrade, E.L.; Pereira, T.C.; Leite, C.E.; de Souza, A.H.; Bogo, M.R.; Morrone, F.B.; Gomez, M.V.; Campos, M.M. Spinal blockage of P/Q- or N-type voltage-gated calcium channels modulates functional and symptomatic changes related to haemorrhagic cystitis in mice. Br. J. Pharmacol. 2015, 172, 924–939. [Google Scholar] [CrossRef]
- Silva, R.B.M.; Greggio, S.; Venturin, G.T.; da Costa, J.C.; Gomez, M.V.; Campos, M.M. Beneficial Effects of the Calcium Channel Blocker CTK 01512-2 in a Mouse Model of Multiple Sclerosis. Mol. Neurobiol. 2018, 55, 9307–9327. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.B.; Kushmerick, C.; Hildebrand, M.E.; Garcia, E.; Stea, A.; Cordeiro, M.N.; Richardson, M.; Gomez, M.V.; Snutch, T.P. Inhibition of high voltage-activated calcium channels by spider toxin PnTx3-6. J. Pharmacol. Exp. Ther. 2005, 314, 1370–1377. [Google Scholar] [CrossRef]
- Scott, D.A.; Wright, C.E.; Angus, J.A. Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur. J. Pharmacol. 2002, 451, 279–286. [Google Scholar] [CrossRef]
- Berecki, G.; Motin, L.; Haythornthwaite, A.; Vink, S.; Bansal, P.; Drinkwater, R.; Wang, C.I.; Moretta, M.; Lewis, R.J.; Alewood, P.F.; et al. Analgesic (omega)-conotoxins CVIE and CVIF selectively and voltage-dependently block recombinant and native N-type calcium channels. Mol. Pharmacol. 2010, 77, 139–148. [Google Scholar] [CrossRef]
- da Silva, J.F.; Castro-Junior, C.J.; Oliveira, S.M.; Dalmolin, G.D.; Silva, C.R.; Vieira, L.B.; Diniz, D.M.; Cordeiro, M.o.N.; Ferreira, J.; Souza, A.H.; et al. Characterization of the antinociceptive effect of PhTx3-4, a toxin from Phoneutria nigriventer, in models of thermal, chemical and incisional pain in mice. Toxicon 2015, 108, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.M.; Silva, C.R.; Trevisan, G.; Villarinho, J.G.; Cordeiro, M.N.; Richardson, M.; Borges, M.H.; Castro, C.J., Jr.; Gomez, M.V.; Ferreira, J. Antinociceptive effect of a novel armed spider peptide Tx3-5 in pathological pain models in mice. Pflugers Arch. 2016, 468, 881–894. [Google Scholar] [CrossRef]
- Tran, L.T.; Gentil, B.J.; Sullivan, K.E.; Durham, H.D. The voltage-gated calcium channel blocker lomerizine is neuroprotective in motor neurons expressing mutant SOD1, but not TDP-43. J. Neurochem. 2014, 130, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Pexton, T.; Moeller-Bertram, T.; Schilling, J.M.; Wallace, M.S. Targeting voltage-gated calcium channels for the treatment of neuropathic pain: A review of drug development. Expert Opin. Investig. Drugs 2011, 20, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S. Recent progress in the discovery and development of N-type calcium channel modulators for the treatment of pain. Prog. Med. Chem. 2014, 53, 147–186. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Feng, Z.P.; Zhang, L.; Pajouhesh, H.; Ding, Y.; Belardetti, F.; Dolphin, D.; Mitscher, L.A.; Snutch, T.P. Scaffold-based design and synthesis of potent N-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2009, 19, 6467–6472. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Feng, Z.P.; Zhang, L.; Jiang, X.; Hendricson, A.; Dong, H.; Tringham, E.; Ding, Y.; Vanderah, T.W.; Porreca, F.; et al. Structure-activity relationships of trimethoxybenzyl piperazine N-type calcium channel inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Sairaman, A.; Cardoso, F.C.; Bispat, A.; Lewis, R.J.; Duggan, P.J.; Tuck, K.L. Synthesis and evaluation of aminobenzothiazoles as blockers of N- and T-type calcium channels. Bioorg. Med. Chem. 2018, 26, 3046–3059. [Google Scholar] [CrossRef]
- Borzenko, A.; Pajouhesh, H.; Morrison, J.L.; Tringham, E.; Snutch, T.P.; Schafer, L.L. Modular, efficient synthesis of asymmetrically substituted piperazine scaffolds as potent calcium channel blockers. Bioorg. Med. Chem. Lett. 2013, 23, 3257–3261. [Google Scholar] [CrossRef]
- Abbadie, C.; McManus, O.B.; Sun, S.Y.; Bugianesi, R.M.; Dai, G.; Haedo, R.J.; Herrington, J.B.; Kaczorowski, G.J.; Smith, M.M.; Swensen, A.M.; et al. Analgesic effects of a substituted N-triazole oxindole (TROX-1), a state-dependent, voltage-gated calcium channel 2 blocker. J. Pharmacol. Exp. Ther. 2010, 334, 545–555. [Google Scholar] [CrossRef]
- Patel, R.; Rutten, K.; Valdor, M.; Schiene, K.; Wigge, S.; Schunk, S.; Damann, N.; Christoph, T.; Dickenson, A.H. Electrophysiological characterization of activation state-dependent CaV2 channel antagonist TROX-1 in spinal nerve injured rats. Neuroscience 2015, 297, 47–57. [Google Scholar] [CrossRef]
- Rahman, W.; Patel, R.; Dickenson, A.H. Electrophysiological evidence for voltage-gated calcium channel 2 (Cav2) modulation of mechano- and thermosensitive spinal neuronal responses in a rat model of osteoarthritis. Neuroscience 2015, 305, 76–85. [Google Scholar] [CrossRef]
- Swensen, A.M.; Herrington, J.; Bugianesi, R.M.; Dai, G.; Haedo, R.J.; Ratliff, K.S.; Smith, M.M.; Warren, V.A.; Arneric, S.P.; Eduljee, C.; et al. Characterization of the substituted N-triazole oxindole TROX-1, a small-molecule, state-dependent inhibitor of CaV2 calcium channels. Mol. Pharmacol. 2012, 81, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Wu, N.; Zhang, C.; Su, R.B.; Lu, X.Q.; Liu, Y.; Yun, L.H.; Zheng, J.Q.; Li, J. Analgesic activity of ZC88, a novel N-type voltage-dependent calcium channel blocker, and its modulation of morphine analgesia, tolerance and dependence. Eur. J. Pharmacol. 2008, 586, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, L.; Zhang, K.; Liu, X.; Dai, W.; Zhang, C.; Yong, Z.; Li, J.; Zheng, J. ZC88, a novel N-type calcium channel blocker from 4-amino-piperidine derivatives state-dependent inhibits Cav2.2 calcium channels. Brain Res. 2015, 1605, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.Z.; Zhao, T.Y.; Wang, Z.Y.; Lu, G.Y.; Zhang, S.Z.; Zhang, C.; Wu, N.; Li, J. Synergistic antinociception between ZC88, an N-type voltage-dependent calcium channel blocker, and ibuprofen in mouse models of visceral and somatic inflammatory pain. Eur. J. Pain 2019, 23, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakagawasai, O.; Fujii, S.; Kameyama, K.; Murakami, S.; Hozumi, S.; Esashi, A.; Taniguchi, R.; Yanagisawa, T.; Tan-no, K.; et al. Antinociceptive action of amlodipine blocking N-type Ca2+ channels at the primary afferent neurons in mice. Eur. J. Pharmacol. 2001, 419, 175–181. [Google Scholar] [CrossRef]
- Koganei, H.; Shoji, M.; Iwata, S. Suppression of formalin-induced nociception by cilnidipine, a voltage-dependent calcium channel blocker. Biol. Pharm. Bull. 2009, 32, 1695–1700. [Google Scholar] [CrossRef]
- Yamamoto, S.; Suzuki, Y.; Ono, H.; Kume, K.; Ohsawa, M. N- and L-type calcium channels blocker cilnidipine ameliorates neuropathic pain. Eur. J. Pharmacol. 2016, 793, 66–75. [Google Scholar] [CrossRef]
- Yamashita, T.; Kamikaseda, S.; Tanaka, A.; Tozaki-Saitoh, H.; Caaveiro, J.M.M.; Inoue, K.; Tsuda, M. New Inhibitory Effects of Cilnidipine on Microglial P2X7 Receptors and IL-1β Release: An Involvement in its Alleviating Effect on Neuropathic Pain. Cells 2021, 10, 434. [Google Scholar] [CrossRef]
- Murakami, M.; Mori, T.; Nakagawasai, O.; Hagiwara, K.; Sakurada, Y.; Mobarakeh, I.I.; Murata, A.; Yamadera, F.; Miyoshi, I.; Tan-No, K.; et al. Inhibitory effect of pranidipine on N-type voltage-dependent Ca2+ channels in mice. Neurosci. Lett. 2004, 367, 118–122. [Google Scholar] [CrossRef]
- Shan, Z.; Cai, S.; Yu, J.; Zhang, Z.; Vallecillo, T.G.M.; Serafini, M.J.; Thomas, A.M.; Pham, N.Y.N.; Bellampalli, S.S.; Moutal, A.; et al. Reversal of Peripheral Neuropathic Pain by the Small-Molecule Natural Product Physalin F via Block of CaV2.3 (R-Type) and CaV2.2 (N-Type) Voltage-Gated Calcium Channels. ACS Chem. Neurosci. 2019, 10, 2939–2955. [Google Scholar] [CrossRef]
- Dong, F.W.; Jiang, H.H.; Yang, L.; Gong, Y.; Zi, C.T.; Yang, D.; Ye, C.J.; Li, H.; Yang, J.; Nian, Y.; et al. Valepotriates From the Roots and Rhizomes of. Front. Pharmacol. 2018, 9, 885. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Chiu, Y.M.; Dai, Z.K.; Wu, B.N. Loganin Ameliorates Painful Diabetic Neuropathy by Modulating Oxidative Stress, Inflammation and Insulin Sensitivity in Streptozotocin-Nicotinamide-Induced Diabetic Rats. Cells 2021, 10, 2688. [Google Scholar] [CrossRef] [PubMed]
- Perez-Reyes, E.; Schneider, T. Molecular biology of calcium channels. Kidney Int. 1995, 48, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Perez-Reyes, E. Three for T: Molecular analysis of the low voltage-activated calcium channel family. Cell. Mol. Life Sci. 1999, 56, 660–669. [Google Scholar] [CrossRef]
- Jacus, M.O.; Uebele, V.N.; Renger, J.J.; Todorovic, S.M. Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J. Neurosci. 2012, 32, 9374–9382. [Google Scholar] [CrossRef]
- Harding, E.K.; Zamponi, G.W. Central and peripheral contributions of T-type calcium channels in pain. Mol. Brain 2022, 15, 39. [Google Scholar] [CrossRef]
- Li, M.; Hansen, J.B.; Huang, L.; Keyser, B.M.; Taylor, J.T. Towards selective antagonists of T-type calcium channels: Design, characterization and potential applications of NNC 55-0396. Cardiovasc. Drug. Rev. 2005, 23, 173–196. [Google Scholar] [CrossRef]
- Matsunami, M.; Kirishi, S.; Okui, T.; Kawabata, A. Chelating luminal zinc mimics hydrogen sulfide-evoked colonic pain in mice: Possible involvement of T-type calcium channels. Neuroscience 2011, 181, 257–264. [Google Scholar] [CrossRef]
- Matsunami, M.; Miki, T.; Nishiura, K.; Hayashi, Y.; Okawa, Y.; Nishikawa, H.; Sekiguchi, F.; Kubo, L.; Ozaki, T.; Tsujiuchi, T.; et al. Involvement of the endogenous hydrogen sulfide/Cav3.2 T-type Ca2+ channel pathway in cystitis-related bladder pain in mice. Br. J. Pharmacol. 2012, 167, 917–928. [Google Scholar] [CrossRef]
- Okubo, K.; Takahashi, T.; Sekiguchi, F.; Kanaoka, D.; Matsunami, M.; Ohkubo, T.; Yamazaki, J.; Fukushima, N.; Yoshida, S.; Kawabata, A. Inhibition of T-type calcium channels and hydrogen sulfide-forming enzyme reverses paclitaxel-evoked neuropathic hyperalgesia in rats. Neuroscience 2011, 188, 148–156. [Google Scholar] [CrossRef]
- Ertel, S.I.; Clozel, J.P. Mibefradil (Ro 40-5967): The first selective T-type Ca2+ channel blocker. Expert Opin. Investig. Drugs 1997, 6, 569–582. [Google Scholar] [CrossRef]
- Bladen, C.; Gadotti, V.M.; Gündüz, M.G.; Berger, N.D.; Şimşek, R.; Şafak, C.; Zamponi, G.W. 1,4-Dihydropyridine derivatives with T-type calcium channel blocking activity attenuate inflammatory and neuropathic pain. Pflugers Arch. 2015, 467, 1237–1247. [Google Scholar] [CrossRef]
- Snutch, T.P.; Zamponi, G.W. Recent advances in the development of T-type calcium channel blockers for pain intervention. Br. J. Pharmacol. 2018, 175, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Teleb, M.; Zhang, F.X.; Huang, J.; Gadotti, V.M.; Farghaly, A.M.; AboulWafa, O.M.; Zamponi, G.W.; Fahmy, H. Synthesis and biological evaluation of novel N3-substituted dihydropyrimidine derivatives as T-type calcium channel blockers and their efficacy as analgesics in mouse models of inflammatory pain. Bioorg. Med. Chem. 2017, 25, 1926–1938. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, M.; Matsui, K.; Fukushi, S.; Okazaki, K.; Sekiguchi, F.; Kawabata, A. Effects of Bepridil and Pimozide, Existing Medicines Capable of Blocking T-Type Ca2+ Channels, on Visceral Pain in Mice. Biol. Pharm. Bull. 2021, 44, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Choe, W.; Messinger, R.B.; Leach, E.; Eckle, V.S.; Obradovic, A.; Salajegheh, R.; Jevtovic-Todorovic, V.; Todorovic, S.M. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol. Pharmacol. 2011, 80, 900–910. [Google Scholar] [CrossRef]
- Hoffmann, T.; Kistner, K.; Joksimovic, S.L.J.; Todorovic, S.M.; Reeh, P.W.; Sauer, S.K. Painful diabetic neuropathy leads to functional CaV3.2 expression and spontaneous activity in skin nociceptors of mice. Exp. Neurol. 2021, 346, 113838. [Google Scholar] [CrossRef]
- Shin, S.M.; Cai, Y.; Itson-Zoske, B.; Qiu, C.; Hao, X.; Xiang, H.; Hogan, Q.H.; Yu, H. Enhanced T-type calcium channel 3.2 activity in sensory neurons contributes to neuropathic-like pain of monosodium iodoacetate-induced knee osteoarthritis. Mol. Pain 2020, 16, 1744806920963807. [Google Scholar] [CrossRef]
- Francois, A.; Kerckhove, N.; Meleine, M.; Alloui, A.; Barrere, C.; Gelot, A.; Uebele, V.N.; Renger, J.J.; Eschalier, A.; Ardid, D.; et al. State-dependent properties of a new T-type calcium channel blocker enhance CaV3.2 selectivity and support analgesic effects. Pain 2013, 154, 283–293. [Google Scholar] [CrossRef]
- Grundy, L.; Tay, C.; Christie, S.; Harrington, A.M.; Castro, J.; Cardoso, F.C.; Lewis, R.J.; Zagorodnyuk, V.; Brierley, S.M. The T-type calcium channel CaV3.2 regulates bladder afferent responses to mechanical stimuli. Pain 2022, 164, 1012–1026. [Google Scholar] [CrossRef]
- Tomita, S.; Sekiguchi, F.; Deguchi, T.; Miyazaki, T.; Ikeda, Y.; Tsubota, M.; Yoshida, S.; Nguyen, H.D.; Okada, T.; Toyooka, N.; et al. Critical role of Ca(v)3.2 T-type calcium channels in the peripheral neuropathy induced by bortezomib, a proteasome-inhibiting chemotherapeutic agent, in mice. Toxicology 2019, 413, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Harding, E.K.; Dedek, A.; Bonin, R.P.; Salter, M.W.; Snutch, T.P.; Hildebrand, M.E. The T-type calcium channel antagonist, Z944, reduces spinal excitability and pain hypersensitivity. Br. J. Pharmacol. 2021, 178, 3517–3532. [Google Scholar] [CrossRef] [PubMed]
- Gambeta, E.; Gandini, M.A.; Souza, I.A.; Zamponi, G.W. CaV3.2 calcium channels contribute to trigeminal neuralgia. Pain 2022, 163, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Lee, M. Z944: A first in class T-type calcium channel modulator for the treatment of pain. J. Peripher. Nerv. Syst. 2014, 19 (Suppl. S2), S11–S12. [Google Scholar] [CrossRef] [PubMed]
- Flatters, S.J.; Bennett, G.J. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain 2004, 109, 150–161. [Google Scholar] [CrossRef]
- Kerckhove, N.; Pereira, B.; Soriot-Thomas, S.; Alchaar, H.; Deleens, R.; Hieng, V.S.; Serra, E.; Lanteri-Minet, M.; Arcagni, P.; Picard, P.; et al. Efficacy and safety of a T-type calcium channel blocker in patients with neuropathic pain: A proof-of-concept, randomized, double-blind and controlled trial. Eur. J. Pain 2018, 22, 1321–1330. [Google Scholar] [CrossRef]
- Kerckhove, N.; Scanzi, J.; Pereira, B.; Ardid, D.; Dapoigny, M. Assessment of the effectiveness and safety of ethosuximide in the treatment of abdominal pain related to irritable bowel syndrome—IBSET: Protocol of a randomised, parallel, controlled, double-blind and multicentre trial. BMJ Open 2017, 7, e015380. [Google Scholar] [CrossRef]
- El-Haggar, S.M.; Hegazy, S.K.; Abd-Elsalam, S.M.A.; Bahaa, M.M. Open-label pilot study of ethosuximide as adjunctive therapy for relieving abdominal pain related to Irritable Bowel Syndrome. J. Clin. Pharm. Ther. 2022, 47, 306–312. [Google Scholar] [CrossRef]
- Kerckhove, N.; Mallet, C.; Pereira, B.; Chenaf, C.; Duale, C.; Dubray, C.; Eschalier, A. Assessment of the effectiveness and safety of Ethosuximide in the Treatment of non-Diabetic Peripheral Neuropathic Pain: EDONOT-protocol of a randomised, parallel, controlled, double-blinded and multicentre clinical trial. BMJ Open 2016, 6, e013530. [Google Scholar] [CrossRef]
- Suzuki, S.; Kawakami, K.; Nishimura, S.; Watanabe, Y.; Yagi, K.; Seino, M.; Miyamoto, K. Zonisamide blocks T-type calcium channel in cultured neurons of rat cerebral cortex. Epilepsy Res. 1992, 12, 21–27. [Google Scholar] [CrossRef]
- Matar, N.; Jin, W.; Wrubel, H.; Hescheler, J.; Schneider, T.; Weiergräber, M. Zonisamide block of cloned human T-type voltage-gated calcium channels. Epilepsy Res. 2009, 83, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.; Singla, S.; Kumar, D.; Menaria, G. Comparison of the Effects of Zonisamide, Ethosuximide and Pregabalin in the Chronic Constriction Injury Induced Neuropathic Pain in Rats. Ann. Med. Health Sci. Res. 2015, 5, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Murakami, T.; Ono, H. Zonisamide suppresses pain symptoms of formalin-induced inflammatory and streptozotocin-induced diabetic neuropathy. J. Pharmacol. Sci. 2008, 107, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Hord, A.H.; Denson, D.D.; Chalfoun, A.G.; Azevedo, M.I. The effect of systemic zonisamide (Zonegran) on thermal hyperalgesia and mechanical allodynia in rats with an experimental mononeuropathy. Anesth. Analg. 2003, 96, 1700–1706. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Hashimoto, K.; Tsuji, S. Successful use of zonisamide for central poststroke pain. J. Pain 2004, 5, 192–194. [Google Scholar] [CrossRef]
- Limmer, A.L.; Holland, L.C.; Loftus, B.D. Zonisamide for Cluster Headache Prophylaxis: A Case Series. Headache 2019, 59, 924–929. [Google Scholar] [CrossRef]
- Chung, J.Y.; Kim, M.W.; Kim, M. Efficacy of zonisamide in migraineurs with nonresponse to topiramate. Biomed. Res. Int. 2014, 2014, 891348. [Google Scholar] [CrossRef]
- Pascual-Gómez, J.; Gracia-Naya, M.; Leira, R.; Mateos, V.; Alvaro-González, L.C.; Hernando, I.; Oterino, A.; Iglesias-Díez, F.; Caminero, A.; García-Moncó, J.C.; et al. Zonisamide in the preventive treatment of refractory migraine. Rev. Neurol. 2010, 50, 129–132. [Google Scholar]
- Drake, M.E., Jr.; Greathouse, N.I.; Renner, J.B.; Armentbright, A.D. Open-label zonisamide for refractory migraine. Clin. Neuropharmacol. 2004, 27, 278–280. [Google Scholar] [CrossRef]
- Atli, A.; Dogra, S. Zonisamide in the treatment of painful diabetic neuropathy: A randomized, double-blind, placebo-controlled pilot study. Pain Med. 2005, 6, 225–234. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Scott, V.E.; McGaraughty, S.; Chu, K.L.; Xu, J.; Niforatos, W.; Milicic, I.; Joshi, S.; Zhang, Q.; Xia, Z. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem. Pharmacol. 2014, 89, 536–544. [Google Scholar] [CrossRef]
- Zhang, Q.; Xia, Z.; Joshi, S.; Scott, V.E.; Jarvis, M.F. Optimization of ADME Properties for Sulfonamides Leading to the Discovery of a T-Type Calcium Channel Blocker, ABT-639. ACS Med. Chem. Lett. 2015, 6, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Picard, E.; Carvalho, F.A.; Agosti, F.; Bourinet, E.; Ardid, D.; Eschalier, A.; Daulhac, L.; Mallet, C. Inhibition of Cav3.2 calcium channels: A new target for colonic hypersensitivity associated with low-grade inflammation. Br. J. Pharmacol. 2019, 176, 950–963. [Google Scholar] [CrossRef] [PubMed]
- Serra, J.; Duan, W.R.; Locke, C.; Solà, R.; Liu, W.; Nothaft, W. Effects of a T-type calcium channel blocker, ABT-639, on spontaneous activity in C-nociceptors in patients with painful diabetic neuropathy: A randomized controlled trial. Pain 2015, 156, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.; Duan, W.R.; An, G.; Thomas, J.W.; Nothaft, W. A randomized double-blind, placebo-, and active-controlled study of T-type calcium channel blocker ABT-639 in patients with diabetic peripheral neuropathic pain. Pain 2015, 156, 2013–2020. [Google Scholar] [CrossRef]
- Chuang, R.S.; Jaffe, H.; Cribbs, L.; Perez-Reyes, E.; Swartz, K.J. Inhibition of T-type voltage-gated calcium channels by a new scorpion toxin. Nat. Neurosci. 1998, 1, 668–674. [Google Scholar] [CrossRef]
- Bladen, C.; Hamid, J.; Souza, I.A.; Zamponi, G.W. Block of T-type calcium channels by protoxins I and II. Mol. Brain 2014, 7, 36. [Google Scholar] [CrossRef]
- Cardoso, F.C.; Castro, J.; Grundy, L.; Schober, G.; Garcia-Caraballo, S.; Zhao, T.; Herzig, V.; King, G.F.; Brierley, S.M.; Lewis, R.J. A spider-venom peptide with multitarget activity on sodium and calcium channels alleviates chronic visceral pain in a model of irritable bowel syndrome. Pain 2021, 162, 569–581. [Google Scholar] [CrossRef]
- Chen, Z.X.; Zhang, H.L.; Gu, Z.L.; Chen, B.W.; Han, R.; Reid, P.F.; Raymond, L.N.; Qin, Z.H. A long-form alpha-neurotoxin from cobra venom produces potent opioid-independent analgesia. Acta Pharmacol. Sin. 2006, 27, 402–408. [Google Scholar] [CrossRef]
- Zhang, H.L.; Han, R.; Chen, Z.X.; Gu, Z.L.; Reid, P.F.; Raymond, L.N.; Qin, Z.H. Analgesic effects of receptin, a chemically modified cobratoxin from Thailand cobra venom. Neurosci. Bull. 2006, 22, 267–273. [Google Scholar]
- Liu, Y.L.; Lin, H.M.; Zou, R.; Wu, J.C.; Han, R.; Raymond, L.N.; Reid, P.F.; Qin, Z.H. Suppression of complete Freund’s adjuvant-induced adjuvant arthritis by cobratoxin. Acta Pharmacol. Sin. 2009, 30, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.N.; Liu, Y.L.; Lin, H.M.; Yang, S.L.; Feng, Y.L.; Reid, P.F.; Qin, Z.H. Involvement of cholinergic system in suppression of formalin-induced inflammatory pain by cobratoxin. Acta Pharmacol. Sin. 2011, 32, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Liang, Q.; Zhu, Q.; Ding, D.; Yin, Q.; Tao, J.; Jiang, X. Nicotinic acetylcholine receptor α7 subunit is involved in the cobratoxin-induced antinociception in an animal model of neuropathic pain. Toxicon 2015, 93, 31–36. [Google Scholar] [CrossRef]
- Xu, L.; Wang, S.; Zhang, L.; Liu, B.; Zheng, S.; Yao, M. Cobratoxin Alleviates Cancer-Induced Bone Pain in Rats via Inhibiting CaMKII Signaling Pathway after Acting on M4 Muscarinic Cholinergic Receptors. ACS Chem. Neurosci. 2022, 13, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y.; Jiang, D.; Reid, P.F.; Jiang, X.; Qin, Z.; Tao, J. Alpha-cobratoxin inhibits T-type calcium currents through muscarinic M4 receptor and Gο-protein βγ subunits-dependent protein kinase A pathway in dorsal root ganglion neurons. Neuropharmacology 2012, 62, 1062–1072. [Google Scholar] [CrossRef]
- Chemin, J.; Monteil, A.; Perez-Reyes, E.; Nargeot, J.; Lory, P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 2001, 20, 7033–7040. [Google Scholar] [CrossRef]
- Ross, H.R.; Gilmore, A.J.; Connor, M. Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonoyl dopamine. Br. J. Pharmacol. 2009, 156, 740–750. [Google Scholar] [CrossRef]
- Bladen, C.; Mirlohi, S.; Santiago, M.; Longworth, M.; Kassiou, M.; Banister, S.; Connor, M. Modulation of human T-type calcium channels by synthetic cannabinoid receptor agonists in vitro. Neuropharmacology 2021, 187, 108478. [Google Scholar] [CrossRef]
- Mirlohi, S.; Bladen, C.; Santiago, M.J.; Arnold, J.C.; McGregor, I.; Connor, M. Inhibition of human recombinant T-type calcium channels by phytocannabinoids in vitro. Br. J. Pharmacol. 2022, 179, 4031–4043. [Google Scholar] [CrossRef]
- Ross, H.R.; Napier, I.; Connor, M. Inhibition of recombinant human T-type calcium channels by Delta9-tetrahydrocannabinol and cannabidiol. J. Biol. Chem. 2008, 283, 16124–16134. [Google Scholar] [CrossRef]
- Gadotti, V.M.; Huang, S.; Zamponi, G.W. The terpenes camphene and alpha-bisabolol inhibit inflammatory and neuropathic pain via Cav3.2 T-type calcium channels. Mol. Brain 2021, 14, 166. [Google Scholar] [CrossRef]
- Harding, E.K.; Souza, I.A.; Gandini, M.A.; Gadotti, V.M.; Ali, M.Y.; Huang, S.; Antunes, F.T.T.; Trang, T.; Zamponi, G.W. Differential regulation of Cav3.2 and Cav2.2 calcium channels by CB1 receptors and cannabidiol. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef]
- You, H.; Gadotti, V.M.; Petrov, R.R.; Zamponi, G.W.; Diaz, P. Functional characterization and analgesic effects of mixed cannabinoid receptor/T-type channel ligands. Mol. Pain 2011, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Gadotti, V.M.; You, H.; Petrov, R.R.; Berger, N.D.; Diaz, P.; Zamponi, G.W. Analgesic effect of a mixed T-type channel inhibitor/CB2 receptor agonist. Mol. Pain 2013, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.D.; Gadotti, V.M.; Petrov, R.R.; Chapman, K.; Diaz, P.; Zamponi, G.W. NMP-7 inhibits chronic inflammatory and neuropathic pain via block of Cav3.2 T-type calcium channels and activation of CB2 receptors. Mol. Pain 2014, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Neumaier, F.; Hescheler, J.; Alpdogan, S. Cav2.3 R-type calcium channels: From its discovery to pathogenic de novo CACNA1E variants: A historical perspective. Pflugers Arch. 2020, 472, 811–816. [Google Scholar] [CrossRef]
- Saegusa, H.; Kurihara, T.; Zong, S.; Minowa, O.; Kazuno, A.; Han, W.; Matsuda, Y.; Yamanaka, H.; Osanai, M.; Noda, T.; et al. Altered pain responses in mice lacking alpha 1E subunit of the voltage-dependent Ca2+ channel. Proc. Natl. Acad. Sci. USA 2000, 97, 6132–6137. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Stephens, G.J. Effects of neuropathy on high-voltage-activated Ca2+ current in sensory neurones. Cell Calcium 2009, 46, 248–256. [Google Scholar] [CrossRef]
- Ghodsi, S.M.; Walz, M.; Schneider, T.; Todorovic, S.M. L-cysteine modulates visceral nociception mediated by the Ca. Pflugers Arch. 2022, 474, 435–445. [Google Scholar] [CrossRef]
- Matthews, E.A.; Bee, L.A.; Stephens, G.J.; Dickenson, A.H. The Cav2.3 calcium channel antagonist SNX-482 reduces dorsal horn neuronal responses in a rat model of chronic neuropathic pain. Eur. J. Neurosci. 2007, 25, 3561–3569. [Google Scholar] [CrossRef]
- Martin, L.; Ibrahim, M.; Gomez, K.; Yu, J.; Cai, S.; Chew, L.A.; Bellampalli, S.S.; Moutal, A.; Largent-Milnes, T.; Porreca, F.; et al. Conotoxin contulakin-G engages a neurotensin receptor 2/R-type calcium channel (Cav2.3) pathway to mediate spinal antinociception. Pain 2022, 163, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.S.; Nieto-Rostro, M.; Rahman, W.; Tran-Van-Minh, A.; Ferron, L.; Douglas, L.; Kadurin, I.; Sri Ranjan, Y.; Fernandez-Alacid, L.; Millar, N.S.; et al. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J. Neurosci. 2009, 29, 4076–4088. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Chen, X.; Kim, M.; Gong, N.; Bhatia, S.; Luo, Z.D. Differential effects of voltage-gated calcium channel blockers on calcium channel alpha-2-delta-1 subunit protein-mediated nociception. Eur. J. Pain 2015, 19, 639–648. [Google Scholar] [CrossRef]
- Li, C.Y.; Zhang, X.L.; Matthews, E.A.; Li, K.W.; Kurwa, A.; Boroujerdi, A.; Gross, J.; Gold, M.S.; Dickenson, A.H.; Feng, G.; et al. Calcium channel α2δ1 subunit mediates spinal hyperexcitability in pain modulation. Pain 2006, 125, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Deng, P.; Matthews, E.A.; Kim, D.S.; Feng, G.; Dickenson, A.H.; Xu, Z.C.; Luo, Z.D. Enhanced pre-synaptic glutamate release in deep-dorsal horn contributes to calcium channel α2δ1 protein-mediated spinal sensitization and behavioral hypersensitivity. Mol. Pain 2009, 5, 6. [Google Scholar] [CrossRef]
- Tuchman, M.; Barrett, J.A.; Donevan, S.; Hedberg, T.G.; Taylor, C.P. Central sensitization and CaVα₂δ ligands in chronic pain syndromes: Pathologic processes and pharmacologic effect. J. Pain 2010, 11, 1241–1249. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Q.; Jin, Z.; Qin, Y.; Meng, F.; Zhao, G. Review of Voltage-gated Calcium Channel α2δ Subunit Ligands for the Treatment of Chronic Neuropathic Pain and Insight into Structure-activity Relationship (SAR) by Pharmacophore Modeling. Curr. Med. Chem. 2022, 29, 5097–5112. [Google Scholar] [CrossRef]
- Chen, J.; Li, L.; Chen, S.R.; Chen, H.; Xie, J.D.; Sirrieh, R.E.; MacLean, D.M.; Zhang, Y.; Zhou, M.H.; Jayaraman, V.; et al. The α2δ-1-NMDA Receptor Complex Is Critically Involved in Neuropathic Pain Development and Gabapentin Therapeutic Actions. Cell Rep. 2018, 22, 2307–2321. [Google Scholar] [CrossRef]
- Patel, R.; Dickenson, A.H. Mechanisms of the gabapentinoids and α2δ-1 calcium channel subunit in neuropathic pain. Pharmacol. Res. Perspect. 2016, 4, e00205. [Google Scholar] [CrossRef]
- Chen, E.Y.; Beutler, S.S.; Kaye, A.D.; Edinoff, A.N.; Khademi, S.H.; Stoltz, A.E.; Rueb, N.R.; Cornett, E.M.; Suh, W.J. Mirogabalin as a Novel Gabapentinoid for the Treatment of Chronic Pain Conditions: An Analysis of Current Evidence. Anesth. Pain Med. 2021, 11, e121402. [Google Scholar] [CrossRef]


{kind=link}
| Peptide | Channels Target | Organism Derived | Analgesic Effect in Pain Rodent Models—Reference |
|---|---|---|---|
| GVIA | Cav2.1, and Cav2.2 | C. geographus | Fukuizumi et al. [25] Murakami et al. [49] Scott et al. [97] |
| MVIIC | Cav2.1, and Cav2.2 | C. magus | Dalmolin et al. [53] |
| SVIB | Cav2.1, and Cav2.2 | C. striatus | - |
| CVIA | Cav2.1, and Cav2.2 | Conus catus | - |
| CVIB | Cav2.1, and Cav2.2 | - | |
| CVIC | Cav2.1, and Cav2.2 | - | |
| CVID | Cav2.1, and Cav2.2 | Scott et al. [97] | |
| CVIF | Cav1., Cav1.3, Cav2.2, and Cav2.3 | Berecki et al. [98] | |
| CnVIIA | Cav2.1, and Cav2.2 | Conus consors | - |
| PhTx3-4 | Cav2.1, and Cav2.2 | P. nigriventer spider | Da Silva et al. [99] |
| PhTx3-5 | Cav1.x, and Cav.2.2 | Oliveira et al. [100] | |
| PhTx3-6 | Cav2.1, Cav2.2, and Cav2.3 | See review by da Silva et al. [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antunes, F.T.T.; Campos, M.M.; Carvalho, V.d.P.R.; da Silva Junior, C.A.; Magno, L.A.V.; de Souza, A.H.; Gomez, M.V. Current Drug Development Overview: Targeting Voltage-Gated Calcium Channels for the Treatment of Pain. Int. J. Mol. Sci. 2023, 24, 9223. https://doi.org/10.3390/ijms24119223
Antunes FTT, Campos MM, Carvalho VdPR, da Silva Junior CA, Magno LAV, de Souza AH, Gomez MV. Current Drug Development Overview: Targeting Voltage-Gated Calcium Channels for the Treatment of Pain. International Journal of Molecular Sciences. 2023; 24(11):9223. https://doi.org/10.3390/ijms24119223
Chicago/Turabian StyleAntunes, Flavia Tasmin Techera, Maria Martha Campos, Vanice de Paula Ricardo Carvalho, Claudio Antonio da Silva Junior, Luiz Alexandre Viana Magno, Alessandra Hubner de Souza, and Marcus Vinicius Gomez. 2023. "Current Drug Development Overview: Targeting Voltage-Gated Calcium Channels for the Treatment of Pain" International Journal of Molecular Sciences 24, no. 11: 9223. https://doi.org/10.3390/ijms24119223
APA StyleAntunes, F. T. T., Campos, M. M., Carvalho, V. d. P. R., da Silva Junior, C. A., Magno, L. A. V., de Souza, A. H., & Gomez, M. V. (2023). Current Drug Development Overview: Targeting Voltage-Gated Calcium Channels for the Treatment of Pain. International Journal of Molecular Sciences, 24(11), 9223. https://doi.org/10.3390/ijms24119223