
Molecular Biology and Therapeutic Targets of Primitive Tracheal Tumors: Focus on Tumors Derived by Salivary Glands and Squamous Cell Carcinoma
 , ,
, , 
Abstract
:1. Background
2. Primitive Tracheal Tumors Derived by Salivary Glands
2.1. Adenoid Cystic Carcinoma
2.1.1. Pathological Features
2.1.2. Molecular Biology of ACC
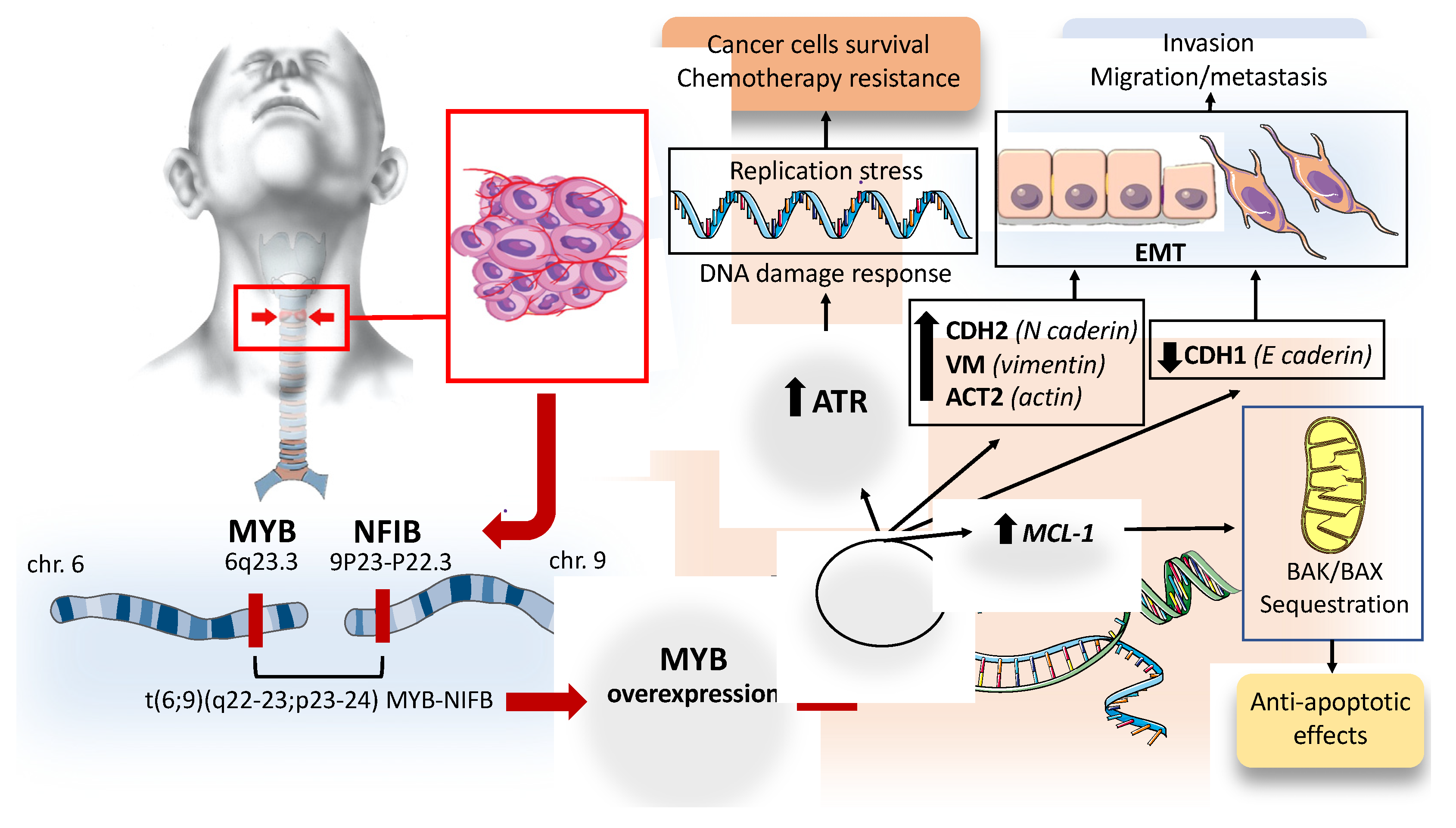
2.1.3. Consequences of Myb Overactivation in ACC
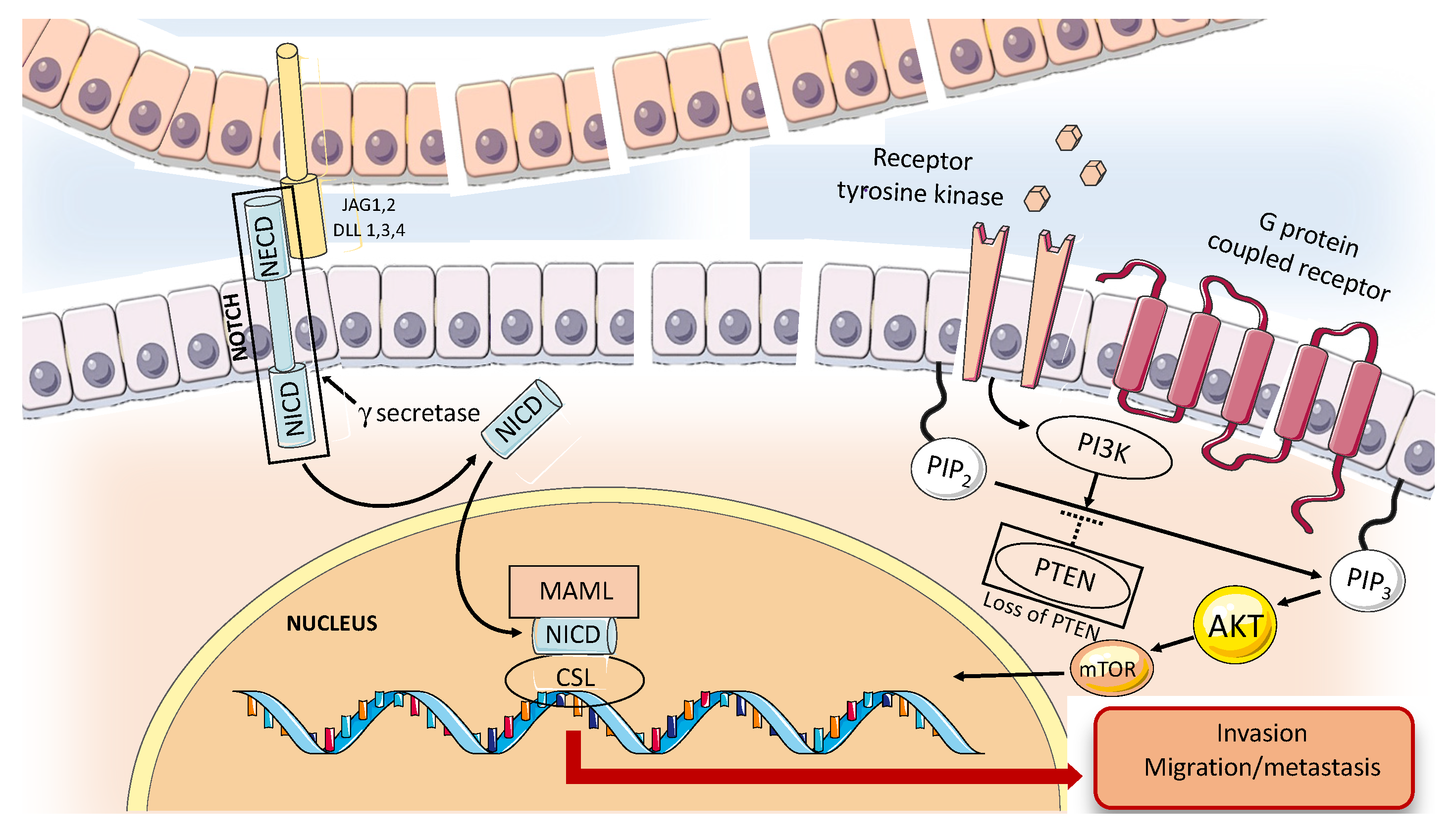
2.1.4. Receptor Tyrosine Kinase and Growth Factors Expression in ACC
2.1.5. Immune Checkpoint Targets in ACC
2.1.6. Current Treatment Strategy for ACC
2.1.7. Molecular Therapeutic Targets for ACC
2.2. Mucoepidermoid Carcinoma of the Trachea
2.3. Primitive Squamous Cell Carcinoma of the Trachea
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AHRF | acute hypoxic respiratory failure |
| ACC | adenoid cystic carcinoma |
| COPD | obstructive pulmonary disease |
| FISH | fluorescent in situ hybridization |
| Cdks | cyclin-dependent kinase |
| NFIB | Nuclear Factor I B |
| CCNBI | Cyclin B1 |
| MAD1L1 | Mitotic Arrest Deficient 1 Like 1 |
| BIRC3 | Baculoviral IAP Repeat Containing 3 |
| HSPA8 | Heat Shock Protein Family A (Hsp70) Member 8 |
| MYC | MYC Proto-Oncogene, BHLH Transcription Factor |
| VEGFA | Vascular Endothelial Growth Factor A |
| FGF2 | Fibroblast Growth Factor 2 |
| SEMA4D | Semaphorin 4D |
| ATR | ataxia telangiectasia and Rad3-related (ATR) |
| VX970 | Berzosertib |
| siRNA | Short Interfering RNA |
| CDH1 | Cadherin 1 |
| CDH2 | Cadherin 2 |
| VIM | Vimentin |
| ACT2 | Actin Alpha 2, Smooth Muscle |
| EMT | epithelial–mesenchymal transition |
| MCL-1 | myeloid cell leukemia-1 |
| BAX | BCL-2-associated X protein |
| MMP7 | Matrix Metallopeptidase 7 |
| MMP9 | Matrix Metallopeptidase 9 |
| ICAM-I | Intercellular Adhesion Molecule 1 |
| SCF | Stem Cell Factor |
| GIST | gastrointestinal stromal tumor |
| MAPK | mitogen activated protein kinase |
| ERK | extracellular signal-regulated kinase |
| PI3K/AKT | phosphatidylinositol 3kinase/protein kinase B |
| PLC-γ | phospholipase-C-γ |
| FISH | fluorescence in situ hybridization |
| EGFR | Epidermal growth factor receptor |
| KRAS | Kristen rat sarcoma viral oncogene homologue |
| VEGF | Vascular Endothelial Growth Factor |
| DLL1 | Delta Like Canonical Notch Ligand 1 |
| JAG1 | Jagged 1 |
| NECD | Notch extracellular domain |
| MAML | Mastermind-like |
| PTEN | Phosphatase and tensin homolog |
| PI3K | phosphatidylinositol 3-kinase |
| TIL | tumor-infiltrating lymphocytes |
| Grb | granzyme B |
| DC | Dendritic cells |
| PD-1 | Programmed Cell Death protein 1 |
| RGMb | Repulsive Guidance Molecule B |
| FLT3 | FMS-like tyrosine kinase 3 |
| FGFR1 | Fibroblast growth factor receptor 1 |
| NCCN | National Comprehensive Cancer Network |
| TP53 | Tumor Protein P53 |
| NFE2L2 | nuclear factor erythroid 2-like 2 |
| CUL3 | cullin 3 |
References
- Macchiarini, P. Primary tracheal tumours. Lancet Oncol. 2006, 7, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Junker, K. Pathology of Tracheal Tumors. Thorac. Surg. Clin. 2014, 24, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Moores, D.; Mane, P. Pathology of Primary Tracheobronchial Malignancies Other than Adenoid Cystic Carcinomas. Thorac. Surg. Clin. 2018, 28, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Gaissert, H.A.; Honings, J.; Gokhale, M. Treatment of Tracheal Tumors. Semin. Thorac. Cardiovasc. Surg. 2009, 21, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Maziak, D.E. Biology of Adenoid Cystic Carcinoma of the Tracheobronchial Tree and Principles of Management. Thorac. Surg. Clin. 2018, 28, 145–148. [Google Scholar] [CrossRef]
- Billroth, T. Beobachtungen űber geschwűlsteder Speicheldrűsen. Virchous Arch. Pathol. Anat. 1859, 17, 357–375. [Google Scholar] [CrossRef]
- Jaso, J.; Malhotra, R. Adenoid cystic carcinoma. Arch. Pathol. Lab. Med. 2011, 135, 511–515. [Google Scholar] [CrossRef]
- Szanto, P.A.; Luna, M.A.; Tortoledo, M.E.; White, R.A. Histologic grading of adenoid cystic carcinoma of the salivary glands. Cancer 1984, 54, 1062–1069. [Google Scholar] [CrossRef]
- da Cruz Perez, D.E.; de Abreu Alves, F.; Nobuko Nishimoto, I.; de Almeida, O.P.; Kowalski, L.P. Prognostic factors in head and neck adenoid cystic carcinoma. Oral Oncol. 2006, 42, 139–146. [Google Scholar] [CrossRef]
- Fordice, J.; Kershaw, C.; El-Naggar, A.; Goepfert, H. Adenoid cystic carcinoma of the head and neck: Predictors of morbidity and mortality. Arch. Otolaryngol. Head Neck Surg. 1999, 125, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Cheuk, W.; Chan, J.K.; Ngan, R.K. Dedifferentiation in adenoid cystic carcinoma of salivary gland: An uncommon complication associated with an accelerated clinical course. Am. J. Surg. Pathol. 1999, 23, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Meis, J.M. “Dedifferentiation” in bone and soft-tissue tumors. A histological indicator of tumor progression. Pathol. Annu. 1991, 26 Pt 1, 37–62. [Google Scholar] [PubMed]
- Seethala, R.R.; Hunt, J.L.; Baloch, Z.W.; Livolsi, V.A.; Leon Barnes, E. Adenoid cystic carcinoma with high-grade transformation: A report of 11 cases and a review of the literature. Am. J. Surg. Pathol. 2007, 31, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.-P.; Hongyo, T.; Aozasa, K.; Chan, J.K. Dedifferentiation of adenoid cystic carcinoma: Report of a case implicating p53 gene mutation. Hum. Pathol. 2001, 32, 1403–1407. [Google Scholar] [CrossRef]
- Sandros, J.; Stenman, G.; Mark, J. Cytogenetic and molecular observations in human and experimental salivary gland tumors. Cancer Genet. Cytogenet. 1990, 44, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Andren, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef] [PubMed]
- Mucenski, M.L.; McLain, K.; Kier, A.B.; Swerdlow, S.H.; Schreiner, C.M.; Miller, T.A.; Pietryga, D.W.; Scott, W.J.; Potter, S. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 1991, 65, 677–689. [Google Scholar] [CrossRef]
- Jiang, J.; Best, S.; Menzel, S.; Silver, N.; Lai, M.I.; Surdulescu, G.L.; Spector, T.D.; Thein, S.L. cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood 2006, 108, 1077–1083. [Google Scholar] [CrossRef] [Green Version]
- Nakata, Y.; Shetzline, S.; Sakashita, C.; Kalota, A.; Rallapalli, R.; Rudnick, S.I.; Zhang, Y.; Emerson, S.G.; Gewirtz, A.M. c-Myb contributes to G2/M cell cycle transition in human hematopoietic cells by direct regulation of cyclin B1 expression. Mol. Cell Biol. 2007, 27, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Cicirò, Y.; Sala, A. MYB oncoproteins: Emerging players and potential therapeutic targets in human cancer. Oncogenesis 2021, 10, 19. [Google Scholar] [CrossRef]
- Mitani, Y.; Li, J.; Rao, P.H.; Zhao, Y.J.; Bell, D.; Lippman, S.M.; Weber, R.S.; Caulin, C.; El-Naggar, A.K. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clin. Cancer Res. 2010, 16, 4722–4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.H.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frerich, C.A.; Sedam, H.N.; Kang, H.; Mitani, Y.; El-Naggar, A.K.; Ness, S.A. N-Terminal Truncated Myb with New Transcriptional Activity Produced Through Use of an Alternative MYB Promoter in Salivary Gland Adenoid Cystic Carcinoma. Cancers 2019, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.K.; Mangiapane, G.; Nevado, P.T.; Tsakaneli, A.; Carlsson, T.; Corda, G.; Nieddu, V.; Abrahamian, C.; Chayka, O.; Rai, L.; et al. ATR is a MYB regulated gene and potential therapeutic target in adenoid cystic carcinoma. Oncogenesis 2020, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.D.; Schleker, T.; Guerra, C.; Garcia, E.O.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Koniaras, K.; Cuddihy, A.R.; Christopoulos, H.; Hogg, A.; O’Connell, M.J. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene 2001, 20, 7453–7463. [Google Scholar] [CrossRef] [Green Version]
- López-Contreras, A.J.; Gutierrez-Martinez, P.; Specks, J.; Rodrigo-Perez, S.; Fernandez-Capetillo, O. An extra allele of Chk1 limits oncogene-induced replicative stress and promotes transformation. J. Exp. Med. 2012, 209, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Fatah, T.M.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R.; et al. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef]
- Hilton, B.A.; Li, Z.; Musich, P.R.; Wang, H.; Cartwright, B.M.; Serrano, M.; Zhou, X.Z.; Lu, K.P.; Zou, Y. ATR Plays a Direct Antiapoptotic Role at Mitochondria, which Is Regulated by Prolyl Isomerase Pin1. Mol. Cell 2015, 60, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.-H.; Zhao, F.; Yang, W.-W.; Chen, C.-W.; Du, Z.-H.; Fu, M.; Ge, X.-Y.; Li, S.-L. MYB promotes the growth and metastasis of salivary adenoid cystic carcinoma. Int. J. Oncol. 2019, 54, 1579–1590. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Yang, C.-Y.; Bai, L. MCL-1 inhibition in cancer treatment. OncoTargets Ther. 2018, 11, 7301–7314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akgul, C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol. Life Sci. 2009, 66, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, F.J.; de Azevedo, J.C.; Lima Ralph, A.C.; Viana Pinheiro, J.d.J.; Morais Freitas, V.; Queiroz Calcagno, D. Salivary glands adenoid cystic carcinoma: A molecular profile update and potential implications. Front. Oncol. 2023, 13. [Google Scholar] [CrossRef]
- Babaei, M.A.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Dev. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besmer, P.; Murphy, J.E.; George, P.C.; Qiu, F.; Bergold, P.J.; Lederman, L.; Snyder, H.W.; Brodeur, D.; Zuckerman, E.E.; Hardy, W.D. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature 1986, 320, 415–421. [Google Scholar] [CrossRef]
- Sheikh, E.; Tran, T.; Vranic, S.; Levy, A.; Bonfil, R.D. Role and Significance of c-KIT Receptor Tyrosine Kinase in Cancer: A Review. Bosn. J. Basic. Med. Sci. 2022, 22, 683–698. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Vila, L.; Liu, H.; Al-Quran, S.Z.; Coco, D.P.; Dong, H.-J.; Liu, C. Identification of c-kit gene mutations in primary adenoid cystic carcinoma of the salivary gland. Mod. Pathol. 2009, 22, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Jeng, Y.-M.; Lin, C.-Y.; Hsu, H.-C. Expression of the c-kit protein is associated with certain subtypes of salivary gland carcinoma. Cancer Lett. 2000, 154, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Holst, V.A.; Marshall, C.; Moskaluk, C.A.; Frierson, H.F. KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Mod. Pathol. 1999, 12, 956–960. [Google Scholar] [PubMed]
- Freier, K.; Flechtenmacher, C.; Walch, A.; Devens, F.; Mühling, J.; Lichter, P.; Joos, S.; Hofele, C. Differential KIT expression in histological subtypes of adenoid cystic carcinoma (ACC) of the salivary gland. Oral. Oncol. 2005, 41, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef]
- Singh, D.; Attri, B.K.; Gill, R.K.; Bariwal, J. Review on EGFR Inhibitors: Critical Updates. Mini Rev. Med. Chem. 2016, 16, 1134–1166. [Google Scholar] [CrossRef] [PubMed]
- Dahse, R.; Driemel, O.; Schwarz, S.; Kromeyer-Hauschild, K.; Berndt, A.; Kosmehl, H. KRAS status and epidermal growth factor receptor expression as determinants for anti-EGFR therapies in salivary gland carcinomas. Oral. Oncol. 2009, 45, 826–829. [Google Scholar] [CrossRef]
- Saida, K.; Murase, T.; Ito, M.; Fujii, K.; Takino, H.; Masaki, A.; Kawakita, D.; Ijichi, K.; Tada, Y.; Kusafuka, K.; et al. Mutation analysis of the EGFR pathway genes, EGFR, RAS, PIK3CA, BRAF, and AKT1, in salivary gland adenoid cystic carcinoma. Oncotarget 2018, 9, 17043–17055. [Google Scholar] [CrossRef] [Green Version]
- Benesova, L.; Minarik, M.; Jancarikova, D.; Belsanova, B.; Pesek, M. Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer. Res. 2010, 30, 1667–1671. [Google Scholar]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Miller, L.E.; Au, V.; Mokhtari, T.E.; Goss, D.; Faden, D.L.; Varvares, M.A. A Contemporary Review of Molecular Therapeutic Targets for Adenoid Cystic Carcinoma. Cancers 2022, 14, 992. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Curr. Vasc. Pharmacol. 2017, 15, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tang, P.; Xu, Z. Clinico-pathological significance of microvessel density and vascular endothelial growth factor expression in adenoid cystic carcinoma of salivary glands. Zhonghua Kou Qiang Yi Xue Za Zhi 2001, 36, 212–214. (In Chinese) [Google Scholar] [PubMed]
- Park, S.; Nam, S.J.; Keam, B.; Kim, T.M.; Jeon, Y.K.; Lee, S.-H.; Hah, J.H.; Kwon, T.-K.; Kim, D.-W.; Sung, M.-W.; et al. VEGF and Ki-67 Overexpression in Predicting Poor Overall Survival in Adenoid Cystic Carcinoma. Cancer Res. Treat. 2016, 48, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobry, C.; Oh, P.; Mansour, M.; Look, A.T.; Aifantis, I. Notch signaling: Switching an oncogene to a tumor suppressor. Blood 2014, 123, 2451–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gragnani, L.; Lorini, S.; Marri, S.; Zignego, A.L. Role of Notch Receptors in Hematologic Malignancies. Cells 2020, 10, 16. [Google Scholar] [CrossRef]
- Parmigiani, E.; Taylor, V.; Giachino, C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells 2020, 9, 2304. [Google Scholar] [CrossRef]
- Chanrion, M.; Kuperstein, I.; Barrière, C.; El Marjou, F.; Cohen, D.; Vignjevic, D.; Stimmer, L.; Paul-Gilloteaux, P.; Bieche, I.; Tavares, S.D.R.; et al. Concomitant Notch activation and p53 deletion trigger epithelial-to-mesenchymal transition and metastasis in mouse gut. Nat. Commun. 2014, 5, 5005. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Zhang, L.; He, C.-S.; Xu, F.; Liu, J.-L.; Hu, Z.-H.; Zhao, L.-P.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Mitani, Y.; Diao, L.; Guijarro, I.; Wang, J.; Zweidler-McKay, P.; Bell, D.; William, W.N., Jr.; Glisson, B.S.; Wick, M.J.; et al. Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors. J. Clin. Oncol. 2017, 35, 352–360. [Google Scholar] [CrossRef]
- Chintakuntlawar, A.V.; Okuno, S.H.; Price, K.A.R. Genomic testing may offer therapeutic opportunity in salivary gland cancers. J. Clin. Oncol. 2015, 33, e17053. [Google Scholar] [CrossRef]
- Su, B.-H.; Qu, J.; Song, M.; Huang, X.-Y.; Hu, X.-M.; Xie, J.; Zhao, Y.; Ding, L.-C.; She, L.; Chen, J.; et al. NOTCH1 signaling contributes to cell growth, anti-apoptosis and metastasis in salivary adenoid cystic carcinoma. Oncotarget 2014, 5, 6885–6895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, N.; Sajjadi, E.; Venetis, K.; Gaudioso, G.; Lopez, G.; Corti, C.; Rocco, E.G.; Criscitiello, C.; Malapelle, U.; Invernizzi, M. PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes 2020, 11, 719. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Du, L.; Wang, R.; Wei, C.; Liu, B.; Zhu, L.; Liu, P.; Liu, Q.; Li, J.; Lu, S.-L.; et al. High frequency of loss of PTEN expression in human solid salivary adenoid cystic carcinoma and its implication for targeted therapy. Oncotarget 2015, 6, 11477–11491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, A.S.; Kannan, K.; Roy, D.M.; Morris, L.G.T.; Ganly, I.; Katabi, N.; Ramaswami, D.; Walsh, L.A.; Eng, S.; Huse, J.T.; et al. The mutational landscape of adenoid cystic carcinoma. Nat. Genet. 2013, 45, 791–798. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Ljunggren, H.-G.; Jonsson, R.; Höglund, P. Seminal immunologic discoveries with direct clinical implications: The 2018 Nobel Prize in Physiology or Medicine honours discoveries in cancer immunotherapy. Scand. J. Immunol. 2018, 88, e12731. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef]
- Mosconi, C.; de Arruda, J.A.A.; de Farias, A.C.R.; Oliveira, G.A.Q.; de Paula, H.M.; Fonseca, F.; Mesquita, R.A.; Silva, T.A.; Mendonça, E.F.; Batista, A.C. Immune microenvironment and evasion mechanisms in adenoid cystic carcinomas of salivary glands. Oral. Oncol. 2019, 88, 95–101. [Google Scholar] [CrossRef]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, V.; Gjini, E.; Liao, X.; Chau, N.G.; Haddad, R.I.; Severgnini, M.; Hammerman, P.; El-Naggar, A.; Freeman, G.J.; Hodi, F.S.; et al. Immune Profiling of Adenoid Cystic Carcinoma: PD-L2 Expression and Associations with Tumor-Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 679–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yu, S.; Zhu, B.; Bedoret, D.; Bu, X.; Francisco, L.M.; Hua, P.; Duke-Cohan, J.S.; Umetsu, D.T.; Sharpe, A.H.; et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J. Exp. Med. 2014, 211, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Tapias, L.F.; Shih, A.; Mino-Kenudson, M.; Muniappan, A.; A Gaissert, H.; Lanuti, M.; Mathisen, D.J.; Ott, H.C. Programmed death ligand 1 and CD8+ immune cell infiltrates in resected primary tracheal malignant neoplasms. Eur. J. Cardio-Thorac. Surg. 2019, 55, 691–698. [Google Scholar] [CrossRef]
- Suzuki, T. What is the best management strategy for adenoid cystic carcinoma of the trachea? Ann. Thorac. Cardiovasc. Surg. 2011, 17, 535–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grillo, H.C.; Mathisen, D.J. Primary tracheal tumors: Treatment and results. Ann. Thorac. Surg. 1990, 49, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Gaissert, H.A.; Grillo, H.C.; Shadmehr, M.; Wright, C.D.; Gokhale, M.; Wain, J.C.; Mathisen, D.J. Long-Term Survival after Resection of Primary Adenoid Cystic and Squamous Cell Carcinoma of the Trachea and Carina. Ann. Thorac. Surg. 2004, 78, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Regnard, J.; Fourquier, P.; Levasseur, P. Results and prognostic factors in resections of primary tracheal tumors: A multicenter retrospective study. J. Thorac. Cardiovasc. Surg. 1996, 111, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Maziak, D.E.; Todd, T.R.; Keshavjee, S.H.; Winton, T.L.; Van Nostrand, P.; Pearson, F. Adenoid cystic carcinoma of the airway: Thirty-two-year experience. J. Thorac. Cardiovasc. Surg. 1996, 112, 1522–1532. [Google Scholar] [CrossRef] [Green Version]
- Mendenhall, W.M.; Morris, C.G.; Amdur, R.J.; Werning, J.W.; Hinerman, R.W.; Villaret, D.B. Radiotherapy alone or combined with surgery for adenoid cystic carcinoma of the head and neck. Head Neck 2004, 26, 154–162. [Google Scholar] [CrossRef]
- van Weert, S.; Bloemena, E.; van der Waal, I.; de Bree, R.; Rietveld, D.H.; Kuik, J.D.; Leemans, C.R. Adenoid cystic carcinoma of the head and neck: A single-center analysis of 105 consecutive cases over a 30-year period. Oral. Oncol. 2013, 49, 824–829. [Google Scholar] [CrossRef]
- Le Péchoux, C.; Baldeyrou, P.; Ferreira, I.; Mahé, M. Cylindromes thoraciques: Thoracic Adenoid cystic carcinomas. Cancer Radiother. 2005, 9, 358–361. [Google Scholar] [CrossRef]
- Högerle, B.A.; Lasitschka, F.; Muley, T.; Bougatf, N.; Herfarth, K.; Adeberg, S.; Eichhorn, M.; Debus, J.; Winter, H.; Rieken, S.; et al. Primary adenoid cystic carcinoma of the trachea: Clinical outcome of 38 patients after interdisciplinary treatment in a single institution. Radiat. Oncol. 2019, 14, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurie, S.A.; Ho, A.L.; Fury, M.G.; Sherman, E.; Pfister, D.G. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: A systematic review. Lancet Oncol. 2011, 12, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Li, Y.; Pinto, H.A.; Jennings, T.; Kies, M.S.; Silverman, P.; Forastiere, A.A. Phase II trial of taxol in salivary gland malignancies (E1394): A trial of the Eastern Cooperative Oncology Group. Head Neck 2006, 28, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.R.; Talmi, Y.; Catane, R.; Symon, Z.; Yosepovitch, A.; Levitt, M. A phase II study of Imatinib for advanced adenoid cystic carcinoma of head and neck salivary glands. Oral. Oncol. 2007, 43, 33–36. [Google Scholar] [CrossRef]
- Hotte, S.J.; Winquist, E.W.; Lamont, E.; MacKenzie, M.; Vokes, E.; Chen, E.X.; Brown, S.; Pond, G.R.; Murgo, A.; Siu, L.L. Imatinib Mesylate in Patients with Adenoid Cystic Cancers of the Salivary Glands Expressing c-kit: A Princess Margaret Hospital Phase II Consortium Study. J. Clin. Oncol. 2005, 23, 585–590. [Google Scholar] [CrossRef]
- Ghosal, N.; Mais, K.; Shenjere, P.; Julyan, P.; Hastings, D.; Ward, T.; Ryder, W.; Bruce, I.; Homer, J.; Slevin, N. Phase II study of cisplatin and imatinib in advanced salivary adenoid cystic carcinoma. Br. J. Oral. Maxillofac. Surg. 2011, 49, 510–515. [Google Scholar] [CrossRef]
- Wong, S.; Karrison, T.; Hayes, D.; Kies, M.; Cullen, K.; Tanvetyanon, T.; Argiris, A.; Takebe, N.; Lim, D.; Saba, N.; et al. Phase II trial of dasatinib for recurrent or metastatic c-KIT expressing adenoid cystic carcinoma and for nonadenoid cystic malignant salivary tumors. Ann. Oncol. 2016, 27, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Chau, N.G.; Hotte, S.J.; Chen, E.X.; Chin, S.F.; Turner, S.; Wang, L.; Siu, L.L. A phase II study of sunitinib in recurrent and/or metastatic adenoid cystic carcinoma (ACC) of the salivary glands: Current progress and challenges in evaluating molecularly targeted agents in ACC. Ann. Oncol. 2012, 23, 1562–1570. [Google Scholar] [CrossRef]
- Jakob, J.A.; Kies, M.S.; Glisson, B.S.; Kupferman, M.E.; Liu, D.D.; Lee, J.J.; El-Naggar, A.K.; Gonzalez-Angulo, A.M.; Blumenschein, G.R. Phase II study of gefitinib in patients with advanced salivary gland cancers. Head Neck 2015, 37, 644–649. [Google Scholar] [CrossRef] [Green Version]
- Locati, L.; Bossi, P.; Perrone, F.; Potepan, P.; Crippa, F.; Mariani, L.; Casieri, P.; Orsenigo, M.; Losa, M.; Bergamini, C.; et al. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: A phase II study. Oral. Oncol. 2009, 45, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Agulnik, M.; Cohen, E.W.; Cohen, R.B.; Chen, E.X.; Vokes, E.E.; Hotte, S.J.; Winquist, E.; Laurie, S.; Hayes, D.N.; Dancey, J.E.; et al. Phase II Study of Lapatinib in Recurrent or Metastatic Epidermal Growth Factor Receptor and/or erbB2 Expressing Adenoid Cystic Carcinoma and Non–Adenoid Cystic Carcinoma Malignant Tumors of the Salivary Glands. J. Clin. Oncol. 2007, 25, 3978–3984. [Google Scholar] [CrossRef] [PubMed]
- Dillon, P.M.; Petroni, G.R.; Horton, B.J.; Moskaluk, C.A.; Fracasso, P.M.; Douvas, M.G.; Varhegyi, N.; Zaja-Milatovic, S.; Thomas, C.Y. A Phase II Study of Dovitinib in Patients with Recurrent or Metastatic Adenoid Cystic Carcinoma. Clin. Cancer Res. 2017, 23, 4138–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locati, L.D.; Galbiati, D.; Calareso, G.; Alfieri, S.; Singer, S.; Cavalieri, S.; Bergamini, C.; Bossi, P.; Orlandi, E.; Resteghini, C.; et al. Patients with adenoid cystic carcinomas of the salivary glands treated with lenvatinib: Activity and quality of life. Cancer 2020, 126, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Tchekmedyian, V.; Sherman, E.J.; Dunn, L.; Tran, C.; Baxi, S.; Katabi, N.; Antonescu, C.R.; Ostrovnaya, I.; Haque, S.S.; Pfister, D.G.; et al. Phase II Study of Lenvatinib in Patients With Progressive, Recurrent or Metastatic Adenoid Cystic Carcinoma. J. Clin. Oncol. 2019, 37, 1529–1537. [Google Scholar] [CrossRef]
- Pfister, D.G.; Spencer, S.; Adelstein, D.; Adkins, D.; Anzai, Y.; Brizel, D.M.; Bruce, J.Y.; Busse, P.M.; Caudell, J.J.; Cmelak, A.J.; et al. Head and Neck Cancers, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2020, 18, 873–898. [Google Scholar] [CrossRef]
- Ho, A.L.; Sherman, E.J.; Fury, M.G.; Baxi, S.S.; Haque, S.; Sima, C.S.; Antonescu, C.R.; Katabi, N.; Pfister, D.G. Phase II study of axitinib in patients with progressive, recurrent/metastatic adenoid cystic carcinoma. J. Clin. Oncol. 2014, 32, 6093. [Google Scholar] [CrossRef]
- Ho, A.L.; Sherman, E.J.; Baxi, S.S.; Haque, S.; Ni, A.; Antonescu, C.R.; Katabi, N.; Morris, L.G.; Chan, T.A.-T.; Pfister, D.G. Phase II study of regorafenib in progressive, recurrent/metastatic adenoid cystic carcinoma. J. Clin. Oncol. 2016, 34, 6096. [Google Scholar] [CrossRef]
- Thomson, D.J.; Silva, P.; Denton, K.; Bonington, S.; Mak, S.K.; Swindell, R.; Homer, J.; Sykes, A.J.; Lee, L.W.; Yap, B.K.; et al. Phase II trial of sorafenib in advanced salivary adenoid cystic carcinoma of the head and neck. Head Neck 2015, 37, 182–187. [Google Scholar] [CrossRef]
- Guigay, J.; Fayette, J.; Even, C.; Cupissol, D.; Rolland, F.; Peyrade, F.; Laguerre, B.; Le Tourneau, C.; Zanetta, S.; Le Moal, L.B.; et al. PACSA: Phase II study of pazopanib in patients with progressive recurrent or metastatic (R/M) salivary gland carcinoma (SGC). J. Clin. Oncol. 2016, 34, 6086. [Google Scholar] [CrossRef]
- Even, C.; Lassen, U.; Merchan, J.; Le Tourneau, C.; Soria, J.C.; Ferte, C.; Ricci, F.; Diener, J.T.; Yuen, E.; Smith, C.; et al. Safety and clinical activity of the Notch inhibitor, crenigacestat (LY3039478), in an open-label phase I trial expansion cohort of advanced or metastatic adenoid cystic carcinoma. Investig. New Drugs 2020, 38, 402–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrarotto, R.; Eckhardt, G.; Patnaik, A.; LoRusso, P.; Faoro, L.; Heymach, J.; Kapoun, A.; Xu, L.; Munster, P. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann. Oncol. 2018, 29, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Miranda, E.L.; Stathis, A.; Hess, D.; Racca, F.; Quon, D.; Rodon, J.; Gadea, O.S.S.; Garcia, J.M.P.; Nuciforo, P.; Vivancos, A.; et al. Phase 1 study of CB-103, a novel first-in-class inhibitor of the CSL-NICD gene transcription factor complex in human cancers. J. Clin. Oncol. 2021, 39, 3020. [Google Scholar] [CrossRef]
- Ayala Pharmaceuticals, I. A Study of AL101 in Patients with Adenoid Cystic Carcinoma (ACC) Bearing Activating Notch Mutations (ACCURACY). Available online: https://clinicaltrials.gov/ct2/show/NCT03691207b (accessed on 28 May 2023).
- Kim, D.-W.; Oh, D.-Y.; Shin, S.H.; Kang, J.H.; Cho, B.C.; Chung, J.-S.; Kim, H.; Park, K.U.; Kwon, J.H.; Han, J.-Y.; et al. A multicenter phase II study of everolimus in patients with progressive unresectable adenoid cystic carcinoma. BMC Cancer 2014, 14, 795. [Google Scholar] [CrossRef] [Green Version]
- Fayette, J.; Even, C.; Digue, L.; Geoffrois, L.; Rolland, F.; Cupissol, D.; Guigay, J.; Le Tourneau, C.; Dillies, A.-F.; Zanetta, S.; et al. NISCAHN: A phase II, multicenter nonrandomized trial aiming at evaluating nivolumab (N) in two cohorts of patients (pts) with recurrent/metastatic (R/M) salivary gland carcinoma of the head and neck (SGCHN), on behalf of the Unicancer Head & Neck Group. J. Clin. Oncol. 2019, 37, 6083. [Google Scholar] [CrossRef]
- Cohen, R.B.; Delord, J.P.; Doi, T.; Piha-Paul, S.A.; Liu, S.V.; Gilbert, J.; Algazi, A.P.; Damian, S.; Hong, R.L.; Le Tourneau, C.; et al. Pembrolizumab for the treatment of advanced salivary gland carcinoma: Findings of the phase 1b KEYNOTE-028 study. Am. J. Clin. Oncol. 2018, 41, 1083. [Google Scholar] [CrossRef]
- Mahmood, U.; Bang, A.; Chen, Y.-H.; Mak, R.H.; Lorch, J.H.; Hanna, G.J.; Nishino, M.; Manuszak, C.; Thrash, E.M.; Sev- ergnini, M.; et al. A Randomized Phase 2 Study of Pembrolizumab with or Without Radiation in Patients with Recurrent or Metastatic Adenoid Cystic Carcinoma. Int. J. Radiat. Oncol. 2020, 109, 134–144. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, S.; Xue, Q.; Mu, J.; Gao, Y.; Tan, F.; Mao, Y.; Wang, D.; Zhao, J.; Gao, S.; et al. Treatment outcomes of patients with tracheobronchial mucoepidermoid carcinoma compared with those with adenoid cystic carcinoma. Eur. J. Surg. Oncol. EJSO 2020, 46, 1888–1895. [Google Scholar] [CrossRef]
- Chen, Z.; Ni, W.; Li, J.-L.; Lin, S.; Zhou, X.; Sun, Y.; Li, J.W.; Leon, M.E.; Hurtado, M.D.; Zolotukhin, S.; et al. The CRTC1-MAML2 fusion is the major oncogenic driver in mucoepidermoid carcinoma. J. Clin. Investig. 2021, 6, e139497. [Google Scholar] [CrossRef]
- Tonon, G.; Modi, S.; Wu, L.; Kubo, A.; Coxon, A.B.; Komiya, T.; O’Neil, K.; Stover, K.; El-Naggar, A.; Griffin, J.D.; et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat. Genet. 2003, 33, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Chen, J.; Gu, Y.; Hu, C.; Li, J.-L.; Lin, S.; Shen, H.; Cao, C.; Gao, R.; Ha, P.K.; et al. Aberrantly activated AREG–EGFR signaling is required for the growth and survival of CRTC1–MAML2 fusion-positive mucoepidermoid carcinoma cells. Oncogene 2014, 33, 3869–3877. [Google Scholar] [CrossRef] [Green Version]
- Canettieri, G.; Coni, S.; Della Guardia, M.; Nocerino, V.; Antonucci, L.; Di Magno, L.; Screaton, R.; Screpanti, I.; Giannini, G.; Gulino, A. The coactivator CRTC1 promotes cell proliferation and transformation via AP-1. Proc. Natl. Acad. Sci. USA 2009, 106, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Amelio, A.L.; Fallahi, M.; Schaub, F.X.; Zhang, M.; Lawani, M.B.; Alperstein, A.S.; Southern, M.R.; Young, B.M.; Wu, L.; Zajac-Kaye, M.; et al. CRTC1/MAML2 gain-of-function interactions with MYC create a gene signature predictive of cancers with CREB-MYC involvement. Proc. Natl. Acad. Sci. USA 2014, 111, E3260–E3268. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, J.; Gao, P.; Nakamura, M.; Cao, Y.; Shen, H.; Griffin, J.D. Transforming activity of MECT1-MAML2 fusion oncoprotein is mediated by constitutive CREB activation. EMBO J. 2005, 24, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, W.; Chen, Z.; Zhou, X.; Yang, R.; Yu, M.; Lu, J.; Kaye, F.J.; Wu, L. Targeting Notch and EGFR signaling in human mucoepidermoid carcinoma. Signal Transduct. Target. Ther. 2021, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; McDermott, J.D.; Schrock, A.B.; Elvin, J.A.; Gay, L.; Karam, S.D.; Raben, D.; Somerset, H.; Ali, S.M.; Ross, J.S.; et al. Comprehensive genomic profiling of salivary mucoepidermoid carcinomas reveals frequent BAP1, PIK3CA, and other actionable genomic alterations. Ann. Oncol. 2017, 28, 748–753. [Google Scholar] [CrossRef]
- Locati, L.; Perrone, F.; Cortelazzi, B.; Bergamini, C.; Bossi, P.; Civelli, E.M.; Morosi, C.; Vullo, S.L.; Imbimbo, M.; Quattrone, P.; et al. A phase II study of sorafenib in recurrent and/or metastatic salivary gland carcinomas: Translational analyses and clinical impact. Eur. J. Cancer 2016, 69, 158–165. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.J.; Lee, J.Y.; Lee, S.-H.; Sun, J.-M.; Park, K.; An, H.J.; Cho, J.Y.; Kang, E.J.; Lee, H.-Y.; et al. Clinical trial of nintedanib in patients with recurrent or metastatic salivary gland cancer of the head and neck: A multicenter phase 2 study (Korean Cancer Study Group HN14-01). Cancer 2017, 123, 1958–1964. [Google Scholar] [CrossRef] [Green Version]
- Gaissert, H.A.; Burns, J. The Compromised Airway: Tumors, Strictures, and Tracheomalacia. Surg. Clin. North. Am. 2010, 90, 1065–1089. [Google Scholar] [CrossRef]
- Urdaneta, A.I.; Yu, J.B.; Wilson, L.D. Population Based Cancer Registry Analysis of Primary Tracheal Carcinoma. Am. J. Clin. Oncol. 2011, 34, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Fan, M.; Sheets, N.C.; Chen, R.C.; Jiang, G.-L.; Marks, L.B. The Use of Radiation Therapy Appears to Improve Outcome in Patients with Malignant Primary Tracheal Tumors: A SEER-Based Analysis. Int. J. Radiat. Oncol. 2012, 84, 464–470. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, G.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.; Cho, B.C.; Castro, G.; Srimuninnimit, V.; Bondarenko, I.; Kubota, K.; Lubiniecki, G.M.; et al. Pembrolizumab (pembro) versus platinum-based chemotherapy (chemo) as first-line therapy for advanced/metastatic NSCLC with a PD-L1 tumor proportion score (TPS) ≥ 1%: Open-label, phase 3 KEYNOTE-042 study. J. Clin. Oncol. 2018, 36, LBA4. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. KEYNOTE-407 Investigators. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Gandara, D.R.; Hammerman, P.S.; Sos, M.L.; Lara, P.N.; Hirsch, F.R. Squamous Cell Lung Cancer: From Tumor Genomics to Cancer Therapeutics. Clin. Cancer Res. 2015, 21, 2236–2243. [Google Scholar] [CrossRef] [Green Version]
- Paik, P.K.; Pillai, R.N.; Lathan, C.S.; Velasco, S.A.; Papadimitrakopoulou, V. New Treatment Options in Advanced Squamous Cell Lung Cancer. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e198–e206. [Google Scholar] [CrossRef]
- Bahleda, R.; Dienstmann, R.; Adamo, B.; Gazzah, A.; Infante, J.R.; Zhong, B.; Platero, S.J.; Smit, H.; Perera, T.; Stuyckens, K.; et al. Phase 1 study of JNJ-42756493, a pan-fibroblast growth factor receptor (FGFR) inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 2501. [Google Scholar] [CrossRef]
- Nogova, L.; Sequist, L.V.; Cassier, P.A.; Hidalgo, M.; Delord, J.-P.; Schuler, M.H.; Lim, W.-T.; Camidge, D.R.; Buettner, R.; Heukamp, L.C.; et al. Targeting FGFR1-amplified lung squamous cell carcinoma with the selective pan-FGFR inhibitor BGJ398. J. Clin. Oncol. 2014, 32, 8034. [Google Scholar] [CrossRef]
- Spoerke, J.M.; O’Brien, C.; Huw, L.; Koeppen, H.; Fridlyand, J.; Brachmann, R.K.; Haverty, P.M.; Pandita, A.; Mohan, S.; Sampath, D.; et al. Phosphoinositide 3-Kinase (PI3K) Pathway Alterations Are Associated with Histologic Subtypes and Are Predictive of Sensitivity to PI3K Inhibitors in Lung Cancer Preclinical Models. Clin. Cancer Res. 2012, 18, 6771–6783. [Google Scholar] [CrossRef] [Green Version]
- Vansteenkiste, J.F.; Canon, J.L.; De Braud, F.; Grossi, F.; De Pas, T.; Gray, J.E.; Su, W.-C.; Felip, E.; Yoshioka, H.; Gridelli, C.; et al. Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: Results from the phase II BASALT-1 study. J. Thorac. Oncol. 2015, 10, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.R.; Fink, A.; et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef] [Green Version]
- Wade, J.L.; Langer, C.J.; Redman, M.; Aggarwal, C.; Bradley, J.D.; Crawford, J.; Miao, J.; Griffin, K.; Herbst, R.S.; Kelly, K.; et al. A phase II study of GDC-0032 (taselisib) for previously treated PI3K positive patients with stage IV squamous cell lung cancer (SqNSCLC): LUNG-MAP sub-study SWOG S1400B. J. Clin. Oncol. 2017, 35, 9054. [Google Scholar] [CrossRef]
- Li, J.; Tan, F.; Wang, Y.; Xue, Q.; Gao, Y.; Mu, J.; Mao, Y.; Zhao, J.; Wang, D.; Feng, X.; et al. Clinical characteristics, surgical treatments, prognosis, and prognostic factors of primary tracheal cancer patients: 20-year data of the National Cancer Center, China. Transl. Lung Cancer Res. 2022, 11, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Genden, E.M.; Laitman, B.M. Human Tracheal Transplantation. Transplantation 2023. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Du, Z.; Fang, Z.; Shi, Y.; Chen, X.; Jin, M.; Liu, K. Survival benefit of radiotherapy and nomogram for patients with primary tracheal malignant tumors: A propensity score-matched SEER database analysis. J. Cancer Res. Clin. Oncol. 2023. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Study | Number of Treated Patients | Subtype | Drug | ORR(%) | mPFS (mo) |
|---|---|---|---|---|---|
| Gilbert et al. [84] | 45 | Salivary gland carcinoma | Paclitaxel | 0 | 4 |
| Pfeffer et al. [85] | 10 | Adenoid cystic carcinoma | Imatinib | 0 | 6 |
| Hotte et al. [86] | 15 | Adenoid cystic carcinoma | Imatinib | 0 | 2.5 |
| Ghosal et al. [87] | 28 | Adenoid cystic carcinoma | Imatinib + Cisplatin | 0 | 15 |
| Wong et al. [88] | 40 | Adenoid cystic carcinoma | Desatinib | 2.5 | 4.8 |
| Chau et al. [89] | 13 | Adenoid cystic carcinoma | Sunitinib | 0 | 7.2 |
| Jakob et al. [90] | 19 | Adenoid cystic carcinoma | Gefitinib | 0 | 4.3 |
| Locati et al. [91] | 30 | Adenoid cystic + non adenoid cystic carcinoma | Cetuximab | 0 | 6 |
| Agulnik et al. [92] | 19 | Adenoid cystic carcinoma | Lapatinib | 0 | 3.5 |
| Dillon et al. [93] | 34 | Adenoid cystic carcinoma | Dovitinib | 0 | 8.2 |
| Locati et al. [94] | 26 | Adenoid cystic carcinoma | Lenvatinib | 0 | 9.1 |
| Tchekmedyian et al. [95] | 32 | Adenoid cystic carcinoma | Lenvatinib | 0 | 17.5 |
| Pfister et al. [96] | 32 | Adenoid cystic carcinoma | Lenvatinib | 15.6 | 17.5 |
| Ho et al. [97] | 33 | Adenoid cystic carcinoma | Axitinib | 0 | 5.7 |
| Ho et al. [98] | 38 | Adenoid cystic carcinoma | Regorafenib | NR | NR |
| Thomson et al. [99] | 23 | Adenoid cystic carcinoma | Sorafenib | 0 | 11.3 |
| Guigay et al. [100] | 46 | Adenoid cystic carcinoma | Pazopanib | 0 | 5.9 |
| Even et al. [101] | 22 | Adenoid cystic carcinoma | NOTCH inhibitor crenigacestat (LY3039478) | NR | 5.3 |
| Ferrarotto et.al. [102] | 48 | Solid tumors (adenoid cystic carcinoma) | Brontictuzumab | NR | 2 |
| Miranda et al. [103] | 41 | Solid tumors and hematological malignancies (adenoid cystic carcinoma) | CB-103 | NR | 5 |
| ACCURACY [104] | 87 | Adenoid cystic carcinoma | AL 101 | 15 | NR |
| Dong-Wan et al. [105] | 34 | Adenoid cystic carcinoma | Everolimus | NR | 11.2 |
| Fayette et al. NISCAHN. [106] | 46 | Salivary gland carcinoma | Nivolumab | 0 | 4.9 |
| KEYNOTE-028 [107] | 26 | Salivary gland carcinoma | Pembrolizumab | 12 | 4 |
| Mahmood et al. [108] | 10 | Adenoid cystic carcinoma | Pembrolizumab | 0 | 4.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchioni, A.; Tonelli, R.; Samarelli, A.V.; Cappiello, G.F.; Andreani, A.; Tabbì, L.; Livrieri, F.; Bosi, A.; Nori, O.; Mattioli, F.; et al. Molecular Biology and Therapeutic Targets of Primitive Tracheal Tumors: Focus on Tumors Derived by Salivary Glands and Squamous Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 11370. https://doi.org/10.3390/ijms241411370
Marchioni A, Tonelli R, Samarelli AV, Cappiello GF, Andreani A, Tabbì L, Livrieri F, Bosi A, Nori O, Mattioli F, et al. Molecular Biology and Therapeutic Targets of Primitive Tracheal Tumors: Focus on Tumors Derived by Salivary Glands and Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(14):11370. https://doi.org/10.3390/ijms241411370
Chicago/Turabian StyleMarchioni, Alessandro, Roberto Tonelli, Anna Valeria Samarelli, Gaia Francesca Cappiello, Alessandro Andreani, Luca Tabbì, Francesco Livrieri, Annamaria Bosi, Ottavia Nori, Francesco Mattioli, and et al. 2023. "Molecular Biology and Therapeutic Targets of Primitive Tracheal Tumors: Focus on Tumors Derived by Salivary Glands and Squamous Cell Carcinoma" International Journal of Molecular Sciences 24, no. 14: 11370. https://doi.org/10.3390/ijms241411370