
Endosome Traffic Modulates Pro-Inflammatory Signal Transduction in CD4+ T Cells—Implications for the Pathogenesis of Systemic Lupus Erythematosus

Abstract
:1. Introduction
2. Endosomal Trafficking and Recycling Pathways
2.1. Endocytic Regulation of Antigen Presentation to CD4+ T Cells
2.2. Contribution of Endosomal Traffic to T Cell Synapse Formation
3. Trafficked Receptors Impact Metabolic Abnormalities in T Cells
3.1. TCR and STIM1 Trafficking Affects Calcium Flux
3.2. Glucose Transporters and Metabolism
3.3. Transferrin Receptor, CD71, and Iron Metabolism
3.4. NAD+ Synthesis and Metabolism
3.4.1. Amino Acid Transporter, CD98
3.4.2. NAD+ Hydrolases, CD38, CD157, and SARM1
3.4.3. CD73
3.5. Role of Endosome Traffic in Toll-like Receptor-Mediated Signaling
4. Proinflammatory Signaling Pathways Impacted by Trafficked Receptors in CD4+ T Cells
4.1. IL-2 and Tregs Are Decreased
4.1.1. CREM and CREB Control IL-2 Production
4.1.2. NFAT and AP-1 Control IL-2 Production
4.2. IL-17 and Th17 Are Increased
4.2.1. JAK/STAT3 Pathway Regulates Th17 Differentiation
4.2.2. mTOR Regulates IL-17 Production and Th17 Differentiation
4.2.3. Histone Modification Regulates Th17 Differentiation and IL-17 Production
4.3. Regulation of Tfh Development

{kind=link}
{kind=link}
{kind=link}
| Recycled Receptors | Responsible Rab GTPase | Downstream Metabolites | Subsequent Signaling/Epigenetic Pathways | Effects on T Cell Subset/Cytokine | References |
|---|---|---|---|---|---|
| CD3ζ (↓) | Rab4A (↑) | Ca2+ (↑) | Ca2+/CaM/PP2A/de-pCREB | Decrease IL-2 (↓) | [10,229] |
| Ca2+/CaM/PP2A/SP-1/CREM (↑) | [10,230,231] | ||||
| Calcineurin/NFAT (↑) (no AP-1) | [174,175,176,177,232] | ||||
| CaMKIV/CREMα | Increase IL-17 (↑) and Th17 (↑) | [10,277] | |||
| CaMKIV/PI3K/AKT/mTORC1 (S6K) | [10,275] | ||||
| Syk/PI3K/PIP3/PDK1/AKT/pTSC2/mTORC1 (S6K) | [10,59,283,288,289,291] | ||||
| CD38(↑)/ CD157 | ? | CREM/ICER/RORγt | [174,175,176,177,302] | ||
| ICER/Sirtuin-2/mTORC1/HIF-1α/RORγt | [174,175,176,177,303] | ||||
| CREM/ICER/Sirtuin-2/p70S6K/HIF-1α/RORγt | [174,175,176,177,239,255] | ||||
| NAD+ (↓) | Sirtuin-1/STAT3 | Increase Th17 | [174,175,176,177,312,313] | ||
| Sirtuin-2/de-Ac c-Jun | Decrease IL-2 | [157,162,239] | |||
| CD98 (↑) | Rab22a | Kynurenine | NAD+ synthesis | - | [158,159,160] |
| CD4 (↓) | Rab4A (↑) | ? | ? | ? | [61] |
| CD71 (TfR) (↑) | Rab4A, Rab5, Rab11a | Fe2+ | TET/DNA hypomethylation | Tfh | [61,93,135,320] |
| GLUT1 (↑) | Rab25 | Glucose | PEP/Ca2+/NFAT (no AP-1) | Decrease IL-2 (↓) | [117,119,232] |
| PMK2/STAT3 | Increase Th17 (↑) | [299,300] |
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3-PEHPC | 2-hydroxy-2-phosphono-3-(pyridin-3-yl) propanoic acid |
| AKT | serine/threonine protein kinase B |
| AMP | adenosine monophosphate |
| AP-1 | activator protein 1 |
| AP-3 | adaptor protein 3 |
| APC | antigen presenting cell |
| Bcl6 | B cell lymphoma 6 |
| CaM | calmodulin |
| CaMKIV | calcium/calmodulin-dependent protein kinase IV |
| CCR6 | C-C-motif chemokine receptor 6 |
| CD | cluster of differentiation |
| CD40L | CD40 ligand |
| cGVHD | chronic graft versus host disease |
| CIE | clathrin-independent endocytosis |
| CME | clathrin-mediated endocytosis |
| CpG | cytosine and guanine separated by a phosphate |
| CRE | cyclic AMP responsive element |
| CREB | cyclic AMP responsive element-binding protein |
| CREM | cyclic AMP element modulator |
| CXCL | chemokine (C-X-C motif) ligand |
| DN T | double negative T cell |
| Drp1 | dynamin-related protein 1 |
| EAE | experimental autoimmune encephalomyelitis |
| EEA1 | early endosomal antigen 1 |
| EGR2 | early growth response 2 |
| ELAVL1 | embryonic lethal vision like protein 1 |
| ERC | endosomal recycling compartment |
| FcεRIγ | Fc epsilon receptor I gamma chain |
| FoxP3 | forkhead box P3 |
| GAP | GTPase-activating protein |
| G-CSF | granulocyte colony stimulating factor |
| GDP | guanosine diphosphate |
| GEF | guanine nucleotide exchange factor |
| Gfi1 | growth factor independent 1 transcriptional repressor |
| GM-CSF | granulocyte monocyte-colony stimulating factor |
| GTP | guanosine triphosphate |
| GTPase | guanosine triphosphatase |
| HIF-1α | hypoxia-inducible factor 1α |
| HRES-1 | HTLV-1 related endogenous retroviral sequence 1 |
| ICER | inducible cyclic AMP early repressor |
| IFN | interferon |
| IFNAR1 | Type I IFN receptor |
| IL | interleukin |
| IL-6R | interleukin 6 receptor |
| IS | immunological synapse |
| ITAM | immunoreceptor tyrosine-based activation motif |
| JAK | Janus kinase |
| LAT | linker for activation of T cells |
| Lck | lymphocyte-specific proteins tyrosine kinase |
| MHC | major histocompatibility complex |
| miRNA | microRNA |
| mRNA | messenger RNA |
| MRL-lpr | Murphy Roths Large-lymphoproliferation |
| mTOR | mechanistic target of rapamycin |
| mTORC | mechanistic target of rapamycin complex |
| NFAT | nuclear factor of activated T cells |
| NF-κB | nuclear factor-κB |
| OXPHOS | oxidative phosphorylation |
| Pam3CSK4 | Pam3-Cys-Ser-Lys4 |
| PDK1 | phosphatidylinositol-dependent kinase 1 |
| PI3K | phosphoinositol-3 kinase |
| PI3P | phosphoinositol-3-phosphate |
| PIP2 | phosphatidylinositol-4,5-biphosphate |
| PIP3 | phosphatidylinositol-3,4,5-triphosphate |
| PKB | serine/threonine protein kinase B |
| PKC | protein kinase C |
| PLC-γ | phospholipase Cγ |
| PP2A | protein phosphatase 2 |
| RORγt | retinoic-acid-receptor-related orphan nuclear receptor γ |
| S6K | ribosomal S6 kinase |
| SH2 | SRC homology domain 2 |
| sIL-6R | soluble IL-6 receptor |
| SLE | systemic lupus erythematosus |
| SLP-76 | SH2-domain-containing leukocyte protein of 76 kDa |
| SMAC | supramolecular activation complex |
| SNPs | single nucleotide polymorphisms |
| SNX27 | sorting nexin 27 |
| SP-1 | specificity protein 1 |
| STAT | signal transducer and activator of transcription |
| Syk | spleen tyrosine kinase |
| TCR | T cell receptor |
| Tfh | follicular helper T cell |
| TGFβ | transforming growth factor β |
| TGN | trans-Golgi network |
| Th17 | helper T 17 cell |
| TLR | toll-like receptor |
| Treg | regulatory T cell |
| TSC2 | tuberous sclerosis complex 2 |
| WASp | Wiscott–Aldrich syndrome protein |
| ZAP-70 | ζ-associated protein kinase 70 |
References
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.B.; Zoncu, R.; Root, D.E.; Sabatini, D.M.; Sawyers, C.L. Identification of an oncogenic RAB protein. Science 2015, 350, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, H.-T.; Wang, Y.-C. Rab-mediated vesicle trafficking in cancer. J. Biomed. Sci. 2016, 23, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, C.E.L.; Tang, B.L. The role of the small GTPase Rab31 in cancer. J. Cell. Mol. Med. 2015, 19, 1–10. [Google Scholar] [CrossRef]
- Wang, S.; Hu, C.; Wu, F.; He, S. Rab25 GTPase: Functional roles in cancer. Oncotarget 2017, 8, 64591–64599. [Google Scholar] [CrossRef] [Green Version]
- Veleri, S.; Punnakkal, P.; Dunbar, G.L.; Maiti, P. Molecular Insights into the Roles of Rab Proteins in Intracellular Dynamics and Neurodegenerative Diseases. Neuromol. Med. 2018, 20, 18–36. [Google Scholar] [CrossRef]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R486. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Rabs, Membrane Dynamics, and Parkinson’s Disease. J. Cell. Physiol. 2017, 232, 1626–1633. [Google Scholar] [CrossRef]
- Prashar, A.; Schnettger, L.; Bernard, E.M.; Gutierrez, M.G. Rab GTPases in Immunity and Inflammation. Front. Cell Infect. Microbiol. 2017, 7, 435. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, D.R.; Telarico, T.; Bonilla, E.; Li, Q.; Banerjee, S.; Middleton, F.A.; Phillips, P.E.; Crow, M.K.; Oess, S.; Muller-Esterl, W.; et al. Activation of mTOR controls the loss of TCRζ in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J. Immunol. 2009, 182, 2063–2073. [Google Scholar] [CrossRef] [Green Version]
- Evnouchidou, I.; Caillens, V.; Koumantou, D.; Saveanu, L. The role of endocytic trafficking in antigen T cell receptor activation. Biomed. J. 2022, 45, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. Oxidative stress and endosome recycling are complementary mechanisms reorganizing the T-cell receptor signaling complex in SLE. Clin. Immunol. 2012, 142, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, L.A.; Tsokos, G.C. The IL-2 Defect in Systemic Lupus Erythematosus Disease Has an Expansive Effect on Host Immunity. J. Biomed. Biotechnol. 2010, 2010, e740619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonelli, M.; Savitskaya, A.; von Dalwigk, K.; Steiner, C.W.; Aletaha, D.; Smolen, J.S.; Scheinecker, C. Quantitative and qualitative deficiencies of regulatory T cells in patients with systemic lupus erythematosus (SLE). Int. Immunol. 2008, 20, 861–868. [Google Scholar] [CrossRef]
- Miyara, M.; Amoura, Z.; Parizot, C.; Badoual, C.; Dorgham, K.; Trad, S.; Nochy, D.; Debré, P.; Piette, J.-C.; Gorochov, G. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J. Immunol. 2005, 175, 8392–8400. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Linker-Israeli, M.; Deans, R.J.; Wallace, D.J.; Prehn, J.; Ozeri-Chen, T.; Klinenberg, J.R. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J. Immunol. 1991, 147, 117–123. [Google Scholar] [CrossRef]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Apostolidis, S.A.; Lieberman, L.A.; Kis-Toth, K.; Crispín, J.C.; Tsokos, G.C. The Dysregulation of Cytokine Networks in Systemic Lupus Erythematosus. J. Interferon Cytokine Res. 2011, 31, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, C.; Tangye, S.G.; Mackay, C.R. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu. Rev. Immunol. 2008, 26, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Mountz, J.D.; Hsu, H.-C.; Ballesteros-Tato, A. Dysregulation of T Follicular Helper Cells in Lupus. J. Immunol. 2019, 202, 1649–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Guo, B.; Wu, H.; Tan, L.; Chang, C.; Lu, Q. Interleukin-17 in systemic lupus erythematosus: A comprehensive review. Autoimmunity 2015, 48, 353–361. [Google Scholar] [CrossRef]
- Linker-Israeli, M.; Bakke, A.C.; Kitridou, R.C.; Gendler, S.; Gillis, S.; Horwitz, D.A. Defective production of interleukin 1 and interleukin 2 in patients with systemic lupus erythematosus (SLE). J. Immunol. 1983, 130, 2651–2655. [Google Scholar] [CrossRef]
- Maxfield, F.R.; McGraw, T.E. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 2004, 5, 121–132. [Google Scholar] [CrossRef]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Bitsikas, V.; Corrêa, I.R., Jr.; Nichols, B.J. Clathrin-independent pathways do not contribute significantly to endocytic flux. eLife 2014, 3, e03970. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Novick, P.; Zerial, M. The diversity of Rab proteins in vesicle transport. Curr. Opin. Cell Biol. 1997, 9, 496–504. [Google Scholar] [CrossRef]
- Barr, F.; Lambright, D.G. Rab GEFs and GAPs. Curr. Opin. Cell Biol. 2010, 22, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Mellman, I.S.; Muller, W.A.; Cohn, Z.A. Cohn Endocytosis and the recycling of plasma membrane. J. Cell Biol. 1983, 96, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 1996, 12, 575–625. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Fedoseienko, A.; Chen, B.; Burstein, E.; Jia, D.; Billadeau, D.D. Endosomal Receptor Trafficking: Retromer and Beyond. Traffic 2018, 19, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sönnichsen, B.; De Renzis, S.; Nielsen, E.; Rietdorf, J.; Zerial, M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 2000, 149, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Sheff, D.R.; Daro, E.A.; Hull, M.; Mellman, I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J. Cell Biol. 1999, 145, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Riederer, M.A.; Soldati, T.; Shapiro, A.D.; Lin, J.; Pfeffer, S.R. Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol. 1994, 125, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the switch in early-to-late endosome transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Bogan, J.S. Regulation of glucose transporter translocation in health and diabetes. Annu. Rev. Biochem. 2012, 81, 507–532. [Google Scholar] [CrossRef]
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, A.; Von Zastrow, M. Signal transduction and endocytosis: Close encounters of many kinds. Nat. Rev. Mol. Cell Biol. 2002, 3, 600–614. [Google Scholar] [CrossRef]
- Jones, M.C.; Caswell, P.T.; Norman, J.C. Endocytic recycling pathways: Emerging regulators of cell migration. Curr. Opin. Cell Biol. 2006, 18, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Gorvel, J.P.; Chavrier, P.; Zerial, M.; Gruenberg, J. rab5 controls early endosome fusion in vitro. Cell 1991, 64, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Parton, R.G.; Mather, I.H.; Stunnenberg, H.; Simons, K.; Hoflack, B.; Zerial, M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 1992, 70, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, H.; Giner, A.; Hoflack, B.; Zerial, M. A GDP/GTP Exchange-stimulatory Activity for the Rab5-RabGDI Complex on Clathrin-coated Vesicles from Bovine Brain. J. Biol. Chem. 1995, 270, 11257–11262. [Google Scholar] [CrossRef] [Green Version]
- Christoforidis, S.; Miaczynska, M.; Ashman, K.; Wilm, M.; Zhao, L.; Yip, S.C.; Waterfield, M.D.; Backer, J.M.; Zerial, M. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1999, 1, 249–252. [Google Scholar] [CrossRef]
- Schu, P.V.; Takegawa, K.; Fry, M.J.; Stack, J.H.; Waterfield, M.D.; Emr, S.D. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 1993, 260, 88–91. [Google Scholar] [CrossRef]
- Patki, V.; Virbasius, J.; Lane, W.S.; Toh, B.H.; Shpetner, H.S.; Corvera, S. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 7326–7330. [Google Scholar] [CrossRef] [Green Version]
- Mills, I.G.; Jones, A.T.; Clague, M.J. Involvement of the endosomal autoantigen EEA1 in homotypic fusion of early endosomes. Curr. Biol. 1998, 8, 881–884. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, A.; Lippé, R.; Christoforidis, S.; Gaullier, J.M.; Brech, A.; Callaghan, J.; Toh, B.H.; Murphy, C.; Zerial, M.; Stenmark, H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 1998, 394, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Christoforidis, S.; McBride, H.M.; Burgoyne, R.D.; Zerial, M. The Rab5 effector EEA1 is a core component of endosome docking. Nature 1999, 397, 621–625. [Google Scholar] [CrossRef]
- Dumas, J.J.; Merithew, E.; Sudharshan, E.; Rajamani, D.; Hayes, S.; Lawe, D.; Corvera, S.; Lambright, D.G. Multivalent endosome targeting by homodimeric EEA1. Mol. Cell 2001, 8, 947–958. [Google Scholar] [CrossRef]
- Lawe, D.C.; Chawla, A.; Merithew, E.; Dumas, J.; Carrington, W.; Fogarty, K.; Lifshitz, L.; Tuft, R.; Lambright, D.; Corvera, S. Sequential roles for phosphatidylinositol 3-phosphate and Rab5 in tethering and fusion of early endosomes via their interaction with EEA1. J. Biol. Chem. 2002, 277, 8611–8617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, H.M.; Rybin, V.; Murphy, C.; Giner, A.; Teasdale, R.; Zerial, M. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell 1999, 98, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Kouranti, I.; Sachse, M.; Arouche, N.; Goud, B.; Echard, A. Rab35 Regulates an Endocytic Recycling Pathway Essential for the Terminal Steps of Cytokinesis. Curr. Biol. 2006, 16, 1719–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, A.; Sharma, D.K.; Marks, D.L.; Pagano, R.E. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol. Biol. Cell 2004, 15, 4500–4511. [Google Scholar] [CrossRef]
- Yudowski, G.A.; Puthenveedu, M.A.; Henry, A.G.; von Zastrow, M. Cargo-Mediated Regulation of a Rapid Rab4-Dependent Recycling Pathway. Mol. Biol. Cell 2009, 20, 2774–2784. [Google Scholar] [CrossRef] [Green Version]
- McCaffrey, M.W.; Bielli, A.; Cantalupo, G.; Mora, S.; Roberti, V.; Santillo, M.; Drummond, F.; Bucci, C. Rab4 affects both recycling and degradative endosomal trafficking. FEBS Lett. 2001, 495, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, S.; Rani, S.; Mahanty, S.; Bissig, C.; Arora, P.; Azevedo, C.; Saiardi, A.; van der Sluijs, P.; Delevoye, C.; van Niel, G.; et al. Rab4A organizes endosomal domains for sorting cargo to lysosome-related organelles. J. Cell Sci. 2018, 131, jcs216226. [Google Scholar] [CrossRef] [Green Version]
- van der Sluijs, P.; Hull, M.; Webster, P.; Mâle, P.; Goud, B.; Mellman, I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 1992, 70, 729–740. [Google Scholar] [CrossRef]
- Nagy, G.; Ward, J.; Mosser, D.D.; Koncz, A.; Gergely, P.; Stancato, C.; Qian, Y.; Fernandez, D.; Niland, B.; Grossman, C.E.; et al. Regulation of CD4 Expression via Recycling by HRES-1/RAB4 Controls Susceptibility to HIV Infection. J. Biol. Chem. 2006, 281, 34574–34591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, R.; Rinderknecht, C.H.; Roh, S.; Lee, A.W.; Harding, J.J.; Burster, T.; Hornell, T.M.C.; Mellins, E.D. Achieving stability through editing and chaperoning: Regulation of MHC class II peptide binding and expression. Immunol. Rev. 2005, 207, 242–260. [Google Scholar] [CrossRef] [PubMed]
- Lagattuta, K.A.; Kang, J.B.; Nathan, A.; Pauken, K.E.; Jonsson, A.H.; Rao, D.A.; Sharpe, A.H.; Ishigaki, K.; Raychaudhuri, S. Repertoire analyses reveal T cell antigen receptor sequence features that influence T cell fate. Nat. Immunol. 2022, 23, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Montesinos, G.; López-Ortega, O.; Piedra-Reyes, J.; Bonifaz, L.C.; Moreno, J. Dynamic Changes in the Intracellular Association of Selected Rab Small GTPases with MHC Class II and DM during Dendritic Cell Maturation. Front. Immunol. 2017, 8, 340. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Lazzarino, D.A.; Blier, P.; Mellman, I. The Monomeric Guanosine Triphosphatase rab4 Controls an Essential Step on the Pathway of Receptor-mediated Antigen Processing in B Cells. J. Exp. Med. 1998, 188, 1769–1774. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, M.; Leimgruber, E.; Seguín-Estévez, Q.; Dunand-Sauthier, I.; Barras, E.; Reith, W. Expression of RAB4B, a protein governing endocytic recycling, is co-regulated with MHC class II genes. Nucleic Acids Res. 2007, 35, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Schorey, J.S.; Bhatnagar, S. Exosome function: From tumor immunology to pathogen biology. Traffic 2008, 9, 871–881. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, I.; Akram, Z.; Iqbal, H.M.N.; Munn, A.L. The regulation of Endosomal Sorting Complex Required for Transport and accessory proteins in multivesicular body sorting and enveloped viral budding—An overview. Int. J. Biol. Macromol. 2019, 127, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.L.; Cheng, Y.; Bryant, B.R.; Schorey, J.S. Exosomes function in antigen presentation during an in vivo Mycobacterium tuberculosis infection. Sci. Rep. 2017, 7, 43578. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Zhao, M.; Wu, H.; Zhang, Y.; Tong, X.; Gao, L.; Zhou, L.; Lu, Q.; Zeng, J. Downregulated Serum Exosomal miR-451a Expression Correlates With Renal Damage and Its Intercellular Communication Role in Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 630112. [Google Scholar] [CrossRef] [PubMed]
- Monks, C.R.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef]
- Anderson, G.; Moore, N.C.; Owen, J.J.T.; Jenkinson, E.J. Cellular Interactions in Thymocyte Development. Annu. Rev. Immunol. 1996, 14, 73–99. [Google Scholar] [CrossRef] [PubMed]
- Krissansen, G.W.; Owen, M.J.; Verbi, W.; Crumpton, M.J. Primary structure of the T3 gamma subunit of the T3/T cell antigen receptor complex deduced from cDNA sequences: Evolution of the T3 gamma and delta subunits. EMBO J. 1986, 5, 1799–1808. [Google Scholar] [CrossRef]
- Kane, L.P.; Lin, J.; Weiss, A. Signal transduction by the TCR for antigen. Curr. Opin. Immunol. 2000, 12, 242–249. [Google Scholar] [CrossRef]
- Freiberg, B.A.; Kupfer, H.; Maslanik, W.; Delli, J.; Kappler, J.; Zaller, D.M.; Kupfer, A. Staging and resetting T cell activation in SMACs. Nat. Immunol. 2002, 3, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.; Millán, J.; Mittelbrunn, M.; Sánchez-Madrid, F.; Alonso, M.A. Recruitment of transferrin receptor to immunological synapse in response to TCR engagement. J. Immunol. 2004, 172, 6709–6714. [Google Scholar] [CrossRef] [Green Version]
- Kao, H.; Lin, J.; Littman, D.R.; Shaw, A.S.; Allen, P.M. Regulated Movement of CD4 In and Out of the Immunological Synapse1. J. Immunol. 2008, 181, 8248–8257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchet, J.; Del Río-Iñiguez, I.; Vázquez-Chávez, E.; Lasserre, R.; Agüera-González, S.; Cuche, C.; McCaffrey, M.W.; Di Bartolo, V.; Alcover, A. Rab11-FIP3 Regulation of Lck Endosomal Traffic Controls TCR Signal Transduction. J. Immunol. 2017, 198, 2967–2978. [Google Scholar] [CrossRef] [Green Version]
- Thill, P.A.; Weiss, A.; Chakraborty, A.K. Phosphorylation of a Tyrosine Residue on Zap70 by Lck and Its Subsequent Binding via an SH2 Domain May Be a Key Gatekeeper of T Cell Receptor Signaling In Vivo. Mol. Cell. Biol. 2016, 36, 2396–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samelson, L.E. Signal transduction mediated by the T cell antigen receptor: The role of adapter proteins. Annu. Rev. Immunol. 2002, 20, 371–394. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.; Zhong, X. Role of diacylglycerol kinases in T cell development and function. Crit. Rev. Immunol. 2013, 33, 97–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, H.; Lasserre, R.; Alcover, A. Orchestrating cytoskeleton and intracellular vesicle traffic to build functional immunological synapses. Immunol. Rev. 2013, 256, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Patrussi, L.; Masi, G.; Onnis, A.; Galgano, D.; Lucherini, O.M.; Pazour, G.J.; Baldari, C.T. Specific recycling receptors are targeted to the immune synapse by the intraflagellar transport system. J. Cell Sci. 2014, 127, 1924–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degli Esposti, M.; Matarrese, P.; Tinari, A.; Longo, A.; Recalchi, S.; Khosravi-Far, R.; Malorni, W.; Misasi, R.; Garofalo, T.; Sorice, M. Changes in membrane lipids drive increased endocytosis following Fas ligation. Apoptosis 2017, 22, 681–695. [Google Scholar] [CrossRef] [Green Version]
- Carpier, J.-M.; Zucchetti, A.E.; Bataille, L.; Dogniaux, S.; Shafaq-Zadah, M.; Bardin, S.; Lucchino, M.; Maurin, M.; Joannas, L.D.; Magalhaes, J.G.; et al. Rab6-dependent retrograde traffic of LAT controls immune synapse formation and T cell activation. J. Exp. Med. 2018, 215, 1245–1265. [Google Scholar] [CrossRef]
- González-Mancha, N.; Rodríguez-Rodríguez, C.; Alcover, A.; Merida, I. Sorting Nexin 27 Enables MTOC and Secretory Machinery Translocation to the Immune Synapse. Front. Immunol. 2022, 12, 814570. [Google Scholar] [CrossRef]
- Temkin, P.; Lauffer, B.; Jäger, S.; Cimermancic, P.; Krogan, N.J.; von Zastrow, M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat. Cell Biol. 2011, 13, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-M.; Tsokos, G.C. The role of CD8+ T-cell systemic lupus erythematosus pathogenesis: An update. Curr. Opin. Rheumatol. 2021, 33, 586–591. [Google Scholar] [CrossRef]
- Yin, Z.; Bai, L.; Li, W.; Zeng, T.; Tian, H.; Cui, J. Targeting T cell metabolism in the tumor microenvironment: An anti-cancer therapeutic strategy. J. Exp. Clin. Cancer Res. 2019, 38, 403. [Google Scholar] [CrossRef] [PubMed]
- Voss, K.; Sewell, A.E.; Krystofiak, E.S.; Gibson-Corley, K.N.; Young, A.C.; Basham, J.H.; Sugiura, A.; Arner, E.N.; Beavers, W.N.; Kunkle, D.E.; et al. Elevated transferrin receptor impairs T cell metabolism and function in systemic lupus erythematosus. Sci. Immunol. 2023, 8, eabq0178. [Google Scholar] [CrossRef] [PubMed]
- Enyedy, E.J.; Nambiar, M.P.; Liossis, S.N.; Dennis, G.; Kammer, G.M.; Tsokos, G.C. Fc epsilon receptor type I gamma chain replaces the deficient T cell receptor zeta chain in T cells of patients with systemic lupus erythematosus. Arthritis Rheum. 2001, 44, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Suzuki, K.; Kondo, T.; Yoshimoto, K.; Tsuzaka, K. CD3 ζ defects in systemic lupus erythematosus. Ann. Rheum. Dis. 2012, 71, i78–i81. [Google Scholar] [CrossRef]
- Takai, T. Fc receptors and their role in immune regulation and autoimmunity. J. Clin. Immunol. 2005, 25, 1–18. [Google Scholar] [CrossRef]
- Liu, C.P.; Lin, W.J.; Huang, M.; Kappler, J.W.; Marrack, P. Development and function of T cells in T cell antigen receptor/CD3 zeta knockout mice reconstituted with Fc epsilon RI gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 616–621. [Google Scholar] [CrossRef] [Green Version]
- Liossis, S.N.; Ding, X.Z.; Dennis, G.J.; Tsokos, G.C. Altered pattern of TCR/CD3-mediated protein-tyrosyl phosphorylation in T cells from patients with systemic lupus erythematosus. Deficient expression of the T cell receptor zeta chain. J. Clin. Investig. 1998, 101, 1448–1457. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.; Warke, V.G.; Nambiar, M.P.; Tsokos, G.C.; Farber, D.L. The FcR gamma subunit and Syk kinase replace the CD3 zeta-chain and ZAP-70 kinase in the TCR signaling complex of human effector CD4 T cells. J. Immunol. 2003, 170, 4189–4195. [Google Scholar] [CrossRef] [Green Version]
- Choi, O.H.; Kim, J.-H. Calcium mobilization via sphingosine kinase in signalling by the FctRI antigen receptor. Nature 1996, 380, 634–636. [Google Scholar] [CrossRef]
- Savina, A.; Furlán, M.; Vidal, M.; Colombo, M.I. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J. Biol. Chem. 2003, 278, 20083–20090. [Google Scholar] [CrossRef] [Green Version]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Zhou, Y.; Srinivasan, P.; Razavi, S.; Seymour, S.; Meraner, P.; Gudlur, A.; Stathopulos, P.B.; Ikura, M.; Rao, A.; Hogan, P.G. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat. Struct. Mol. Biol. 2013, 20, 973–981. [Google Scholar] [CrossRef] [Green Version]
- de Souza, L.B.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Fast endocytic recycling determines TRPC1-STIM1 clustering in ER-PM junctions and plasma membrane function of the channel. Biochim. Biophys. Acta 2015, 1853, 2709–2721. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Panicker, S.; Lau, K.-Y.; Apparsundaram, S.; Patel, V.A.; Chen, S.-L.; Soto, R.; Jung, J.K.C.; Ravindran, P.; Okuhara, D.; et al. Characterization of a novel CRAC inhibitor that potently blocks human T cell activation and effector functions. Mol. Immunol. 2013, 54, 355–367. [Google Scholar] [CrossRef]
- Wang, Y.; Huynh, W.; Skokan, T.D.; Lu, W.; Weiss, A.; Vale, R.D. CRACR2a is a calcium-activated dynein adaptor protein that regulates endocytic traffic. J. Cell Biol. 2019, 218, 1619–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Rice, L.; Shrimpton, J.; Lawless, D.; Walker, K.; Carter, C.; McKeown, L.; Anwar, R.; Doody, G.M.; Srikanth, S.; et al. Biallelic mutations in calcium release activated channel regulator 2A (CRACR2A) cause a primary immunodeficiency disorder. Elife 2021, 10, e72559. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, X.; Cornaby, C.; Li, W.; Morel, L. Metabolic regulation of pathogenic autoimmunity: Therapeutic targeting. Curr. Opin. Immunol. 2019, 61, 10–16. [Google Scholar] [CrossRef]
- Sharabi, A.; Tsokos, G.C. T cell metabolism: New insights in systemic lupus erythematosus pathogenesis and therapy. Nat. Rev. Rheumatol. 2020, 16, 100–112. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine Stimulation Promotes Glucose Uptake via Phosphatidylinositol-3 Kinase/Akt Regulation of Glut1 Activity and Trafficking. MBoC 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.W.; Agarwal, R.; Mitra, S.; Lee, J.-S.; Carey, M.; Gray, J.W.; Mills, G.B. Rab25 increases cellular ATP and glycogen stores protecting cancer cells from bioenergetic stress. EMBO Mol. Med. 2012, 4, 125–141. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, T.; Sato, T.; Furukawa, K.; Morimoto, S.; Endo, Y.; Umeda, M.; Sumiyoshi, R.; Fukui, S.; Kawashiri, S.; Iwamoto, N.; et al. Promotion of Calcium/Calmodulin-Dependent Protein Kinase 4 by GLUT1-Dependent Glycolysis in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 766–772. [Google Scholar] [CrossRef]
- Li, W.; Qu, G.; Choi, S.-C.; Cornaby, C.; Titov, A.; Kanda, N.; Teng, X.; Wang, H.; Morel, L. Targeting T Cell Activation and Lupus Autoimmune Phenotypes by Inhibiting Glucose Transporters. Front. Immunol. 2019, 10, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.; Hanczko, R.; Telarico, T.; Oaks, Z.; Landas, S. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol. Med. 2011, 17, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Banki, K.; Hutter, E.; Colombo, E.; Gonchoroff, N.J.; Perl, A. Glutathione levels and sensitivity to apoptosis are regulated by changes in transaldolase expression. J. Biol. Chem. 1996, 271, 32994–33001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banki, K.; Hutter, E.; Gonchoroff, N.J.; Perl, A. Elevation of Mitochondrial Transmembrane Potential and Reactive Oxygen Intermediate Levels Are Early Events and Occur Independently from Activation of Caspases in Fas Signaling1. J. Immunol. 1999, 162, 1466–1479. [Google Scholar] [CrossRef]
- Gergely, P.; Grossman, C.; Niland, B.; Puskas, F.; Neupane, H.; Allam, F.; Banki, K.; Phillips, P.E.; Perl, A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Gergely, P.; Niland, B.; Gonchoroff, N.; Pullmann, R.; Phillips, P.E.; Perl, A. Persistent Mitochondrial Hyperpolarization, Increased Reactive Oxygen Intermediate Production, and Cytoplasmic Alkalinization Characterize Altered IL-10 Signaling in Patients with Systemic Lupus Erythematosus. J. Immunol. 2002, 169, 1092–1101. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.; Gergely, P.; Nagy, G.; Koncz, A.; Banki, K. Mitochondrial hyperpolarization: A checkpoint of T-cell life, death and autoimmunity. Trends Immunol. 2004, 25, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mTOR in T cells of lupus patients. Arthiritis Rhem. 2012, 64, 2937–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caza, T.N.; Fernandez, D.R.; Talaber, G.; Oaks, Z.; Haas, M.; Madaio, M.P.; Lai, Z.-W.; Miklossy, G.; Singh, R.R.; Chudakov, D.M.; et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann. Rheum. Dis. 2014, 73, 1888–1897. [Google Scholar] [CrossRef]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Wincup, C.; Sawford, N.; Rahman, A. Pathological mechanisms of abnormal iron metabolism and mitochondrial dysfunction in systemic lupus erythematosus. Expert. Rev. Clin. Immunol. 2021, 17, 957–967. [Google Scholar] [CrossRef]
- Kleven, M.D.; Jue, S.; Enns, C.A. The Transferrin Receptors, TfR1 and TfR2 Bind Transferrin through Differing Mechanisms. Biochemistry 2018, 57, 1552–1559. [Google Scholar] [CrossRef]
- Zhao, M.; Li, M.-Y.; Gao, X.-F.; Jia, S.-J.; Gao, K.-Q.; Zhou, Y.; Zhang, H.-H.; Huang, Y.; Wang, J.; Wu, H.-J.; et al. Downregulation of BDH2 modulates iron homeostasis and promotes DNA demethylation in CD4+ T cells of systemic lupus erythematosus. Clin. Immunol. 2018, 187, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Song, Y.; Wu, J.; Lu, S.; Min, X.; Liu, L.; Hu, L.; Zheng, M.; Du, P.; Yu, Y.; et al. Iron-dependent epigenetic modulation promotes pathogenic T cell differentiation in lupus. J. Clin. Investig. 2022, 132, e152345. [Google Scholar] [CrossRef]
- Jabara, H.H.; Boyden, S.E.; Chou, J.; Ramesh, N.; Massaad, M.J.; Benson, H.; Bainter, W.; Fraulino, D.; Rahimov, F.; Sieff, C.; et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat. Genet. 2016, 48, 74–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aljohani, A.H.; Al-Mousa, H.; Arnaout, R.; Al-Dhekri, H.; Mohammed, R.; Alsum, Z.; Nicolas-Jilwan, M.; Alrogi, F.; Al-Muhsen, S.; Alazami, A.M.; et al. Clinical and Immunological Characterization of Combined Immunodeficiency Due to TFRC Mutation in Eight Patients. J. Clin. Immunol. 2020, 40, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Rossatti, P.; Redpath, G.M.I.; Ziegler, L.; Samson, G.P.B.; Clamagirand, C.D.; Legler, D.F.; Rossy, J. Rapid increase in transferrin receptor recycling promotes adhesion during T cell activation. BMC Biol. 2022, 20, 189. [Google Scholar] [CrossRef]
- Voulgarelis, M.; Kokori, S.I.; Ioannidis, J.P.; Tzioufas, A.G.; Kyriaki, D.; Moutsopoulos, H.M. Anaemia in systemic lupus erythematosus: Aetiological profile and the role of erythropoietin. Ann. Rheum. Dis. 2000, 59, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Zhao, S.; Chen, B.; Huang, Y.; Guo, C.; Li, M.; Ye, B.; Wang, S.; Zhang, H.; Yang, N. Iron controls T helper cell pathogenicity by promoting glucose metabolism in autoimmune myopathy. Clin. Transl. Med. 2022, 12, e999. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Jukarainen, S.; Heinonen, S.; Rämö, J.T.; Rinnankoski-Tuikka, R.; Rappou, E.; Tummers, M.; Muniandy, M.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; et al. Obesity Is Associated With Low NAD+/SIRT Pathway Expression in Adipose Tissue of BMI-Discordant Monozygotic Twins. J. Clin. Endocrinol. Metab. 2016, 101, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Gujar, A.D.; Le, S.; Mao, D.D.; Dadey, D.Y.A.; Turski, A.; Sasaki, Y.; Aum, D.; Luo, J.; Dahiya, S.; Yuan, L.; et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc. Natl. Acad. Sci. USA 2016, 113, E8247–E8256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, B.E.; Sharif, T.; Martell, E.; Dai, C.; Kim, Y.; Lee, P.W.K.; Gujar, S.A. NAD+ salvage pathway in cancer metabolism and therapy. Pharmacol. Res. 2016, 114, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, E.; Suarez-Fueyo, A.; Bradley, S.J.; Mizui, M.; Marin, A.V.; Mulki, L.; Krishfield, S.; Malavasi, F.; Yoon, J.; Ho Sui, S.J.; et al. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep. 2020, 30, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, H.A.; Veech, R.L. Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv. Enzym. Regul. 1969, 7, 397–413. [Google Scholar] [CrossRef]
- Yang, Y.; Sauve, A.A. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.A.; Madsen, A.S.; Olsen, C.A.; Hirschey, M.D. Metabolic control by sirtuins and other enzymes that sense NAD+, NADH, or their ratio. Biochim. Biophys. Acta 2017, 1858, 991–998. [Google Scholar] [CrossRef]
- Fletcher, R.S.; Lavery, G.G. The emergence of the nicotinamide riboside kinases in the regulation of NAD+ metabolism. J. Mol. Endocrinol. 2018, 61, R107–R121. [Google Scholar] [CrossRef] [Green Version]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef]
- Nacarelli, T.; Zhang, R. NAD+ metabolism controls inflammation during senescence. Mol. Cell Oncol. 2019, 6, 1605819. [Google Scholar] [CrossRef]
- Poljsak, B. NAD+ in Cancer Prevention and Treatment: Pros and Cons. J. Clin. Exp. Oncol. 2018, 2016, 2. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef] [PubMed]
- De Flora, A.; Zocchi, E.; Guida, L.; Franco, L.; Bruzzone, S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N. Y. Acad. Sci. 2004, 1028, 176–191. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Ijichi, H.; Ichiyama, A.; Hayaishi, O. Studies on the biosynthesis of nicotinamide adenine dinucleotide. 3. Comparative in vivo studies on nicotinic acid, nicotinamide, and quinolinic acid as precursors of nicotinamide adenine dinucleotide. J. Biol. Chem. 1966, 241, 3701–3707. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Sig Transduct. Target. 2021, 6, 2. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Neyens, D.; Ramsay, G.; Taylor, P.M.; Cantrell, D.A. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat. Commun. 2018, 9, 1981. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Sepp, N.; Kowald, E.; Kind, S.; Schmuth, M.; Fuchs, D. Degradation of tryptophan in patients with systemic lupus erythematosus. Adv. Exp. Med. Biol. 1999, 467, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Sepp, N.; Kowald, E.; Ortner, U.; Wirleitner, B.; Fritsch, P.; Baier-Bitterlich, G.; Fuchs, D. Enhanced tryptophan degradation in systemic lupus erythematosus. Immunobiology 2000, 201, 621–630. [Google Scholar] [CrossRef]
- Oaks, Z.; Perl, A. Metabolic control of the epigenome in systemic Lupus erythematosus. Autoimmunity 2014, 47, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Choi, S.-C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4+ T Cell Metabolism Reverses Lupus. Sci. Transl. Med. 2015, 7, 274ra18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado-Báez, L.; Cole, N.B.; Krämer, H.; Donaldson, J.G. Microtubule-dependent endosomal sorting of clathrin-independent cargo by Hook1. J. Cell Biol. 2013, 201, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.-C.; Brown, J.; Gong, M.; Ge, Y.; Zadeh, M.; Li, W.; Croker, B.P.; Michailidis, G.; Garrett, T.J.; Mohamadzadeh, M.; et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci. Transl. Med. 2020, 12, eaax2220. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Abboud, G.; Ma, L.; Choi, S.-C.; Kanda, N.; Zeumer-Spataro, L.; Lee, J.; Peng, W.; Cagmat, J.; Faludi, T.; et al. Microbiota-mediated skewing of tryptophan catabolism modulates CD4+ T cells in lupus-prone mice. iScience 2022, 25, 104241. [Google Scholar] [CrossRef]
- Perl, A.; Hanczko, R.; Lai, Z.W.; Oaks, Z.; Kelly, R.; Borsuk, R.; Asara, J.M.; Phillips, P.E. Comprehensive metabolome analyses reveal N-acetylcysteine-responsive accumulation of kynurenine in systemic lupus erythematosus: Implications for activation of the mechanistic target of rapamycin. Metabolomics 2015, 11, 1157–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Z.W.; Borsuk, R.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Francis, L.; Tily, H.; Bartos, A.; Faraone, S.V.; et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J. Immunol. 2013, 191, 2236–2246. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.-W.; Kelly, R.; Winans, T.; Marchena, I.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Tily, H.; Francis, L.; et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: A single-arm, open-label, phase 1/2 trial. Lancet 2018, 391, 1186–1196. [Google Scholar] [CrossRef]
- Kato, H.; Perl, A. The IL-21-mtor axis blocks treg differentiation and function by suppression of autophagy in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2018, 70, 427. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.-T.; Gomez, T.S.; Sackey, B.K.; Billadeau, D.D.; Burd, C.G. Rab GTPase regulation of retromer-mediated cargo export during endosome maturation. Mol. Biol. Cell 2012, 23, 2505–2515. [Google Scholar] [CrossRef]
- Curnock, R.; Calcagni, A.; Ballabio, A.; Cullen, P.J. TFEB controls retromer expression in response to nutrient availability. J. Cell Biol. 2019, 218, 3954–3966. [Google Scholar] [CrossRef]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Katz, F.; Povey, S.; Parkar, M.; Schneider, C.; Sutherland, R.; Stanley, K.; Solomon, E.; Greaves, M. Chromosome assignment of monoclonal antibody-defined determinants on human leukemic cells. Eur. J. Immunol. 1983, 13, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, O.; Tanaka, H.; Itoh, M.; Ishihara, K.; Hirano, T. Genomic structure of human BST-1. Immunol. Lett. 1996, 54, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Hon, C.L. ADP-ribosyl Cyclase and CD38 Catalyze the Synthesis of a Calcium-mobilizing Metabolite from NADP+(∗). J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [Green Version]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84B, 207–217. [Google Scholar] [CrossRef]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Bhan, A.K.; Reinherz, E.L.; Poppema, S.; McCluskey, R.T.; Schlossman, S.F. Location of T cell and major histocompatibility complex antigens in the human thymus. J. Exp. Med. 1980, 152, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Ortolan, E.; Augeri, S.; Fissolo, G.; Musso, I.; Funaro, A. CD157: From immunoregulatory protein to potential therapeutic target. Immunol. Lett. 2019, 205, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.O.; Ishihara, K.; Denno, K.; Kobune, Y.; Itoh, M.; Muraoka, O.; Kaisho, T.; Sasaki, T.; Ochi, T.; Hirano, T. Elevated levels of the soluble form of bone marrow stromal cell antigen 1 in the sera of patients with severe rheumatoid arthritis. Arthritis Rheum. 1996, 39, 629–637. [Google Scholar] [CrossRef]
- Khandelwal, P.; Lane, A.; Chaturvedi, V.; Owsley, E.; Davies, S.M.; Marmer, D.; Filipovich, A.H.; Jordan, M.B.; Marsh, R.A. Peripheral Blood CD38 Bright CD8+ Effector Memory T Cells Predict Acute Graft-versus-Host Disease. Biol. Blood Marrow Transplant. 2015, 21, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Joosse, M.E.; Menckeberg, C.L.; de Ruiter, L.F.; Raatgeep, H.C.; van Berkel, L.A.; Simons-Oosterhuis, Y.; Tindemans, I.; Muskens, A.M.; Hendriks, R.W.; Hoogenboezem, R.M.; et al. Frequencies of circulating regulatory TIGIT+CD38+ effector T cells correlate with the course of inflammatory bowel disease. Mucosal Immunol. 2019, 12, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavón, E.J.; Zumaquero, E.; Rosal-Vela, A.; Khoo, K.-M.; Cerezo-Wallis, D.; García-Rodríguez, S.; Carrascal, M.; Abian, J.; Graeff, R.; Callejas-Rubio, J.-L.; et al. Increased CD38 expression in T cells and circulating anti-CD38 IgG autoantibodies differentially correlate with distinct cytokine profiles and disease activity in systemic lupus erythematosus patients. Cytokine 2013, 62, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, E.N. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 1993, 261, 1330–1333. [Google Scholar] [CrossRef]
- Zielinska, W.; Barata, H.; Chini, E.N. Metabolism of cyclic ADP-ribose: Zinc is an endogenous modulator of the cyclase/NAD glycohydrolase ratio of a CD38-like enzyme from human seminal fluid. Life Sci. 2004, 74, 1781–1790. [Google Scholar] [CrossRef]
- Kar, A.; Mehrotra, S.; Chatterjee, S. CD38: T Cell Immuno-Metabolic Modulator. Cells 2020, 9, 1716. [Google Scholar] [CrossRef] [PubMed]
- Funaro, A.; Reiniš, M.; Trubiani, O.; Santi, S.; Di Primio, R.; Malavasi, F. CD38 Functions Are Regulated Through an Internalization Step1. J. Immunol. 1998, 160, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Mittelbrunn, M.; de la Fuente, H.; Pérez-Martínez, M.; García-Pérez, A.; Ariza-Veguillas, A.; Malavasi, F.; Zubiaur, M.; Sánchez-Madrid, F.; Sancho, J. Antigen-induced clustering of surface CD38 and recruitment of intracellular CD38 to the immunologic synapse. Blood 2008, 111, 3653–3664. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Turcotte, M.; Stagg, J. Targeting CD73 and downstream adenosine receptor signaling in triple-negative breast cancer. Expert Opin. Ther. Targets 2014, 18, 863–881. [Google Scholar] [CrossRef]
- Koszałka, P.; Gołuńska, M.; Stanisławowski, M.; Urban, A.; Stasiłojć, G.; Majewski, M.; Wierzbicki, P.; Składanowski, A.C.; Bigda, J. CD73 on B16F10 melanoma cells in CD73-deficient mice promotes tumor growth, angiogenesis, neovascularization, macrophage infiltration and metastasis. Int. J. Biochem. Cell Biol. 2015, 69, 1–10. [Google Scholar] [CrossRef]
- Turcotte, M.; Spring, K.; Pommey, S.; Chouinard, G.; Cousineau, I.; George, J.; Chen, G.M.; Gendoo, D.M.A.; Haibe-Kains, B.; Karn, T.; et al. CD73 Is Associated with Poor Prognosis in High-Grade Serous Ovarian Cancer. Cancer Res. 2015, 75, 4494–4503. [Google Scholar] [CrossRef] [Green Version]
- Hesse, J.; Siekierka-Harreis, M.; Steckel, B.; Alter, C.; Schallehn, M.; Honke, N.; Schnieringer, M.-L.; Wippich, M.; Braband, R.; Schneider, M.; et al. Profound inhibition of CD73-dependent formation of anti-inflammatory adenosine in B cells of SLE patients. EBioMedicine 2021, 73, 103616. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, X.; Li, X.; Wang, G.; Ma, Y.; Zhao, S.; Zheng, S. Expression of FOXP3 in CD4+ CD39+ T cells of patients with systemic lupus erythematosus and dynamic observation of treatment with glucocorticoid. Zhonghua Yi Xue Za Zhi 2009, 89, 1636–1638. [Google Scholar]
- Li, D.; Li, X.; Zhang, J.; Hu, S.; Xiao, B.; Chen, W.; Zeng, X. The expression of CD73 in CD4+ regulatory T cells in patients with new-onset systemic lupus erythematosus. Zhonghua Nei Ke Za Zhi 2010, 49, 772–775. [Google Scholar] [PubMed]
- Grozio, A.; Sociali, G.; Sturla, L.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 2013, 288, 25938–25949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sociali, G.; Raffaghello, L.; Magnone, M.; Zamporlini, F.; Emionite, L.; Sturla, L.; Bianchi, G.; Vigliarolo, T.; Nahimana, A.; Nencioni, A.; et al. Antitumor effect of combined NAMPT and CD73 inhibition in an ovarian cancer model. Oncotarget 2016, 7, 2968–2984. [Google Scholar] [CrossRef] [Green Version]
- Garavaglia, S.; Bruzzone, S.; Cassani, C.; Canella, L.; Allegrone, G.; Sturla, L.; Mannino, E.; Millo, E.; De Flora, A.; Rizzi, M. The high-resolution crystal structure of periplasmic Haemophilus influenzae NAD nucleotidase reveals a novel enzymatic function of human CD73 related to NAD metabolism. Biochem. J. 2012, 441, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.J.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef] [Green Version]
- Wilk, A.; Hayat, F.; Cunningham, R.; Li, J.; Garavaglia, S.; Zamani, L.; Ferraris, D.M.; Sykora, P.; Andrews, J.; Clark, J.; et al. Extracellular NAD+ enhances PARP-dependent DNA repair capacity independently of CD73 activity. Sci. Rep. 2020, 10, 651. [Google Scholar] [CrossRef] [Green Version]
- Romio, M.; Reinbeck, B.; Bongardt, S.; Hüls, S.; Burghoff, S.; Schrader, J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am. J. Physiol. Cell Physiol. 2011, 301, C530–C539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.S.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 expression on extracellular vesicles derived from CD4+CD25+Foxp3+ T cells contributes to their regulatory function. Eur. J. Immunol. 2013, 43, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, Y.; Chen, P.; Shi, X.; Liu, Y.; Shi, L.; Cong, P.; Mao, S.; Tong, C.; Du, C.; et al. Solute carrier family 12 member 8 (SLC12A8) is a potential biomarker and related to tumor immune cell infiltration in bladder cancer. Bioengineered 2021, 12, 4946–4961. [Google Scholar] [CrossRef]
- Castro, C.; Oyamada, H.A.A.; Cafasso, M.O.S.D.; Lopes, L.M.; Monteiro, C.; Sacramento, P.M.; Alves-Leon, S.V.; da Fontoura Galvão, G.; Hygino, J.; de Souza, J.P.B.M.; et al. Elevated proportion of TLR2- and TLR4-expressing Th17-like cells and activated memory B cells was associated with clinical activity of cerebral cavernous malformations. J. Neuroinflamm. 2022, 19, 28. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.-Y.; Huffel, C.V.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.-J. Toll-like receptors and danger signaling in kidney injury. J. Am. Soc. Nephrol. 2010, 21, 1270–1274. [Google Scholar] [CrossRef] [Green Version]
- Brandt, K.J.; Fickentscher, C.; Kruithof, E.K.; de Moerloose, P. TLR2 Ligands Induce NF-κB Activation from Endosomal Compartments of Human Monocytes. PLoS ONE 2013, 8, e80743. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [Green Version]
- Husebye, H.; Aune, M.H.; Stenvik, J.; Samstad, E.; Skjeldal, F.; Halaas, O.; Nilsen, N.J.; Stenmark, H.; Latz, E.; Lien, E.; et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity 2010, 33, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lou, J.; Ouyang, C.; Chen, W.; Liu, Y.; Liu, X.; Cao, X.; Wang, J.; Lu, L. Ras-related protein Rab10 facilitates TLR4 signaling by promoting replenishment of TLR4 onto the plasma membrane. Proc. Natl. Acad. Sci. USA 2010, 107, 13806–13811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petnicki-Ocwieja, T.; Sharma, B.; Powale, U.; Pathak, D.; Tan, S.; Hu, L.T. An AP-3-dependent pathway directs phagosome fusion with Rab8 and Rab11 vesicles involved in TLR2 signaling. Traffic 2022, 23, 558–567. [Google Scholar] [CrossRef]
- Liu, Y.; Liao, J.; Zhao, M.; Wu, H.; Yung, S.; Chan, T.M.; Yoshimura, A.; Lu, Q. Increased expression of TLR2 in CD4+ T cells from SLE patients enhances immune reactivity and promotes IL-17 expression through histone modifications. Eur. J. Immunol. 2015, 45, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Nyirenda, M.H.; Sanvito, L.; Darlington, P.J.; O’Brien, K.; Zhang, G.-X.; Constantinescu, C.S.; Bar-Or, A.; Gran, B. TLR2 Stimulation Drives Human Naive and Effector Regulatory T Cells into a Th17-Like Phenotype with Reduced Suppressive Function. J. Immunol. 2011, 187, 2278–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Choi, S.J.; Ji, J.D.; Song, G.G. Association between toll-like receptor polymorphisms and systemic lupus erythematosus: A meta-analysis update. Lupus 2016, 25, 593–601. [Google Scholar] [CrossRef]
- Wang, C.-M.; Chang, S.-W.; Wu, Y.-J.J.; Lin, J.-C.; Ho, H.-H.; Chou, T.-C.; Yang, B.; Wu, J.; Chen, J.-Y. Genetic variations in Toll-like receptors (TLRs 3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population. Sci. Rep. 2014, 4, 3792. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Chai, Q.; Zhao, Y.; Li, P.; Qiao, J.; Huang, J. Increased activation of toll-like receptors-7 and -8 of peripheral blood mononuclear cells and upregulated serum cytokines in patients with pediatric systemic lupus erythematosus. Int. J. Clin. Exp. Med. 2015, 8, 20472–20480. [Google Scholar]
- Dominguez-Villar, M.; Gautron, A.-S.; de Marcken, M.; Keller, M.J.; Hafler, D.A. TLR7 induces anergy in human CD4+ T cells. Nat. Immunol. 2015, 16, 118–128. [Google Scholar] [CrossRef]
- Li, L.; Liu, X.; Sanders, K.L.; Edwards, J.L.; Ye, J.; Si, F.; Gao, A.; Huang, L.; Hsueh, E.C.; Ford, D.A.; et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab. 2019, 29, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Hedrich, C.M. Epigenetics in SLE. Curr. Rheumatol. Rep. 2017, 19, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcocer-Varela, J.; Alarcón-Segovia, D. Decreased production of and response to interleukin-2 by cultured lymphocytes from patients with systemic lupus erythematosus. J. Clin. Investig. 1982, 69, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Zhang, N.; Yopp, A.C.; Chen, D.; Mao, M.; Chen, D.; Zhang, H.; Ding, Y.; Bromberg, J.S. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25—Precursors. Am. J. Transplant. 2004, 4, 1614–1627. [Google Scholar] [CrossRef]
- Malek, T.R.; Yu, A.; Vincek, V.; Scibelli, P.; Kong, L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity 2002, 17, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomou, E.E.; Juang, Y.T.; Gourley, M.F.; Kammer, G.M.; Tsokos, G.C. Molecular basis of deficient IL-2 production in T cells from patients with systemic lupus erythematosus. J. Immunol. 2001, 166, 4216–4222. [Google Scholar] [CrossRef] [Green Version]
- Juang, Y.-T.; Wang, Y.; Solomou, E.E.; Li, Y.; Mawrin, C.; Tenbrock, K.; Kyttaris, V.C.; Tsokos, G.C. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J. Clin. Investig. 2005, 115, 996–1005. [Google Scholar] [CrossRef]
- Sunahori, K.; Juang, Y.-T.; Kyttaris, V.C.; Tsokos, G.C. Promoter Hypomethylation Results in Increased Expression of Protein Phosphatase 2A in T Cells from Patients with Systemic Lupus Erythematosus. J. Immunol. 2011, 186, 4508–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.A.; Noeldner, P.K.; Reece, K.; Wadzinski, B.E.; Means, A.R. Regulation and function of the calcium/calmodulin-dependent protein kinase IV/protein serine/threonine phosphatase 2A signaling complex. J. Biol. Chem. 2004, 279, 31708–31716. [Google Scholar] [CrossRef] [Green Version]
- Katsiari, C.G.; Kyttaris, V.C.; Juang, Y.-T.; Tsokos, G.C. Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. J. Clin. Investig. 2005, 115, 3193–3204. [Google Scholar] [CrossRef] [Green Version]
- Juang, Y.-T.; Rauen, T.; Wang, Y.; Ichinose, K.; Benedyk, K.; Tenbrock, K.; Tsokos, G.C. Transcriptional Activation of the cAMP-responsive Modulator Promoter in Human T Cells Is Regulated by Protein Phosphatase 2A-mediated Dephosphorylation of SP-1 and Reflects Disease Activity in Patients with Systemic Lupus Erythematosus. J. Biol. Chem. 2011, 286, 1795–1801. [Google Scholar] [CrossRef] [Green Version]
- Vassilopoulos, D.; Kovacs, B.; Tsokos, G.C. TCR/CD3 complex-mediated signal transduction pathway in T cells and T cell lines from patients with systemic lupus erythematosus. J. Immunol. 1995, 155, 2269–2281. [Google Scholar] [CrossRef]
- Moulton, V.R.; Tsokos, G.C. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis Res. 2011, 13, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyttaris, V.C.; Wang, Y.; Juang, Y.-T.; Weinstein, A.; Tsokos, G.C. Increased Levels of NF-ATc2 Differentially Regulate CD154 and IL-2 Genes in T Cells from Patients with Systemic Lupus Erythematosus. J. Immunol. 2007, 178, 1960–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Kyttaris, V.C.; Juang, Y.-T.; Tenbrock, K.; Weinstein, A.; Tsokos, G.C. Cyclic adenosine 5′-monophosphate response element modulator is responsible for the decreased expression of c-fos and activator protein-1 binding in T cells from patients with systemic lupus erythematosus. J. Immunol. 2004, 173, 3557–3563. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, N.S.; Laoide, B.M.; Schlotter, F.; Sassone-Corsi, P. Transcriptional antagonist cAMP-responsive element modulator (CREM) down-regulates c-fos cAMP-induced expression. Proc. Natl. Acad. Sci. USA 1991, 88, 5448–5452. [Google Scholar] [CrossRef] [Green Version]
- Tenbrock, K.; Tsokos, G.C. Transcriptional Regulation of Interlekin 2 in Sle T Cells. Int. Rev. Immunol. 2004, 23, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [Green Version]
- Hisada, R.; Yoshida, N.; Umeda, M.; Burbano, C.; Bhargava, R.; Scherlinger, M.; Kono, M.; Kyttaris, V.C.; Krishfield, S.; Tsokos, G.C. The deacetylase SIRT2 contributes to autoimmune disease pathogenesis by modulating IL-17A and IL-2 transcription. Cell Mol. Immunol. 2022, 19, 738–750. [Google Scholar] [CrossRef]
- Berg, V.; Modak, M.; Brell, J.; Puck, A.; Künig, S.; Jutz, S.; Steinberger, P.; Zlabinger, G.J.; Stöckl, J. Iron Deprivation in Human T Cells Induces Nonproliferating Accessory Helper Cells. ImmunoHorizons 2020, 4, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhou, X.; Cui, D.; Zhang, W.; Lai, J.; Li, X.; Ruan, Y.; Xie, Y.; Shi, M.; Xiao, Y.; et al. The role of Stim1 in the progression of lupus nephritis in mice. Int. J. Clin. Exp. Pathol. 2020, 13, 3021–3032. [Google Scholar] [PubMed]
- Shi, S.; Zhao, Q.; Ke, C.; Long, S.; Zhang, F.; Zhang, X.; Li, Y.; Liu, X.; Hu, H.; Yin, S. Loureirin B Exerts its Immunosuppressive Effects by Inhibiting STIM1/Orai1 and KV1.3 Channels. Front. Pharmacol. 2021, 12, 685092. [Google Scholar] [CrossRef]
- Nambiar, M.P.; Fisher, C.U.; Warke, V.G.; Krishnan, S.; Mitchell, J.P.; Delaney, N.; Tsokos, G.C. Reconstitution of deficient T cell receptor zeta chain restores T cell signaling and augments T cell receptor/CD3-induced interleukin-2 production in patients with systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Kyttaris, V.; Li, Y.; Juang, Y.-T.; Wang, Y.; Tsokos, G.C. Increased expression of STAT3 in SLE T cells contributes to enhanced chemokine-mediated cell migration. Autoimmunity 2007, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.; Tato, C.M.; Davidson, T.S.; Kanno, Y.; Chen, Z.; Yao, Z.; Blank, R.B.; Meylan, F.; Siegel, R.; Hennighausen, L.; et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007, 26, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.K.; Ho, C.Y.; Li, E.K.; Lam, C.W. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus 2000, 9, 589–593. [Google Scholar] [CrossRef]
- Crispín, J.C.; Oukka, M.; Bayliss, G.; Cohen, R.A.; Van Beek, C.A.; Stillman, I.E.; Kyttaris, V.C.; Juang, Y.-T.; Tsokos, G.C. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 2008, 181, 8761–8766. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.; Lee, W.-W.; Lee, S.-H.; Kim, S.H.; Kang, S.W.; Craft, J.; Kang, I. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res. 2010, 12, R53. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Chu, Y.; Yang, X.; Gao, D.; Zhu, L.; Yang, X.; Wan, L.; Li, M. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 1472–1483. [Google Scholar] [CrossRef]
- Schwarzenberger, P.; La Russa, V.; Miller, A.; Ye, P.; Huang, W.; Zieske, A.; Nelson, S.; Bagby, G.J.; Stoltz, D.; Mynatt, R.L.; et al. IL-17 stimulates granulopoiesis in mice: Use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J. Immunol. 1998, 161, 6383–6389. [Google Scholar] [CrossRef]
- Schwarzenberger, P.; Huang, W.; Ye, P.; Oliver, P.; Manuel, M.; Zhang, Z.; Bagby, G.; Nelson, S.; Kolls, J.K. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J. Immunol. 2000, 164, 4783–4789. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenberger, P.; Huang, W.; Oliver, P.; Byrne, P.; La Russa, V.; Zhang, Z.; Kolls, J.K. IL-17 Mobilizes Peripheral Blood Stem Cells with Short- and Long-Term Repopulating Ability in Mice. J. Immunol. 2001, 167, 2081–2086. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Huang, W.; Zhong, Q.; Schwarzenberger, P. IL-17 receptor knockout mice have enhanced myelotoxicity and impaired hemopoietic recovery following gamma irradiation. J. Immunol. 2006, 176, 6186–6193. [Google Scholar] [CrossRef] [Green Version]
- Von Vietinghoff, S.; Ley, K. IL-17A Controls IL-17F Production and Maintains Blood Neutrophil Counts in Mice. J. Immunol. 2009, 183, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Mitsdoerffer, M.; Lee, Y.; Jäger, A.; Kim, H.-J.; Korn, T.; Kolls, J.K.; Cantor, H.; Bettelli, E.; Kuchroo, V.K. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc. Natl. Acad. Sci. USA 2010, 107, 14292–14297. [Google Scholar] [CrossRef] [Green Version]
- Crispín, J.C.; Liossis, S.-N.C.; Kis-Toth, K.; Lieberman, L.A.; Kyttaris, V.C.; Juang, Y.-T.; Tsokos, G.C. Pathogenesis of human systemic lupus erythematosus: Recent advances. Trends Mol. Med. 2010, 16, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riol-Blanco, L.; Lazarevic, V.; Awasthi, A.; Mitsdoerffer, M.; Wilson, B.S.; Croxford, A.; Waisman, A.; Kuchroo, V.K.; Glimcher, L.H.; Oukka, M. IL-23 Receptor Regulates Unconventional IL-17–Producing T Cells That Control Bacterial Infections. J. Immunol. 2010, 184, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17–producing CD4 + effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Crispín, J.C.; Tsokos, G.C. Human TCR-αβ+ CD4− CD8− T Cells Can Derive from CD8+ T Cells and Display an Inflammatory Effector Phenotype. J. Immunol. 2009, 183, 4675–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsokos, G.C. Systemic Lupus Erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [Green Version]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the TH17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-β are required for differentiation of human TH17 cells. Nature 2008, 454, 350–352. [Google Scholar] [CrossRef] [Green Version]
- Iezzi, G.; Sonderegger, I.; Ampenberger, F.; Schmitz, N.; Marsland, B.J.; Kopf, M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 876–881. [Google Scholar] [CrossRef] [Green Version]
- Javierre, B.M.; Richardson, B. A new epigenetic challenge: Systemic lupus erythematosus. Adv. Exp. Med. Biol. 2011, 711, 117–136. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Niewold, T.B. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J. Autoimmun. 2015, 64, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T. Interleukin 6 and its Receptor: Ten Years Later. Int. Rev. Immunol. 1998, 16, 249–284. [Google Scholar] [CrossRef] [PubMed]
- Harbour, S.N.; DiToro, D.F.; Witte, S.J.; Zindl, C.L.; Gao, M.; Schoeb, T.R.; Jones, G.W.; Jones, S.A.; Hatton, R.D.; Weaver, C.T. TH17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity. Sci. Immunol. 2020, 5, eaaw2262. [Google Scholar] [CrossRef]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef]
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10, 3668. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Cimica, V.; Chen, H.-C.; Iyer, J.K.; Reich, N.C. Dynamics of the STAT3 transcription factor: Nuclear import dependent on Ran and importin-β1. PLoS ONE 2011, 6, e20188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Chen, Y.; Tato, C.M.; Laurence, A.; Joyce-Shaikh, B.; Blumenschein, W.M.; McClanahan, T.K.; O’Shea, J.J.; Cua, D.J. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol. 2009, 10, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Koga, T.; Hedrich, C.M.; Mizui, M.; Yoshida, N.; Otomo, K.; Lieberman, L.A.; Rauen, T.; Crispín, J.C.; Tsokos, G.C. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J. Clin. Investig. 2014, 124, 2234–2245. [Google Scholar] [CrossRef] [Green Version]
- Katsuyama, T.; Tsokos, G.C.; Moulton, V.R. Aberrant T Cell Signaling and Subsets in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1088. [Google Scholar] [CrossRef] [Green Version]
- Hedrich, C.M.; Crispin, J.C.; Rauen, T.; Ioannidis, C.; Apostolidis, S.A.; Lo, M.S.; Kyttaris, V.C.; Tsokos, G.C. cAMP response element modulator α controls IL2 and IL17A expression during CD4 lineage commitment and subset distribution in lupus. Proc. Natl. Acad. Sci. USA 2012, 109, 16606–16611. [Google Scholar] [CrossRef] [Green Version]
- Richardson, B. Primer: Epigenetics of autoimmunity. Nat. Clin. Pract. Rheumatol. 2007, 3, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14, R139–R147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurebayashi, Y.; Nagai, S.; Ikejiri, A.; Ohtani, M.; Ichiyama, K.; Baba, Y.; Yamada, T.; Egami, S.; Hoshii, T.; Hirao, A.; et al. PI3K-Akt-mTORC1-S6K1/2 Axis Controls Th17 Differentiation by Regulating Gfi1 Expression and Nuclear Translocation of RORγ. Cell Rep. 2012, 1, 360–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.-B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, D.; Bonilla, E.; Mirza, N.; Niland, B.; Perl, A. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthiritis Rhem. 2006, 54, 2983–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Zhao, C.; Zhang, B.; Wang, X.; Wang, Y.; An, J.; Chen, J. Restoring T-helper 17 cell/regulatory T-cell balance and decreasing disease activity by rapamycin and all-trans retinoic acid in patients with systemic lupus erythematosus. Lupus 2019, 28, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Singh, R. Central role of PI3K–SYK interaction in fibrinogen-induced lamellipodia and filopodia formation in platelets. FEBS Open. Bio 2016, 6, 1285–1296. [Google Scholar] [CrossRef]
- Simioni, C.; Martelli, A.M.; Zauli, G.; Vitale, M.; McCubrey, J.A.; Capitani, S.; Neri, L.M. Targeting the phosphatidylinositol 3-kinase/Akt/mechanistic target of rapamycin signaling pathway in B-lineage acute lymphoblastic leukemia: An update. J. Cell. Physiol. 2018, 233, 6440–6454. [Google Scholar] [CrossRef] [Green Version]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Henske, E.P.; Jóźwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Prim. 2016, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Perl, A. Mechanistic Target of Rapamycin Complex 1 Expands Th17 and IL-4+ CD4− CD8− Double-Negative T Cells and Contracts Regulatory T Cells in Systemic Lupus Erythematosus. J. Immunol. 2014, 192, 4134–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, S.; Basu, A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1199. [Google Scholar] [CrossRef] [Green Version]
- GFI1 Growth Factor Independent 1 Transcriptional Repressor [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/2672 (accessed on 1 June 2021).
- Ichiyama, K.; Hashimoto, M.; Sekiya, T.; Nakagawa, R.; Wakabayashi, Y.; Sugiyama, Y.; Komai, K.; Saba, I.; Moroy, T.; Yoshimura, A. Gfi1 negatively regulates Th17 differentiation by inhibiting ROR t activity. Int. Immunol. 2009, 21, 881–889. [Google Scholar] [CrossRef] [Green Version]
- EGR2 Early Growth Response 2 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/1959 (accessed on 1 June 2021).
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Kono, M. New insights into the metabolism of Th17 cells. Immunol. Med. 2022, 46, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, L.E.A.; Prado, D.S.; Veras, F.P.; Fonseca, M.M.; Toller-Kawahisa, J.E.; Rosa, M.H.; Públio, G.A.; Martins, T.V.; Ramalho, F.S.; Waisman, A.; et al. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J. Exp. Med. 2020, 217, e20190613. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Maeda, K.; Stocton-Gavanescu, I.; Pan, W.; Umeda, M.; Katsuyama, E.; Burbano, C.; Orite, S.Y.K.; Vukelic, M.; Tsokos, M.G.; et al. Pyruvate kinase M2 is requisite for Th1 and Th17 differentiation. JCI Insight 2019, 4, e127395. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N.; Comte, D.; Mizui, M.; Otomo, K.; Rosetti, F.; Mayadas, T.N.; Crispín, J.C.; Bradley, S.J.; Koga, T.; Kono, M.; et al. ICER is requisite for Th17 differentiation. Nat. Commun. 2016, 7, 12993. [Google Scholar] [CrossRef] [Green Version]
- Dang, E.V.; Barbi, J.; Yang, H.-Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.-R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Ma, Y.; Cole, S.M.; Zander, C.; Chen, K.-H.; Karras, J.; Pope, R.M. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood 2003, 102, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calò, V.; Migliavacca, M.; Bazan, V.; Macaluso, M.; Buscemi, M.; Gebbia, N.; Russo, A. STAT proteins: From normal control of cellular events to tumorigenesis. J. Cell Physiol. 2003, 197, 157–168. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Yoon, M.-S.; Chen, J. Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation. J. Biol. Chem. 2009, 284, 35425–35432. [Google Scholar] [CrossRef] [Green Version]
- Kshirsagar, S.; Riedl, M.; Billing, H.; Tönshoff, B.; Thangavadivel, S.; Steuber, C.; Staude, H.; Wechselberger, G.; Edelbauer, M. Akt-dependent enhanced migratory capacity of Th17 cells from children with lupus nephritis. J. Immunol. 2014, 193, 4895–4903. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, J.; Boily, G.; Bazinet, S.; Saliba, S.; He, X.; Jardine, K.; Kennedy, C.; Staines, W.; Rousseaux, C.; Mueller, R.; et al. sirt1-null mice develop an autoimmune-like condition. Exp. Cell Res. 2008, 314, 3069–3074. [Google Scholar] [CrossRef]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Guan, Y.; Chatterjee, D.; Chin, Y.E. Stat3 Dimerization Regulated by Reversible Acetylation of a Single Lysine Residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Limagne, E.; Thibaudin, M.; Euvrard, R.; Berger, H.; Chalons, P.; Végan, F.; Humblin, E.; Boidot, R.; Rébé, C.; Derangère, V.; et al. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep. 2017, 19, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Hu, Y.; Yang, C.; Zhu, S.; Wang, X.; Zhang, Z.; Deng, H. Decreased NAD Activates STAT3 and Integrin Pathways to Drive Epithelial-Mesenchymal Transition. Mol. Cell. Proteom. 2018, 17, 2005–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Alvarez, B.; Rodriguez, R.M.; Fraga, M.F.; López-Larrea, C. DNA methylation: A promising landscape for immune system-related diseases. Trends Genet. 2012, 28, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Tsagaratou, A.; Lio, C.-W.J.; Yue, X.; Rao, A. TET Methylcytosine Oxidases in T Cell and B Cell Development and Function. Front. Immunol. 2017, 8, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballestar, E.; Sawalha, A.H.; Lu, Q. Clinical value of DNA methylation markers in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2020, 16, 514–524. [Google Scholar] [CrossRef]
- Sunahori, K.; Nagpal, K.; Hedrich, C.M.; Mizui, M.; Fitzgerald, L.M.; Tsokos, G.C. The catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1 protein pathway in T-cells from controls and systemic lupus erythematosus patients. J. Biol. Chem. 2013, 288, 21936–21944. [Google Scholar] [CrossRef] [Green Version]
- Tamanaha, E.; Guan, S.; Marks, K.; Saleh, L. Distributive Processing by the Iron(II)/α-Ketoglutarate-Dependent Catalytic Domains of the TET Enzymes Is Consistent with Epigenetic Roles for Oxidized 5-Methylcytosine Bases. J. Am. Chem. Soc. 2016, 138, 9345–9348. [Google Scholar] [CrossRef] [Green Version]
- Izmirly, P.M.; Parton, H.; Wang, L.; McCune, W.J.; Lim, S.S.; Drenkard, C.; Ferucci, E.D.; Dall’Era, M.; Gordon, C.; Helmick, C.G.; et al. Prevalence of Systemic Lupus Erythematosus in the United States: Estimates From a Meta-Analysis of the Centers for Disease Control and Prevention National Lupus Registries. Arthritis Rheumatol. 2021, 73, 991–996. [Google Scholar] [CrossRef]
- Hughes, T.; Adler, A.; Merrill, J.T.; Kelly, J.A.; Kaufman, K.M.; Williams, A.; Langefeld, C.D.; Gilkeson, G.S.; Sanchez, E.; Martin, J.; et al. Analysis of autosomal genes reveals gene–sex interactions and higher total genetic risk in men with systemic lupus erythematosus. Ann. Rheum. Dis. 2012, 71, 694–699. [Google Scholar] [CrossRef]
- Sawalha, A.H.; Wang, L.; Nadig, A.; Somers, E.C.; McCune, W.J.; Michigan Lupus Cohort; Hughes, T.; Merrill, J.T.; Scofield, R.H.; Strickland, F.M.; et al. Sex-specific differences in the relationship between genetic susceptibility, T cell DNA demethylation and lupus flare severity. J. Autoimmun. 2012, 38, J216–J222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Coit, P.; Sawalha, A.H. Sex-based comparison of CD4+ T cell DNA methylation in lupus reveals proinflammatory epigenetic changes in men. Clin. Immunol. 2022, 243, 109116. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Baltimore, D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009, 10, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafasla, P.; Skliris, A.; Kontoyiannis, D.L. Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nat. Immunol. 2014, 15, 492–502. [Google Scholar] [CrossRef]
- Rothamel, K.; Arcos, S.; Kim, B.; Reasoner, C.; Lisy, S.; Mukherjee, N.; Ascano, M. ELAVL1 primarily couples mRNA stability with the 3′ UTRs of interferon-stimulated genes. Cell Rep. 2021, 35, 109178. [Google Scholar] [CrossRef]
- Mancini, M.; Magnani, E.; Macchi, F.; Bonapace, I.M. The multi-functionality of UHRF1: Epigenome maintenance and preservation of genome integrity. Nucleic Acids Res. 2021, 49, 6053–6068. [Google Scholar] [CrossRef]
- Obata, Y.; Furusawa, Y.; Endo, T.A.; Sharif, J.; Takahashi, D.; Atarashi, K.; Nakayama, M.; Onawa, S.; Fujimura, Y.; Takahashi, M.; et al. The epigenetic regulator Uhrf1 facilitates the proliferation and maturation of colonic regulatory T cells. Nat. Immunol. 2014, 15, 571–579. [Google Scholar] [CrossRef]
- Liu, L.; Hu, L.; Yang, L.; Jia, S.; Du, P.; Min, X.; Wu, J.; Wu, H.; Long, H.; Lu, Q.; et al. UHRF1 downregulation promotes T follicular helper cell differentiation by increasing BCL6 expression in SLE. Clin. Epigenetics 2021, 13, 31. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Hu, N.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Lei, W.; Zhang, G.; Zhou, Y.; Su, Y.; Lu, Q. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 2008, 35, 804–810. [Google Scholar]
- Mishra, N.; Reilly, C.M.; Brown, D.R.; Ruiz, P.; Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Investig. 2003, 111, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.T.; Kanno, Y.; Cannons, J.L.; Handon, R.; Bible, P.; Elkahloun, A.G.; Anderson, S.M.; Wei, L.; Sun, H.; O’Shea, J.J.; et al. Functional and epigenetic studies reveal multistep differentiation and plasticity of in vitro-generated and in vivo-derived follicular T helper cells. Immunity 2011, 35, 622–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poholek, A.C.; Hansen, K.; Hernandez, S.G.; Eto, D.; Chandele, A.; Weinstein, J.S.; Dong, X.; Odegard, J.M.; Kaech, S.M.; Dent, A.L.; et al. In vivo regulation of Bcl6 and T follicular helper cell development. J. Immunol. 2010, 185, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Nakayamada, S.; Poholek, A.C.; Lu, K.T.; Takahashi, H.; Kato, M.; Iwata, S.; Hirahara, K.; Cannons, J.L.; Schwartzberg, P.L.; Vahedi, G.; et al. Type I Interferon induces binding of STAT1 to Bcl6: Divergent Roles of STAT-family transcription factors in the TFH cell genetic program. J. Immunol. 2014, 192, 2156–2166. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Antao, O.Q.; Song, W.; Sanchez, G.M.; Zembrzuski, K.; Koumpouras, F.; Lemenze, A.; Craft, J.; Weinstein, J.S. Type 1 interferon activated STAT4 regulates T follicular helper (Tfh)-cell dependent cytokine and immunoglobulin production in lupus. Arthritis Rheumatol. 2021, 73, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Girard, E.; Chmiest, D.; Fournier, N.; Johannes, L.; Paul, J.-L.; Vedie, B.; Lamaze, C. Rab7 Is Functionally Required for Selective Cargo Sorting at the Early Endosome. Traffic 2014, 15, 309–326. [Google Scholar] [CrossRef]
- Burki, T.K. FDA approval for anifrolumab in patients with lupus. Lancet Rheumatol. 2021, 3, e689. [Google Scholar] [CrossRef]
- Bayeva, M.; Khechaduri, A.; Puig, S.; Chang, H.-C.; Patial, S.; Blackshear, P.J.; Ardehali, H. mTOR Regulates Cellular Iron Homeostasis through Tristetraprolin. Cell Metab. 2012, 16, 645–657. [Google Scholar] [CrossRef] [Green Version]


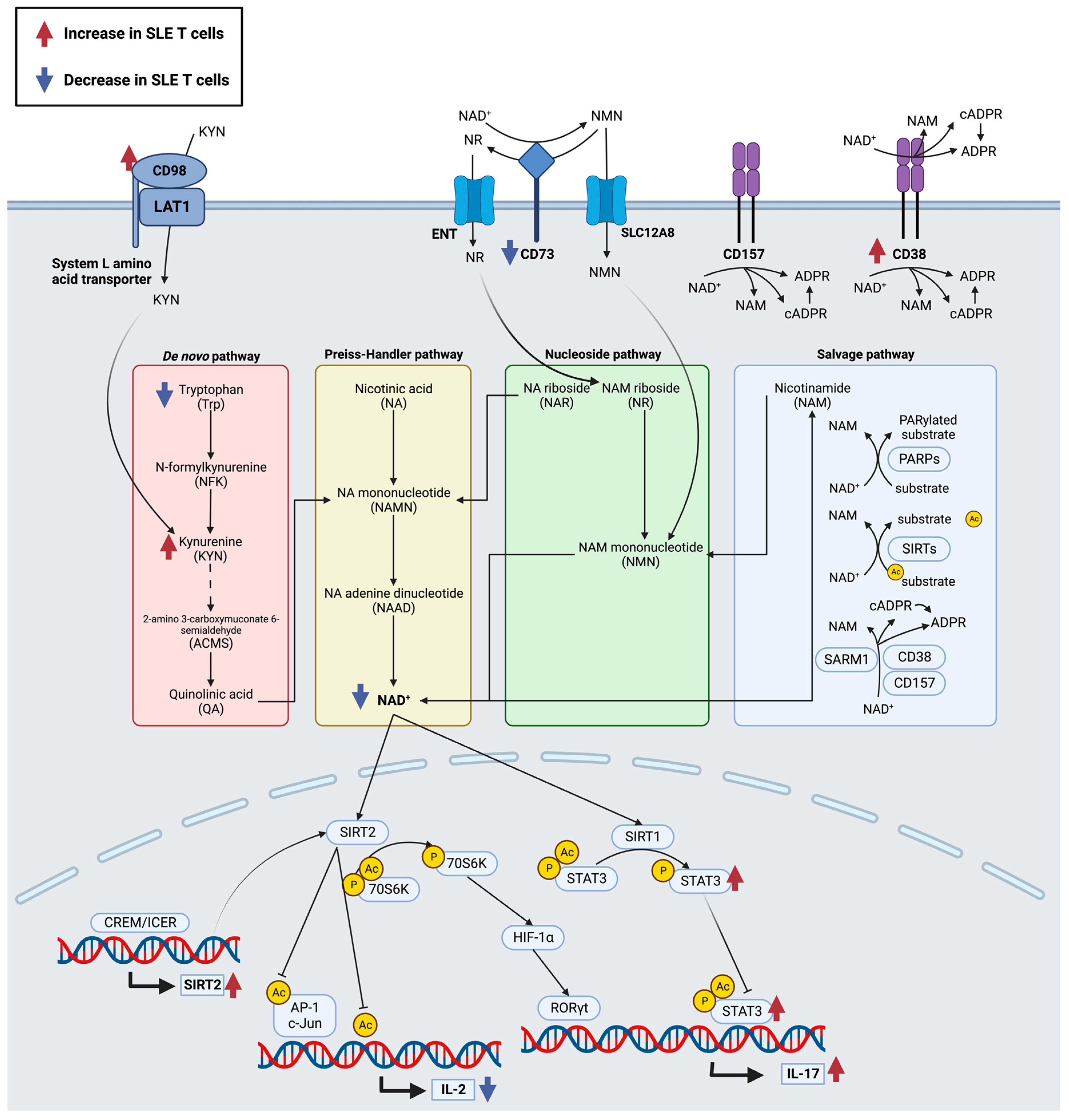
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.S.; Perl, A. Endosome Traffic Modulates Pro-Inflammatory Signal Transduction in CD4+ T Cells—Implications for the Pathogenesis of Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2023, 24, 10749. https://doi.org/10.3390/ijms241310749
Park JS, Perl A. Endosome Traffic Modulates Pro-Inflammatory Signal Transduction in CD4+ T Cells—Implications for the Pathogenesis of Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2023; 24(13):10749. https://doi.org/10.3390/ijms241310749
Chicago/Turabian StylePark, Joy S., and Andras Perl. 2023. "Endosome Traffic Modulates Pro-Inflammatory Signal Transduction in CD4+ T Cells—Implications for the Pathogenesis of Systemic Lupus Erythematosus" International Journal of Molecular Sciences 24, no. 13: 10749. https://doi.org/10.3390/ijms241310749