
Low-Density Lipoprotein Receptor Is a Key Driver of Aggressiveness in Thyroid Tumor Cells

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
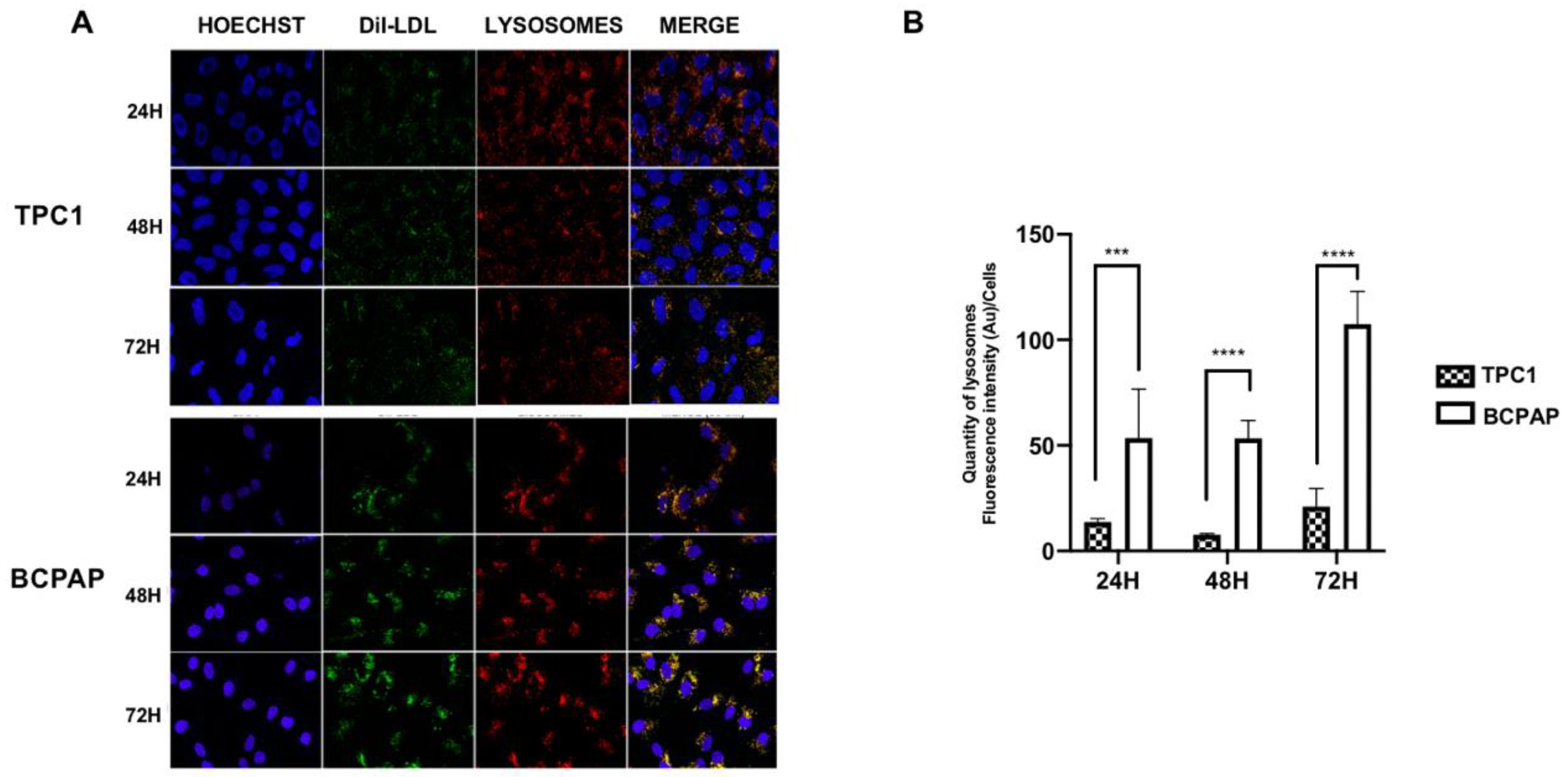
2.1. Differential Regulation of Low-Density Lipoprotein Receptor (LDLR) Protein Expression and Low-Density Lipoprotein (LDL) Uptake in Papillary Thyroid Carcinoma (PTC) Cell Lines with Different Mutational Status after LDL Incubation
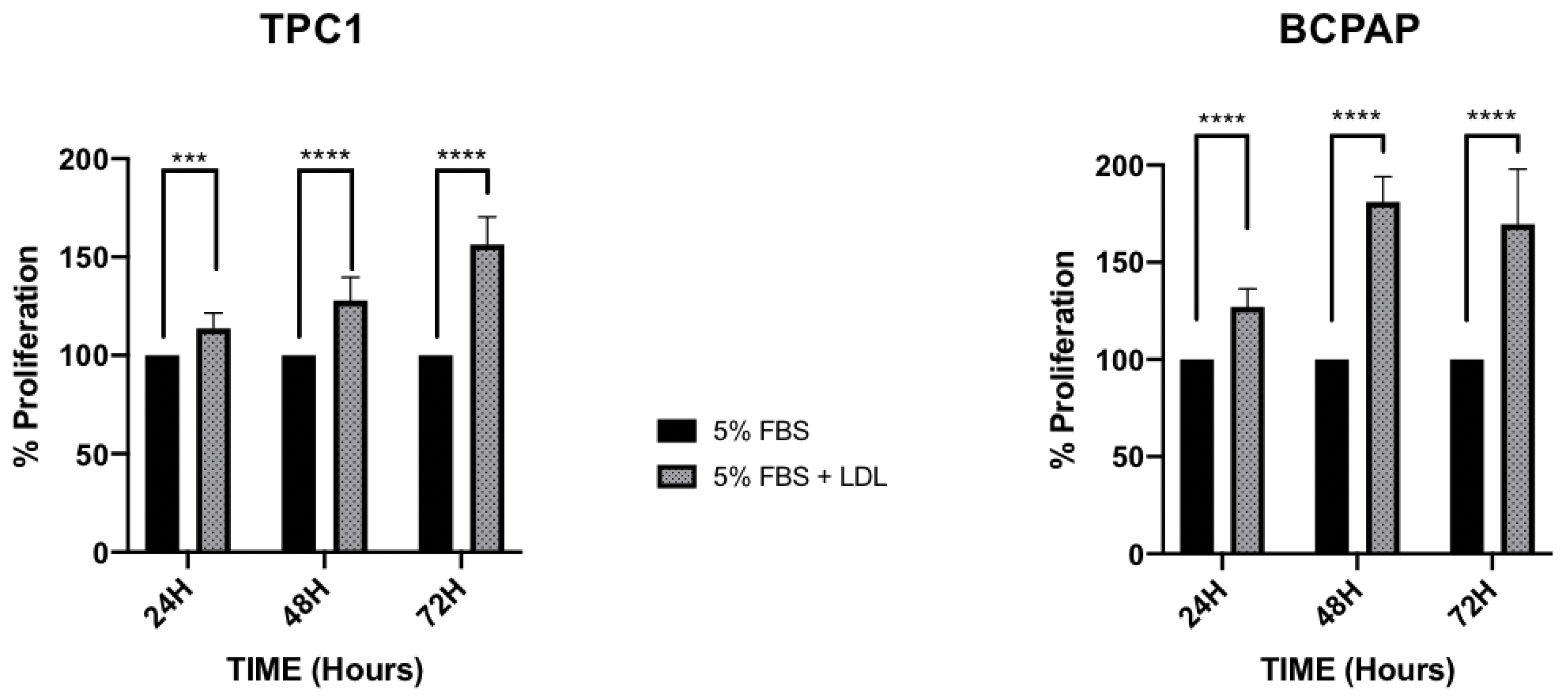
2.2. LDL Promotes Higher Proliferation Levels and Decreases Adhesion Capacity in the BCPAP Cell Line, in Comparison to the Constant Proliferation and Cellular Migration Promoted by LDL in the TPC1 Cell Line
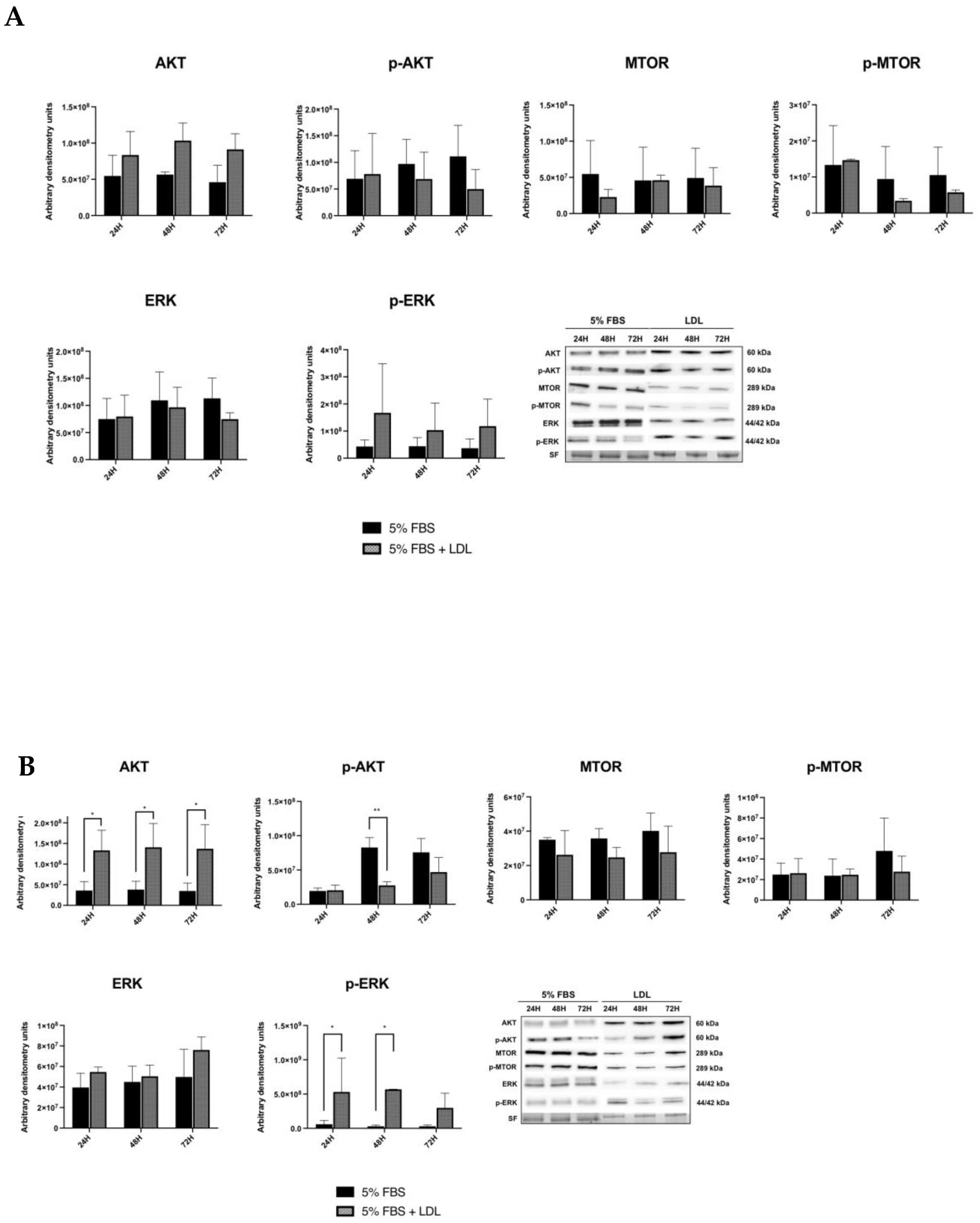
2.3. LDL Enhances the RAS/RAF/MAPK (MEK)/ERK Pathway
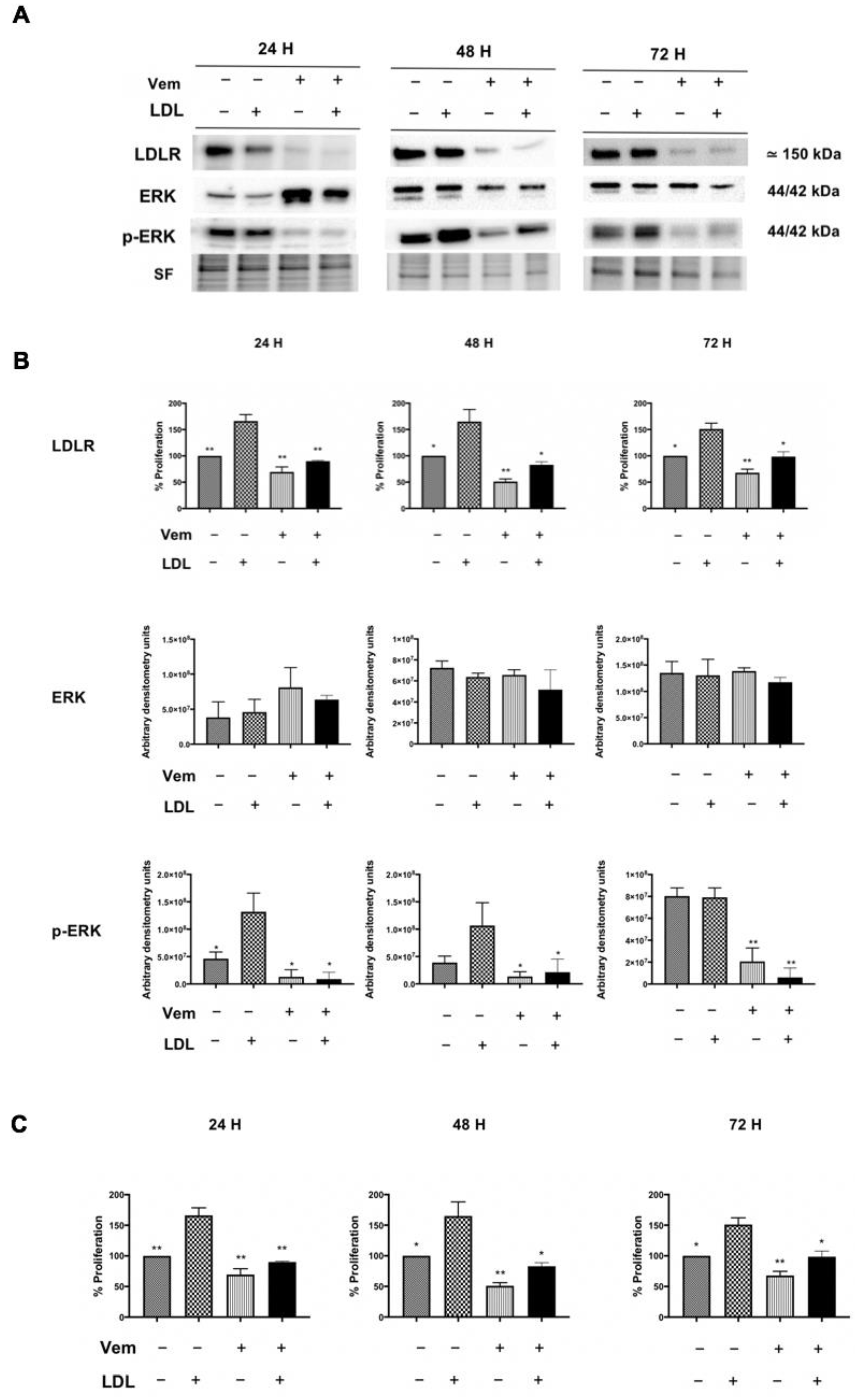
2.4. Vemurafenib Reduces Cellular Proliferation Promoted by LDL, Downregulating LDLR Expression and Impairing RAS/RAF/MAPK (MEK)/ERK Pathway Activation in BCPAP Cell Line
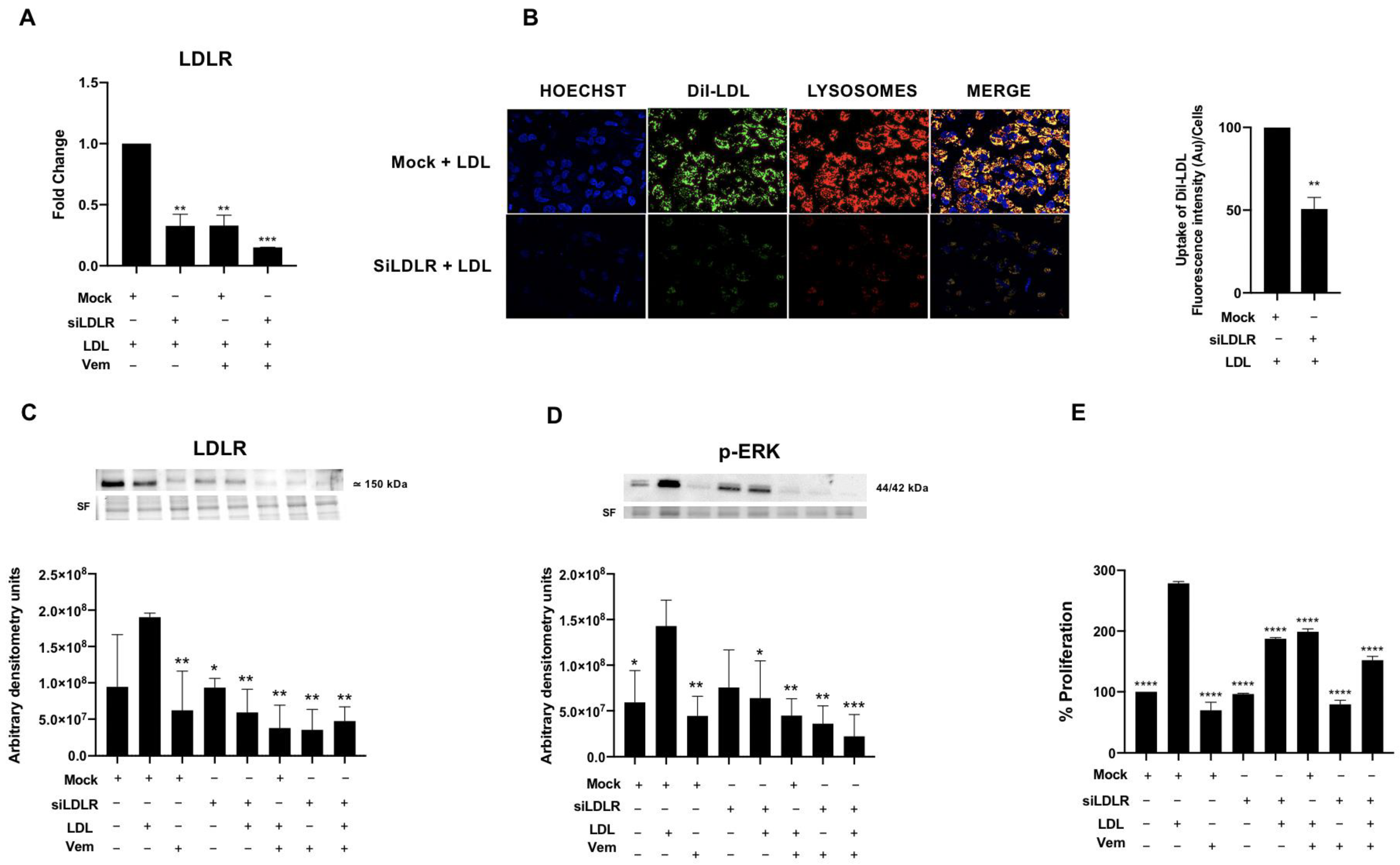
2.5. LDLR Is Required for LDL-Mediated Induction of RAS/RAF/MAPK (MEK)/ERK Pathway, LDL-Uptake and Cell Proliferation in the BCPAP Cell Line
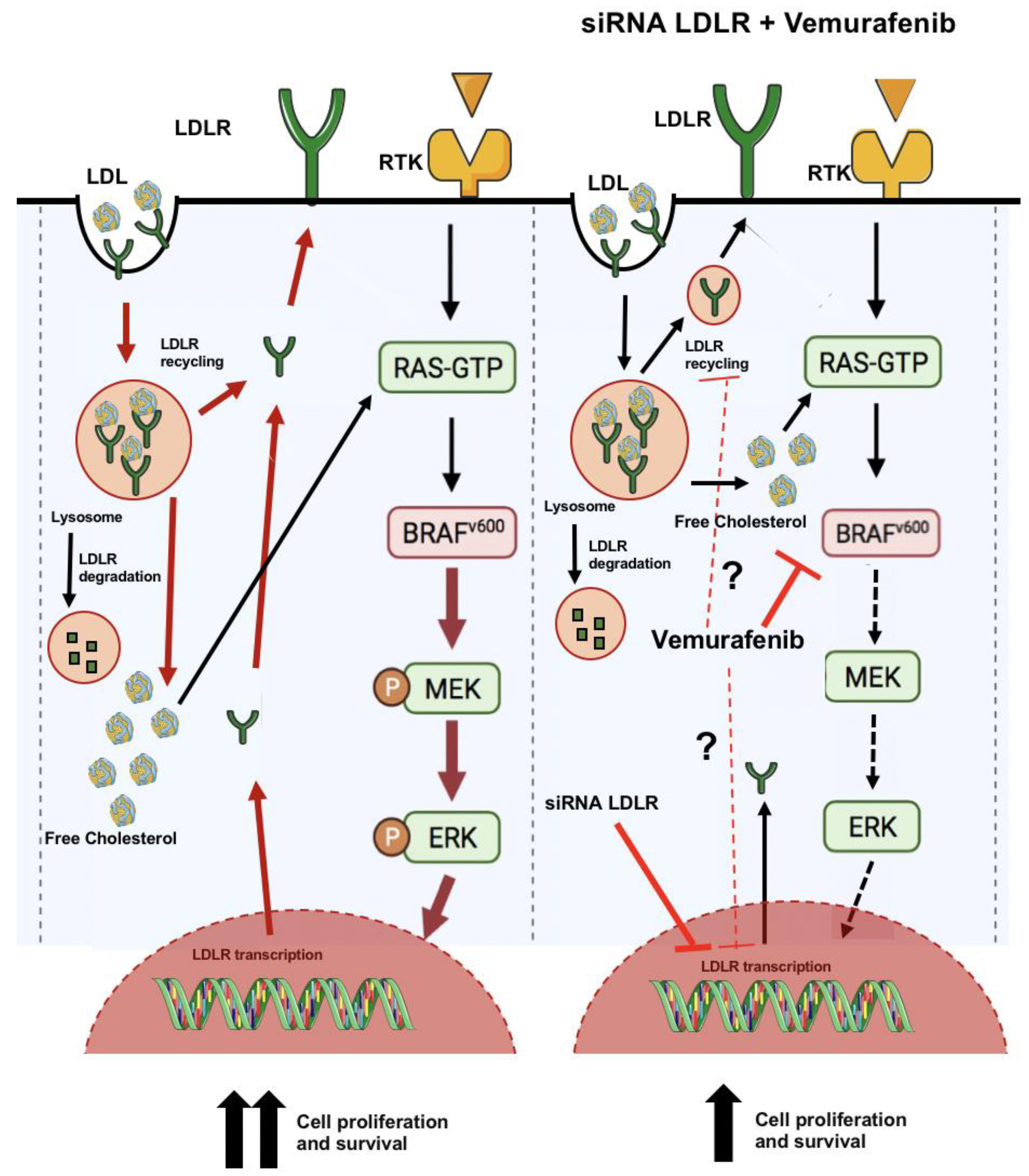
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Human LDL Isolation
4.3. Labeling LDL with DiI
4.4. Analysis of Lipoprotein Uptake by a Fluorometric Assay
4.5. Analysis of Lipoprotein Uptake by Confocal Microscopy
4.6. Quantitative Real-Time PCR
4.7. Protein Extraction and Western Blot
4.8. MTT Assay
4.9. Migration and Adhesion Assay
4.10. Transient Transfection Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noone, A.-M.; Cronin, K.A.; Altekruse, S.F.; Howlader, N.; Lewis, D.R.; Petkov, V.I.; Penberthy, L. Cancer Incidence and Survival Trends by Subtype Using Data from the Surveillance Epidemiology and End Results Program, 1992–2013. Cancer Epidemiol. Biomark. Prev. 2017, 26, 632–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coca-Pelaz, A.; Shah, J.P.; Hernandez-Prera, J.C.; Ghossein, R.A.; Rodrigo, J.P.; Hartl, D.M.; Olsen, K.D.; Shaha, A.R.; Zafereo, M.; Suarez, C.; et al. Papillary Thyroid Cancer—Aggressive Variants and Impact on Management: A Narrative Review. Adv. Ther. 2020, 37, 3112–3128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, J.; Dang, Q.; Wang, X.; Chen, J.; Lin, X.; Yang, N.; Du, J.; Shi, H.; Liu, Y.; et al. Ribosomal Protein RPL5 Regulates Colon Cancer Cell Proliferation and Migration through MAPK/ERK Signaling Pathway. BMC Mol. Cell Biol. 2022, 23, 48. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, X.; Zhang, C.; Xue, L.; Yang, L. Expression and Clinical Significance of MAPK and EGFR in Triple-negative Breast Cancer. Oncol. Lett. 2020, 19, 1842–1848. [Google Scholar] [CrossRef]
- Scully, T.; Kase, N.; Gallagher, E.J.; LeRoith, D. Regulation of Low-Density Lipoprotein Receptor Expression in Triple Negative Breast Cancer by EGFR-MAPK Signaling. Sci. Rep. 2021, 11, 17927. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Nucera, C.; Goldfarb, M.; Hodin, R.; Parangi, S. Role of B-RafV600E in Differentiated Thyroid Cancer and Preclinical Validation of Compounds against B-RafV600E. Biochim. Biophys. Acta BBA Rev. Cancer 2009, 1795, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, J.A.; Bogatchenko, M.; Pusztaszeri, M.; Forest, V.-I.; Hier, M.P.; Yang, J.W.; Tamilia, M.; Payne, R.J. BRAF V600E Mutation Is Associated with Aggressive Features in Papillary Thyroid Carcinomas ≤ 1.5 cm. J. Otolaryngol. Head Neck Surg. 2021, 50, 63. [Google Scholar] [CrossRef]
- Anand, N.; Agrawal, T.; Gupta, A.; Shukla, S.; Pradhan, R.; Husain, N. Diagnostic Efficacy of Brafv600e Immunocytochemistry in Thyroid Aspirates in Bethesda Category Iv and Papillary Thyroid Carcinoma. J. Cytol. 2021, 38, 113. [Google Scholar] [CrossRef]
- Rahman, S.T.; Pandeya, N.; Neale, R.E.; McLeod, D.S.A.; Bain, C.J.; Baade, P.D.; Youl, P.H.; Allison, R.; Leonard, S.; Jordan, S.J. Obesity Is Associated with BRAF V600E -Mutated Thyroid Cancer. Thyroid 2020, 30, 1518–1527. [Google Scholar] [CrossRef]
- Feigelson, H.S.; Bodelon, C.; Powers, J.D.; Curtis, R.E.; Buist, D.S.M.; Veiga, L.H.S.; Bowles, E.J.A.; Berrington de Gonzalez, A.; Gierach, G.L. Body Mass Index and Risk of Second Cancer among Women with Breast Cancer. JNCI J. Natl. Cancer Inst. 2021, 113, 1156–1160. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.; Xu, C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, A.; Jia, Z.; Qiao, C.; Wang, M.; Ding, X. Cholesterol Metabolism in Drug-resistant Cancer (Review). Int. J. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Matrone, A.; Ferrari, F.; Santini, F.; Elisei, R. Obesity as a Risk Factor for Thyroid Cancer. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, C.M.; Pfeiffer, R.M.; Sosa, J.A.; Shiels, M.S. Impact of Overweight and Obesity on US Papillary Thyroid Cancer Incidence Trends (1995–2015). JNCI J. Natl. Cancer Inst. 2020, 112, 810–817. [Google Scholar] [CrossRef]
- Mayengbam, S.S.; Singh, A.; Pillai, A.D.; Bhat, M.K. Influence of Cholesterol on Cancer Progression and Therapy. Transl. Oncol. 2021, 14, 101043. [Google Scholar] [CrossRef]
- Revilla, G.; de Pons, M.P.; Baila-Rueda, L.; García-León, A.; Santos, D.; Cenarro, A.; Magalhaes, M.; Blanco, R.M.; Moral, A.; Ignacio Pérez, J.; et al. Cholesterol and 27-Hydroxycholesterol Promote Thyroid Carcinoma Aggressiveness. Sci. Rep. 2019, 9, 10260. [Google Scholar] [CrossRef] [Green Version]
- Siemianowicz, K.; Gminski, J.; Stajszczyk, M.; Wojakowski, W.; Goss, M.; Machalski, M.; Telega, A.; Brulinski, K.; Magiera-Molendowska, H. Serum LDL Cholesterol Concentration and Lipoprotein Electrophoresis Pattern in Patients with Small Cell Lung Cancer. Int. J. Mol. Med. 2000, 5, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Go, G.-W.; Mani, A. Low-Density Lipoprotein Receptor (LDLR) Family Orchestrates Cholesterol Homeostasis. Yale J. Biol. Med. 2012, 85, 19–28. [Google Scholar]
- Gallagher, E.J.; Zelenko, Z.; Neel, B.A.; Antoniou, I.M.; Rajan, L.; Kase, N.; LeRoith, D. Elevated Tumor LDLR Expression Accelerates LDL Cholesterol-Mediated Breast Cancer Growth in Mouse Models of Hyperlipidemia. Oncogene 2017, 36, 6462–6471. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, L.; Lian, J.; Schauer, S.; Vesely, P.W.; Kratky, D.; Hoefler, G.; Lehner, R. Tumor-Induced Hyperlipidemia Contributes to Tumor Growth. Cell Rep. 2016, 15, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol Uptake Disruption, in Association with Chemotherapy, Is a Promising Combined Metabolic Therapy for Pancreatic Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.D. Role of Mitogen-Activated Protein Kinases and Protein Kinase C in Regulating Low-Density Lipoprotein Receptor Expression. Gene Expr. 2002, 10, 153–164. [Google Scholar] [CrossRef]
- Abidi, P.; Zhang, F.; Li, C.; Liu, J. Blockage of the ERK Signaling Pathway Abrogates the SCAP Ligand-Induced Transcriptional Activation of the LDL Receptor Gene in HepG2 Cells. Int. J. Mol. Med. 2005, 16, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.-F.; Zhu, N.; Zhao, T.-J.; Li, H.-F.; Gu, J.; Liao, D.-F.; Qin, L. Involvement of LDL and Ox-LDL in Cancer Development and Its Therapeutical Potential. Front. Oncol. 2022, 12, 803473. [Google Scholar] [CrossRef]
- Le, A.; Udupa, S.; Zhang, C. The Metabolic Interplay between Cancer and Other Diseases. Trends Cancer 2019, 5, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Gyamfi, J.; Kim, J.; Choi, J. Cancer as a Metabolic Disorder. Int. J. Mol. Sci. 2022, 23, 1155. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; De Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and Cancer: Emerging Roles in Pathogenesis, Diagnosis and Therapeutic Intervention. Adv. Drug Deliv. Rev. 2020, 159, 245–293. [Google Scholar] [CrossRef]
- Labbé, D.P.; Zadra, G.; Yang, M.; Reyes, J.M.; Lin, C.Y.; Cacciatore, S.; Ebot, E.M.; Creech, A.L.; Giunchi, F.; Fiorentino, M.; et al. High-Fat Diet Fuels Prostate Cancer Progression by Rewiring the Metabolome and Amplifying the MYC Program. Nat. Commun. 2019, 10, 4358. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F. Dysregulated Lipid Metabolism in Cancer. World J. Biol. Chem. 2012, 3, 167. [Google Scholar] [CrossRef]
- Gan, X.; Shen, F.; Deng, X.; Feng, J.; Lu, J.; Cai, W.; Peng, L.; Zheng, W.; Wang, W.; Huang, P.; et al. Prognostic Implications of the BRAF-V600E Mutation in Papillary Thyroid Carcinoma Based on a New Cut-off Age Stratification. Oncol. Lett. 2020, 19, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Wang, H.; Liang, Z.; Liang, J.; Li, F.; Lin, Y. BRAFV600E Mutation Associated with Non–Radioiodine-Avid Status in Distant Metastatic Papillary Thyroid Carcinoma. Clin. Nucl. Med. 2014, 39, 675–679. [Google Scholar] [CrossRef]
- Li, J.; Liang, J.; Zhao, T.; Lin, Y. Noninferior Response in BRAFV600E Mutant Nonmetastatic Papillary Thyroid Carcinoma to Radioiodine Therapy. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catar, R.; Chen, L.; Zhao, H.; Wu, D.; Kamhieh-Milz, J.; Lücht, C.; Zickler, D.; Krug, A.W.; Ziegler, C.G.; Morawietz, H.; et al. Native and Oxidized Low-Density Lipoproteins Increase the Expression of the LDL Receptor and the LOX-1 Receptor, Respectively, in Arterial Endothelial Cells. Cells 2022, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, P.; Xuan, J.; Zhu, C.; Liu, J.; Shan, L.; Du, Q.; Ren, Y.; Ye, J. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell. Physiol. Biochem. 2017, 42, 729–742. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Velarde, V.; Klein, R.L.; Joyce, K.C.; Phillips, K.D.; Mayfield, R.K.; Lyons, T.J.; Jaffa, A.A. Native and Modified LDL Activate Extracellular Signal-Regulated Kinases in Mesangial Cells. Diabetes 2000, 49, 2160–2169. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues Dos Santos, C.; Domingues, G.; Matias, I.; Matos, J.; Fonseca, I.; De Almeida, J.M.; Dias, S. LDL-Cholesterol Signaling Induces Breast Cancer Proliferation and Invasion. Lipids Health Dis. 2014, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging Roles of Lipid Metabolism in Cancer Metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid Metabolism in Cancer Progression and Therapeutic Strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef]
- Matsushita, Y.; Nakagawa, H.; Koike, K. Lipid Metabolism in Oncology: Why It Matters, How to Research, and How to Treat. Cancers 2021, 13, 474. [Google Scholar] [CrossRef] [PubMed]
- Valvo, V.; Iesato, A.; Kavanagh, T.R.; Priolo, C.; Zsengeller, Z.; Pontecorvi, A.; Stillman, I.E.; Burke, S.D.; Liu, X.; Nucera, C. Fine-Tuning Lipid Metabolism by Targeting Mitochondria-Associated Acetyl-CoA-Carboxylase 2 in BRAF V600E Papillary Thyroid Carcinoma. Thyroid 2021, 31, 1335–1358. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gu, D.; Lee, S.S.-Y.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating Cholesterol Esterification Suppresses Growth and Metastasis of Pancreatic Cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashkovan, M.; Albero, R.; Gianni, F.; Perez-Duran, P.; Miller, H.I.; Mackey, A.L.; Paietta, E.M.; Tallman, M.S.; Rowe, J.M.; Litzow, M.R.; et al. Intracellular Cholesterol Pools Regulate Oncogenic Signaling and Epigenetic Circuitries in Early T-Cell Precursor Acute Lymphoblastic Leukemia. Cancer Discov. 2022, 12, 856–871. [Google Scholar] [CrossRef]
- Zhao, Q.; Gao, S.M.; Wang, M.C. Molecular Mechanisms of Lysosome and Nucleus Communication. Trends Biochem. Sci. 2020, 45, 978–991. [Google Scholar] [CrossRef]
- Bartel, K.; Pein, H.; Popper, B.; Schmitt, S.; Janaki-Raman, S.; Schulze, A.; Lengauer, F.; Koeberle, A.; Werz, O.; Zischka, H.; et al. Connecting Lysosomes and Mitochondria—A Novel Role for Lipid Metabolism in Cancer Cell Death. Cell Commun. Signal. 2019, 17, 87. [Google Scholar] [CrossRef] [Green Version]
- Machado, E.R.; Annunziata, I.; Van De Vlekkert, D.; Grosveld, G.C.; d’Azzo, A. Lysosomes and Cancer Progression: A Malignant Liaison. Front. Cell Dev. Biol. 2021, 9, 642494. [Google Scholar] [CrossRef]
- Velarde, V.; Jenkins, A.J.; Christopher, J.; Lyons, T.J.; Jaffa, A.A. Activation of MAPK by Modified Low-Density Lipoproteins in Vascular Smooth Muscle Cells. J. Appl. Physiol. 2001, 91, 1412–1420. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Liang, J.; Lu, Y.; Guo, H.; German, P.; Bai, S.; Jonasch, E.; Yang, X.; Mills, G.B.; Ding, Z. Site-Specific Activation of AKT Protects Cells from Death Induced by Glucose Deprivation. Oncogene 2014, 33, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Fathy, M.; Awale, S.; Nikaido, T. Phosphorylated Akt Protein at Ser473 Enables HeLa Cells to Tolerate Nutrient-Deprived Conditions. Asian Pac. J. Cancer Prev. 2017, 18, 3255. [Google Scholar] [CrossRef]
- Xing, J.; Liu, R.; Xing, M.; Trink, B. The BRAFT1799A Mutation Confers Sensitivity of Thyroid Cancer Cells to the BRAFV600E Inhibitor PLX4032 (RG7204). Biochem. Biophys. Res. Commun. 2011, 404, 958–962. [Google Scholar] [CrossRef] [Green Version]
- Theodosakis, N.; Langdon, C.G.; Micevic, G.; Krykbaeva, I.; Means, R.E.; Stern, D.F.; Bosenberg, M.W. Inhibition of Isoprenylation Synergizes with MAPK Blockade to Prevent Growth in Treatment-resistant Melanoma, Colorectal, and Lung Cancer. Pigment Cell Melanoma Res. 2019, 32, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Bose, C.; Bhuvaneswaran, C.; Udupa, K.B. Altered Mitogen-Activated Protein Kinase Signal Transduction in Human Skin Fibroblasts during In Vitro Aging: Differential Expression of Low-Density Lipoprotein Receptor. J. Gerontol. A. Biol. Sci. Med. Sci. 2004, 59, B126–B135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Borkhuu, O.; Bao, W.; Yang, Y.-T. Signaling Pathways in Thyroid Cancer and Their Therapeutic Implications. J. Clin. Med. Res. 2016, 8, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Julve, J.; Escolà-Gil, J.C.; Rotllan, N.; Fiévet, C.; Vallez, E.; de la Torre, C.; Ribas, V.; Sloan, J.H.; Blanco-Vaca, F. Human Apolipoprotein A-II Determines Plasma Triglycerides by Regulating Lipoprotein Lipase Activity and High-Density Lipoprotein Proteome. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the Concentration of Low-Density Lipoprotein Cholesterol in Plasma, without Use of the Preparative Ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Teupser, D.; Thiery, J.; Walli, A.K.; Seidel, D. Quantitative Spectrofluorometry of DiI-Lipoprotein Uptake. Biochim. Biophys Acta 1996, 1303, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.C.; Posch, A. The Design of a Quantitative Western Blot Experiment. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Neris, R.L.S.; Dobles, A.M.C.; Gomes, A.V. Western Blotting Using In-Gel Protein Labeling as a Normalization Control: Advantages of Stain-Free Technology. In Proteomic Profiling; Posch, A., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; Volume 2261, pp. 443–456. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of Feedback Inhibition of HER3 Transcription by RAF and MEK Inhibitors Attenuates Their Antitumor Effects in BRAF-Mutant Thyroid Carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef] [Green Version]
- Byeon, H.K.; Na, H.J.; Yang, Y.J.; Ko, S.; Yoon, S.O.; Ku, M.; Yang, J.; Kim, J.W.; Ban, M.J.; Kim, J.-H.; et al. Acquired Resistance to BRAF Inhibition Induces Epithelial-to-Mesenchymal Transition in BRAF (V600E) Mutant Thyroid Cancer by c-Met-Mediated AKT Activation. Oncotarget 2017, 8, 596–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Revilla, G.; Ruiz-Auladell, L.; Vallverdú, N.F.; Santamaría, P.; Moral, A.; Pérez, J.I.; Li, C.; Fuste, V.; Lerma, E.; Corcoy, R.; et al. Low-Density Lipoprotein Receptor Is a Key Driver of Aggressiveness in Thyroid Tumor Cells. Int. J. Mol. Sci. 2023, 24, 11153. https://doi.org/10.3390/ijms241311153
Revilla G, Ruiz-Auladell L, Vallverdú NF, Santamaría P, Moral A, Pérez JI, Li C, Fuste V, Lerma E, Corcoy R, et al. Low-Density Lipoprotein Receptor Is a Key Driver of Aggressiveness in Thyroid Tumor Cells. International Journal of Molecular Sciences. 2023; 24(13):11153. https://doi.org/10.3390/ijms241311153
Chicago/Turabian StyleRevilla, Giovanna, Lara Ruiz-Auladell, Núria Fucui Vallverdú, Paula Santamaría, Antonio Moral, José Ignacio Pérez, Changda Li, Victoria Fuste, Enrique Lerma, Rosa Corcoy, and et al. 2023. "Low-Density Lipoprotein Receptor Is a Key Driver of Aggressiveness in Thyroid Tumor Cells" International Journal of Molecular Sciences 24, no. 13: 11153. https://doi.org/10.3390/ijms241311153