

Deep Characterization and Comparison of Different Retrovirus-like Particles Preloaded with CRISPR/Cas9 RNPs
 , , and
, , and
Abstract
:1. Introduction
2. Results
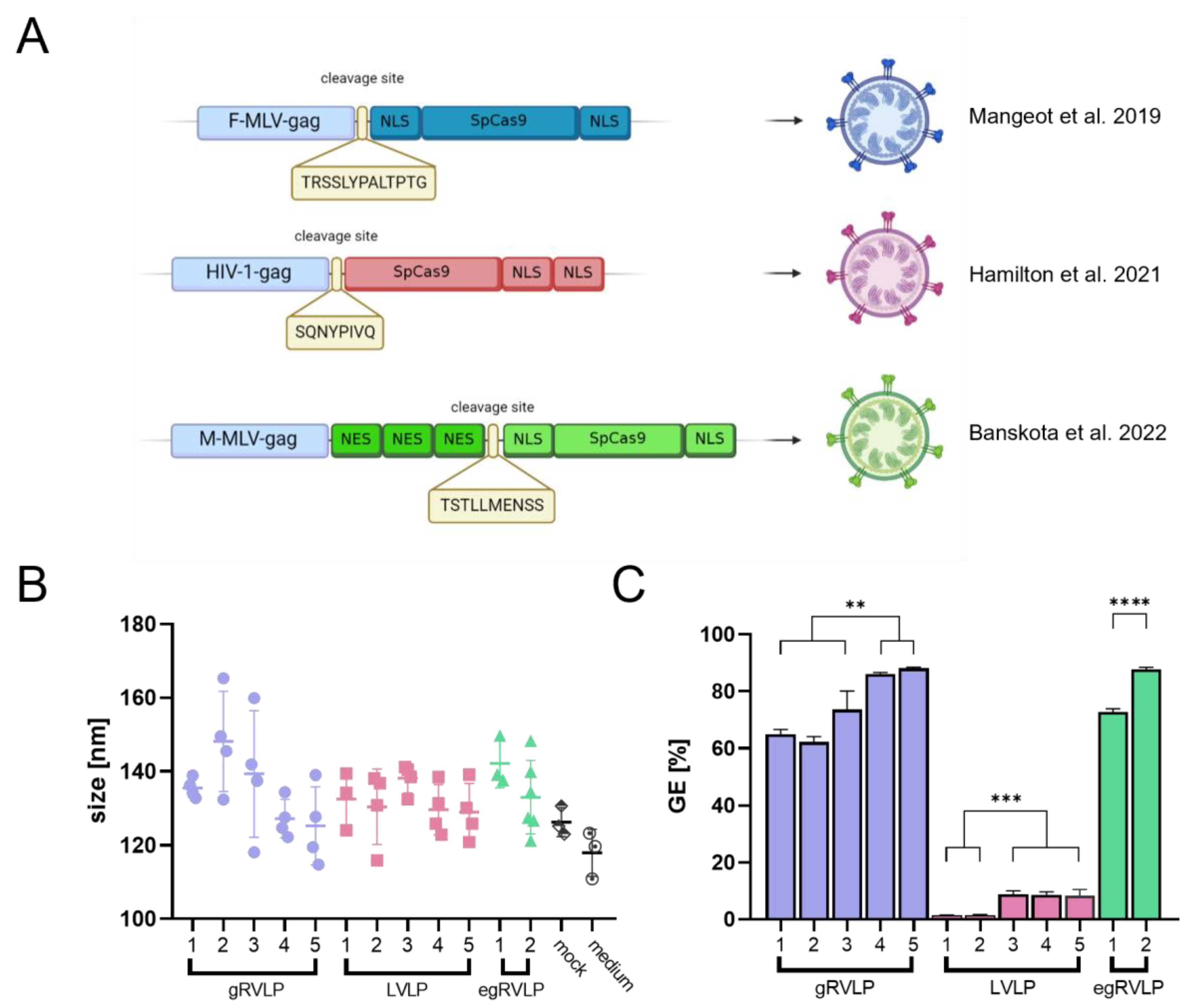
2.1. Establishing Analogous Production Conditions for Three Different Retrovirus-like Particles
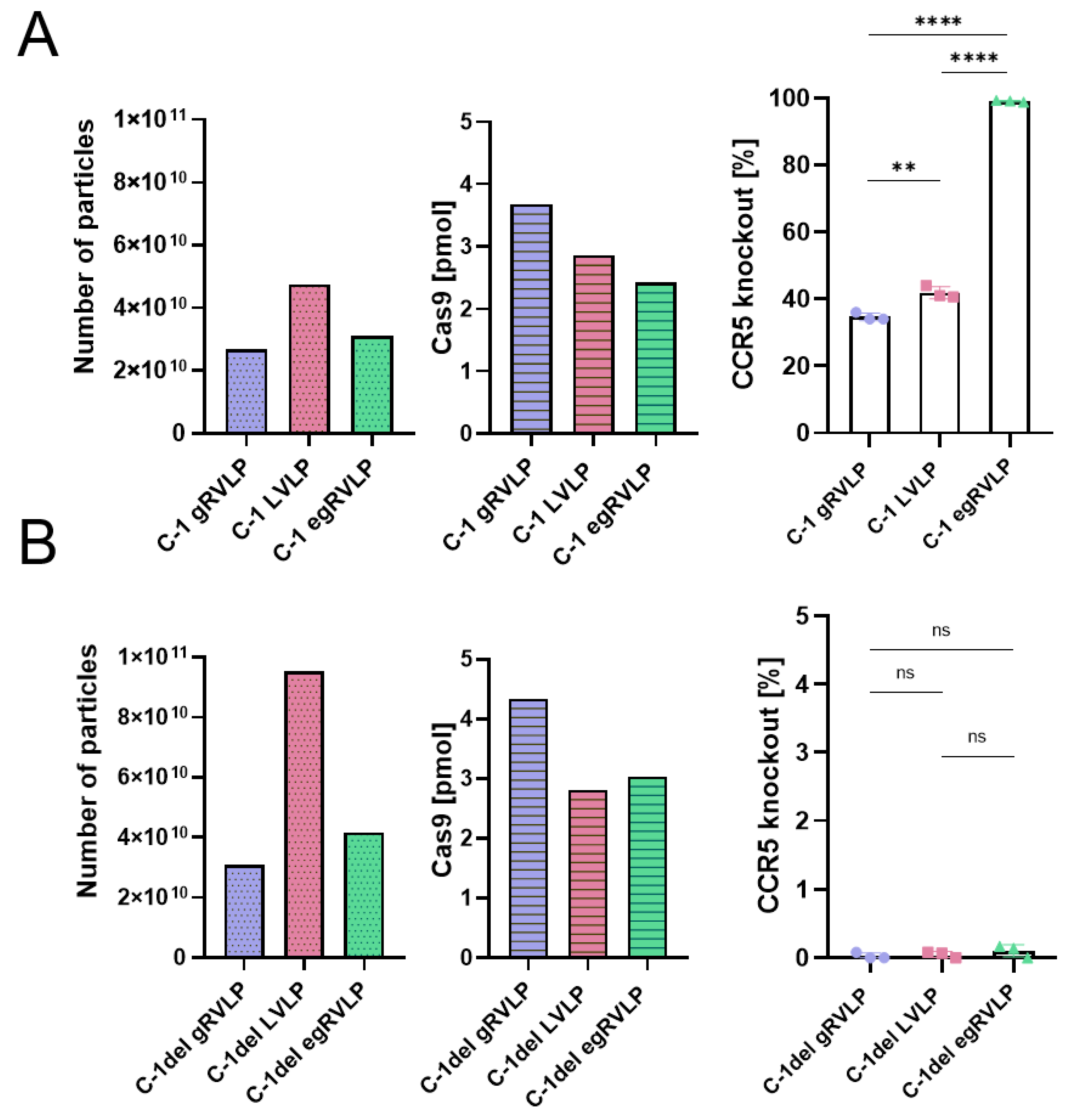
2.2. Analogously Produced Cas9-Preloaded Retrovirus-like Particles (RVLPs) Mediate Different Genome-Editing Efficiencies
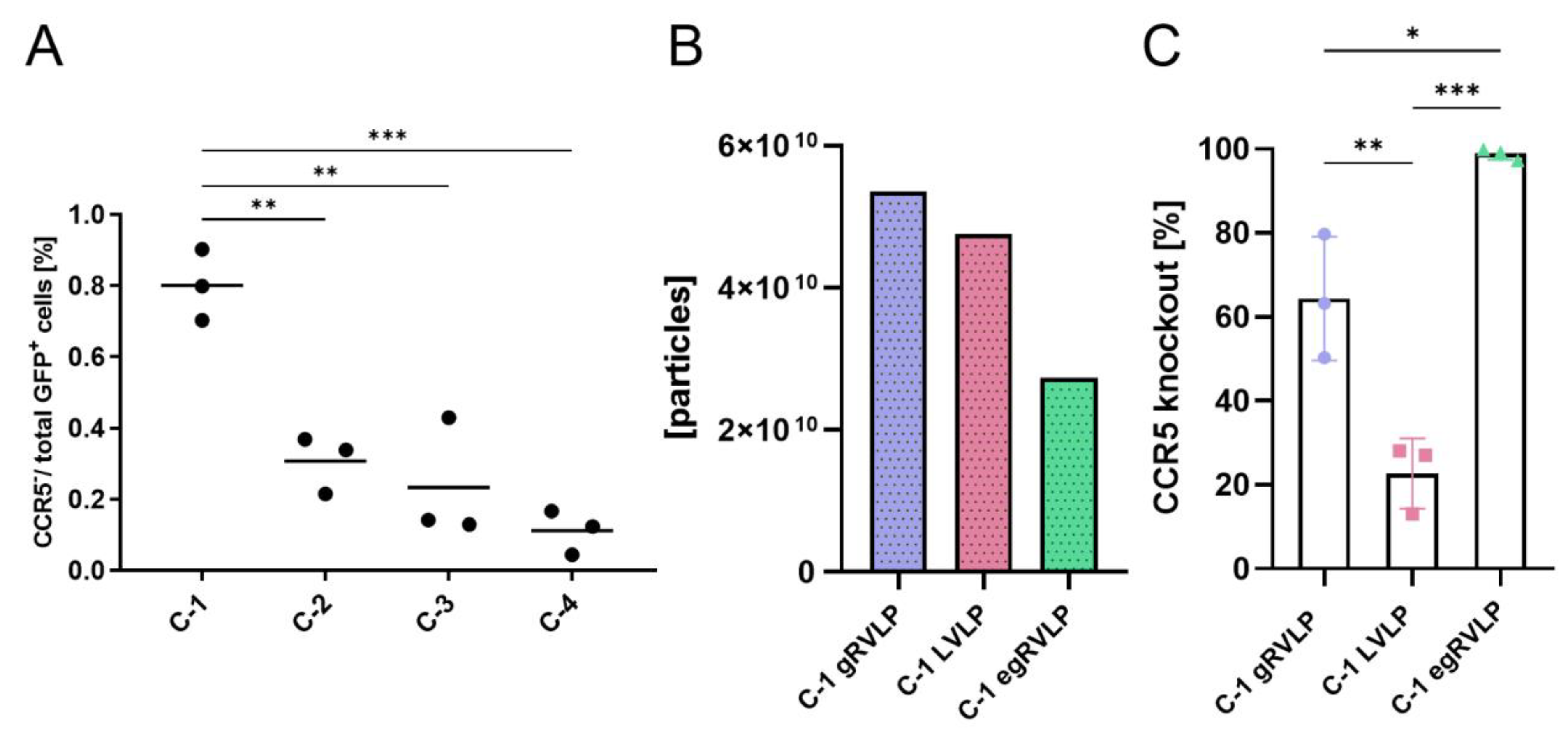
2.3. Efficient Genome Editing of C-C-Motive-Chemokine-Receptor 5 (CCR5) Using RVLPs
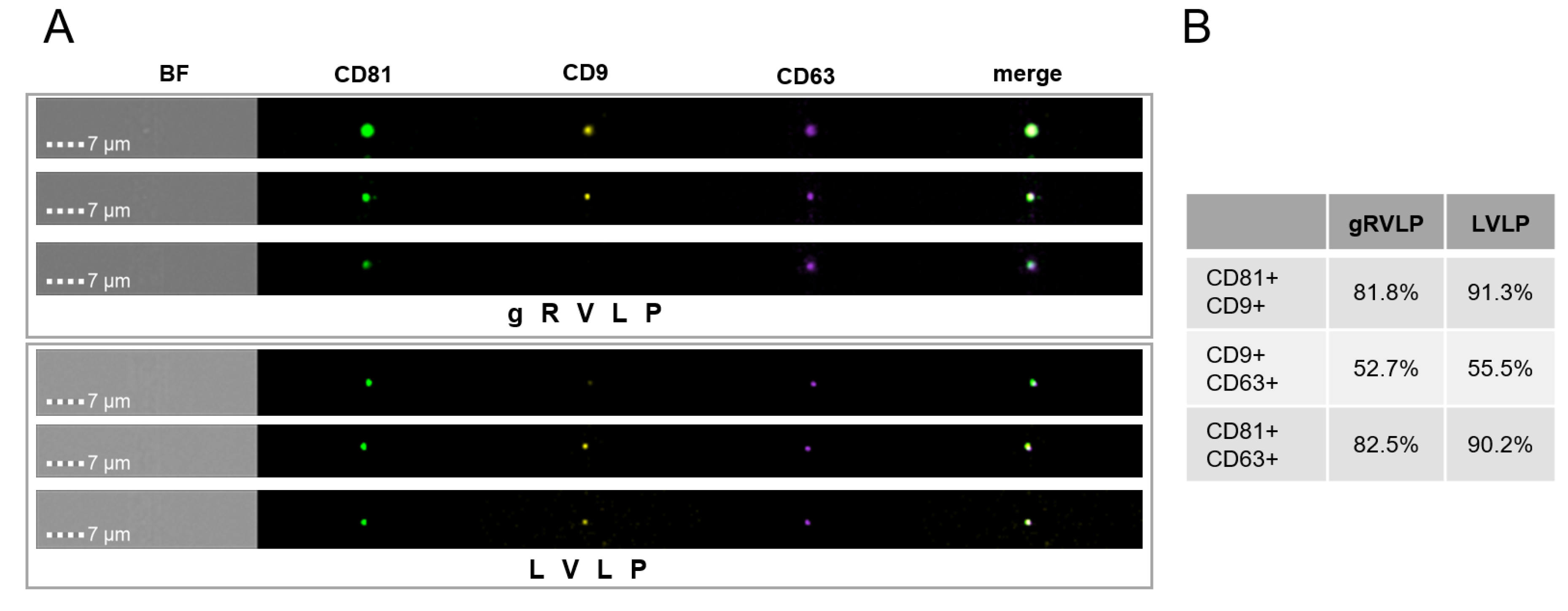
2.4. No Major Impact of RVLP Type on Particle Morphology and Tetraspanin Composition
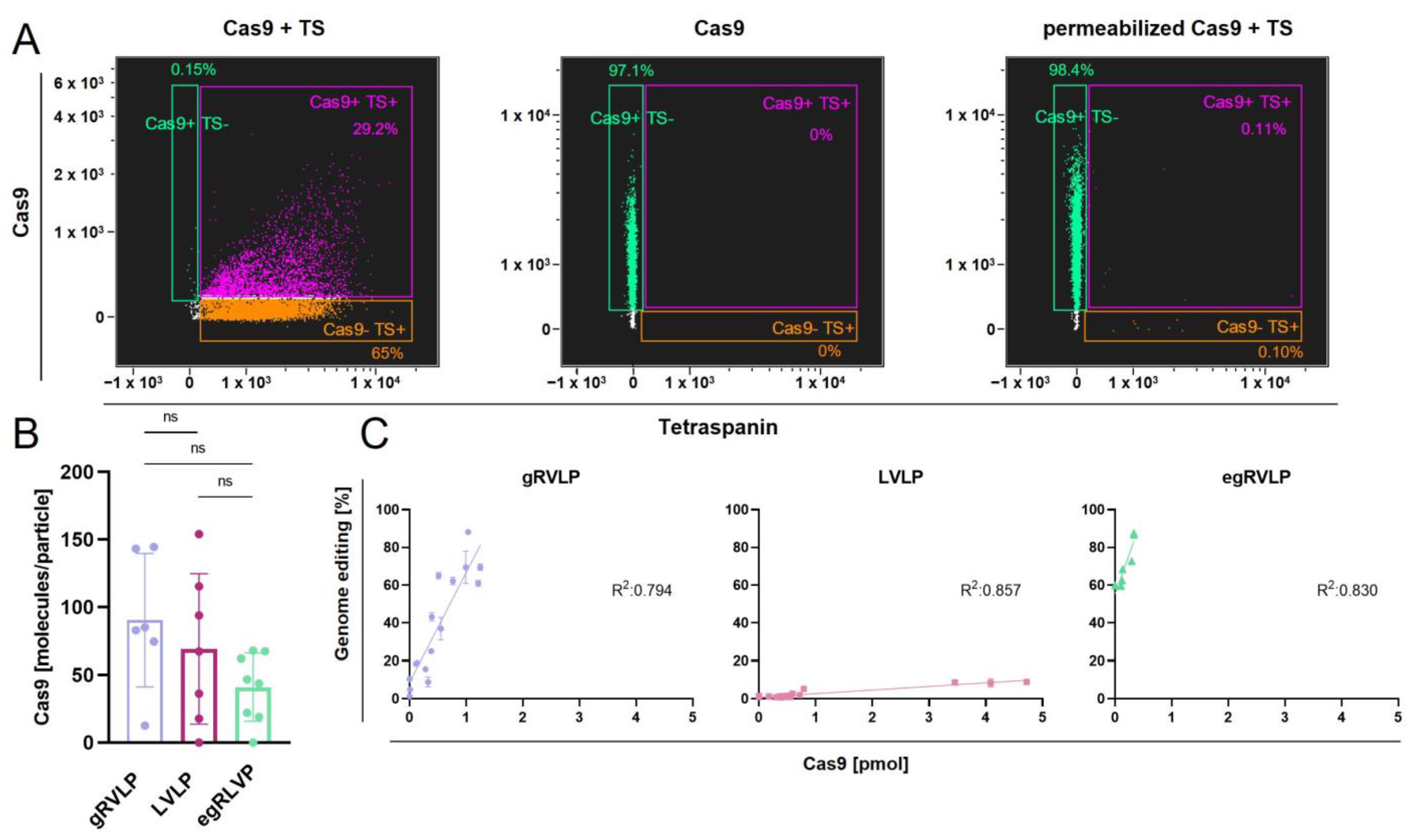
2.5. RVLPs’ Ability to Mediate Genome Editing Correlates with Their Cas9 Content, but Varies between the Tested Retrovirus-like Particles
2.6. Efficient Gene Editing in Human Induced Pluripotent Stem Cells (hiPSC) Using VLPs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Molecular Cloning
4.3. RVLP Production and Cell Transduction
4.4. Flow Cytometry (FC)
4.5. Nanoparticle Tracking Analysis (NTA)
4.6. ELISA
4.7. Imaging Flow Cytometry (IFC)
4.8. Digital PCR (dPCR)
4.9. Quantification and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Bak, R.O.; Gomez-Ospina, N.; Porteus, M.H. Gene Editing on Center Stage. Trends Genet. 2018, 34, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Yarnall, M.T.N.; Ioannidi, E.I.; Schmitt-Ulms, C.; Krajeski, R.N.; Lim, J.; Villiger, L.; Zhou, W.; Jiang, K.; Garushyants, S.K.; Roberts, N.; et al. Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat. Biotechnol. 2022, 41, 500–512. [Google Scholar] [CrossRef]
- Bendixen, L.; Jensen, T.I.; Bak, R.O. CRISPR-Cas-mediated transcriptional modulation: The therapeutic promises of CRISPRa and CRISPRi. Mol. Ther. 2023, 31, 1920–1937. [Google Scholar] [CrossRef]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Cromer, M.K. CRISPR nuclease off-target activity and mitigation strategies. Front. Genome Ed. 2022, 4, 1112956. [Google Scholar] [CrossRef] [PubMed]
- Głów, D.; Meyer, S.; García Roldán, I.; Akingunsade, L.M.; Riecken, K.; Fehse, B. LATE-a novel sensitive cell-based assay for the study of CRISPR/Cas9-related long-term adverse treatment effects. Mol. Ther. Methods Clin. Dev. 2021, 22, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Nabel, G.J. Development of optimized vectors for gene therapy. Proc. Natl. Acad. Sci. USA 1999, 96, 324–326. [Google Scholar] [CrossRef]
- Raguram, A.; Banskota, S.; Liu, D.R. Therapeutic in vivo delivery of gene editing agents. Cell 2022, 185, 2806–2827. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.W. AAV vectors, insertional mutagenesis, and cancer. Mol. Ther. 2007, 15, 1740–1743. [Google Scholar] [CrossRef]
- Stephen, S.L.; Montini, E.; Sivanandam, V.G.; Al-Dhalimy, M.; Kestler, H.A.; Finegold, M.; Grompe, M.; Kochanek, S. Chromosomal integration of adenoviral vector DNA in vivo. J. Virol. 2010, 84, 9987–9994. [Google Scholar] [CrossRef] [Green Version]
- Mashel, T.V.; Tarakanchikova, Y.V.; Muslimov, A.R.; Zyuzin, M.V.; Timin, A.S.; Lepik, K.V.; Fehse, B. Overcoming the delivery problem for therapeutic genome editing: Current status and perspective of non-viral methods. Biomaterials 2020, 258, 120282. [Google Scholar] [CrossRef] [PubMed]
- Luther, D.C.; Lee, Y.W.; Nagaraj, H.; Scaletti, F.; Rotello, V.M. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: Advances and challenges. Expert Opin. Drug Deliv. 2018, 15, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Taha, E.A.; Lee, J.; Hotta, A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J. Control. Release 2022, 342, 345–361. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Wang, M.; Zuris, J.A.; Meng, F.; Rees, H.; Sun, S.; Deng, P.; Han, Y.; Gao, X.; Pouli, D.; Wu, Q.; et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. USA 2016, 113, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- Mock, U.; Riecken, K.; Berdien, B.; Qasim, W.; Chan, E.; Cathomen, T.; Fehse, B. Novel lentiviral vectors with mutated reverse transcriptase for mRNA delivery of TALE nucleases. Sci. Rep. 2014, 4, 6409. [Google Scholar] [CrossRef] [Green Version]
- Segel, M.; Lash, B.; Song, J.; Ladha, A.; Liu, C.C.; Jin, X.; Mekhedov, S.L.; Macrae, R.K.; Koonin, E.V.; Zhang, F. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 2021, 373, 882–889. [Google Scholar] [CrossRef]
- He, J.; Yu, L.; Lin, X.; Liu, X.; Zhang, Y.; Yang, F.; Deng, W. Virus-like Particles as Nanocarriers for Intracellular Delivery of Biomolecules and Compounds. Viruses 2022, 14, 1905. [Google Scholar] [CrossRef]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef]
- Mangeot, P.E.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massouridès, E.; Sohier, T.J.M.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.R.; Tsuchida, C.A.; Nguyen, D.N.; Shy, B.R.; McGarrigle, E.R.; Sandoval Espinoza, C.R.; Carr, D.; Blaeschke, F.; Marson, A.; Doudna, J.A. Targeted delivery of CRISPR-Cas9 and transgenes enables complex immune cell engineering. Cell Rep. 2021, 35, 109207. [Google Scholar] [CrossRef] [PubMed]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 2022, 185, 250–265.e16. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebas, P.; Jadlowsky, J.K.; Shaw, P.A.; Tian, L.; Esparza, E.; Brennan, A.L.; Kim, S.; Naing, S.Y.; Richardson, M.W.; Vogel, A.N.; et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Investig. 2021, 131, e144486. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef]
- Mock, U.; Machowicz, R.; Hauber, I.; Horn, S.; Abramowski, P.; Berdien, B.; Hauber, J.; Fehse, B. mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res. 2015, 43, 5560–5571. [Google Scholar] [CrossRef]
- Schwarze, L.I.; Głów, D.; Sonntag, T.; Uhde, A.; Fehse, B. Optimisation of a TALE nuclease targeting the HIV co-receptor CCR5 for clinical application. Gene Ther. 2021, 28, 588–601. [Google Scholar] [CrossRef]
- Timin, A.S.; Muslimov, A.R.; Lepik, K.V.; Epifanovskaya, O.S.; Shakirova, A.I.; Mock, U.; Riecken, K.; Okilova, M.V.; Sergeev, V.S.; Afanasyev, B.V.; et al. Efficient gene editing via non-viral delivery of CRISPR-Cas9 system using polymeric and hybrid microcarriers. Nanomedicine 2018, 14, 97–108. [Google Scholar] [CrossRef]
- Fehse, B.; Kustikova, O.S.; Bubenheim, M.; Baum, C. Pois(s)on—It’s a question of dose. Gene Ther. 2004, 11, 879–881. [Google Scholar] [CrossRef]
- Koliha, N.; Wiencek, Y.; Heider, U.; Jüngst, C.; Kladt, N.; Krauthäuser, S.; Johnston, I.C.; Bosio, A.; Schauss, A.; Wild, S. A novel multiplex bead-based platform highlights the diversity of extracellular vesicles. J. Extracell. Vesicles 2016, 5, 29975. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.L.; Maire, C.L.; Reimer, R.; Dührsen, L.; Kolbe, K.; Holz, M.; Schneider, E.; Rissiek, A.; Babayan, A.; Hille, C.; et al. Imaging flow cytometry facilitates multiparametric characterization of extracellular vesicles in malignant brain tumours. J. Extracell. Vesicles 2019, 8, 1588555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Guerrero, A.; Abrey Recalde, M.J.; Mangeot, P.E.; Costa, C.; Bernadin, O.; Périan, S.; Fusil, F.; Froment, G.; Martinez-Turtos, A.; Krug, A.; et al. Baboon Envelope Pseudotyped “Nanoblades” Carrying Cas9/gRNA Complexes Allow Efficient Genome Editing in Human T, B, and CD34+ Cells and Knock-in of AAV6-Encoded Donor DNA in CD34+ Cells. Front. Genome Ed. 2021, 3, 604371. [Google Scholar] [CrossRef]
- Jones, S.K., Jr.; Hawkins, J.A.; Johnson, N.V.; Jung, C.; Hu, K.; Rybarski, J.R.; Chen, J.S.; Doudna, J.A.; Press, W.H.; Finkelstein, I.J. Massively parallel kinetic profiling of natural and engineered CRISPR nucleases. Nat. Biotechnol. 2021, 39, 84–93. [Google Scholar] [CrossRef]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarczyk, S.J.; Sitaraman, K.; Young, H.A.; Hughes, S.H.; Chatterjee, D.K. Protein delivery using engineered virus-like particles. Proc. Natl. Acad. Sci. USA 2011, 108, 16998–17003. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.A.; Lytvyn, V.; Deforet, F.; Gilbert, R.; Gaillet, B. Virus-Like Particles Derived from HIV-1 for Delivery of Nuclear Proteins: Improvement of Production and Activity by Protein Engineering. Mol. Biotechnol. 2017, 59, 9–23. [Google Scholar] [CrossRef]
- Yao, X.; Lyu, P.; Yoo, K.; Yadav, M.K.; Singh, R.; Atala, A.; Lu, B. Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. J. Extracell. Vesicles 2021, 10, e12076. [Google Scholar] [CrossRef]
- Lainšček, D.; Kadunc, L.; Keber, M.M.; Bratkovič, I.H.; Romih, R.; Jerala, R. Delivery of an Artificial Transcription Regulator dCas9-VPR by Extracellular Vesicles for Therapeutic Gene Activation. ACS Synth. Biol. 2018, 7, 2715–2725. [Google Scholar] [CrossRef]
- Głów, D.; Maire, C.L.; Schwarze, L.I.; Lamszus, K.; Fehse, B. CRISPR-to-Kill (C2K)-Employing the Bacterial Immune System to Kill Cancer Cells. Cancers 2021, 13, 6306. [Google Scholar] [CrossRef] [PubMed]
- Lyu, P.; Javidi-Parsijani, P.; Atala, A.; Lu, B. Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient ‘hit-and-run’ genome editing. Nucleic Acids Res. 2019, 47, e99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendel, A.; Fine, E.J.; Bao, G.; Porteus, M.H. Quantifying on- and off-target genome editing. Trends Biotechnol. 2015, 33, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Knopp, Y.; Geis, F.K.; Heckl, D.; Horn, S.; Neumann, T.; Kuehle, J.; Meyer, J.; Fehse, B.; Baum, C.; Morgan, M.; et al. Transient Retrovirus-Based CRISPR/Cas9 All-in-One Particles for Efficient, Targeted Gene Knockout. Mol. Ther. Nucleic Acids 2018, 13, 256–274. [Google Scholar] [CrossRef] [Green Version]
- Mock, U.; Hauber, I.; Fehse, B. Digital PCR to assess gene-editing frequencies (GEF-dPCR) mediated by designer nucleases. Nat. Protoc. 2016, 11, 598–615. [Google Scholar] [CrossRef]
- Schambach, A.; Galla, M.; Modlich, U.; Will, E.; Chandra, S.; Reeves, L.; Colbert, M.; Williams, D.A.; von Kalle, C.; Baum, C. Lentiviral vectors pseudotyped with murine ecotropic envelope: Increased biosafety and convenience in preclinical research. Exp. Hematol. 2006, 34, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Bartsch, U.; Stocking, C.; Fehse, B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. 2008, 16, 698–706. [Google Scholar] [CrossRef]
- Schambach, A.; Bohne, J.; Chandra, S.; Will, E.; Margison, G.P.; Williams, D.A.; Baum, C. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol. Ther. 2006, 13, 391–400. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Beyer, W.R.; Westphal, M.; Ostertag, W.; von Laer, D. Oncoretrovirus and lentivirus vectors pseudotyped with lymphocytic choriomeningitis virus glycoprotein: Generation, concentration, and broad host range. J. Virol. 2002, 76, 1488–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Nicola, D.; Riecken, K.; Fehse, B.; Perry, V.H. In-vivo RGB marking and multicolour single-cell tracking in the adult brain. Sci. Rep. 2014, 4, 7520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inglis, H.C.; Danesh, A.; Shah, A.; Lacroix, J.; Spinella, P.C.; Norris, P.J. Techniques to improve detection and analysis of extracellular vesicles using flow cytometry. Cytometry A 2015, 87, 1052–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gRVLP | LVLP | egRVLP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 | 1 | 2 | |
| gRNA [µg] | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 0.7 | 1.5 |
| GagPol [µg] | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 1.2 | 0.9 | 0.6 | 0.3 | 1.5 | 0.5 | 0.9 |
| GagCas9 [µg] | 0.3 | 0.6 | 1 | 2 | 3 | 0.3 | 0.9 | 1.2 | 1.5 | 3 | 0.2 | 3 |
| VSV-G [µg] | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.06 | 0.15 |
| Ratio | 1:3 | 2:3 | 10:9 | 20:9 | 10:3 | 1:4 | 1:1 | 2:1 | 5:1 | 2:1 | 2:5 | 10:3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wichmann, M.; Maire, C.L.; Nuppenau, N.; Habiballa, M.; Uhde, A.; Kolbe, K.; Schröder, T.; Lamszus, K.; Fehse, B.; Głów, D. Deep Characterization and Comparison of Different Retrovirus-like Particles Preloaded with CRISPR/Cas9 RNPs. Int. J. Mol. Sci. 2023, 24, 11399. https://doi.org/10.3390/ijms241411399
Wichmann M, Maire CL, Nuppenau N, Habiballa M, Uhde A, Kolbe K, Schröder T, Lamszus K, Fehse B, Głów D. Deep Characterization and Comparison of Different Retrovirus-like Particles Preloaded with CRISPR/Cas9 RNPs. International Journal of Molecular Sciences. 2023; 24(14):11399. https://doi.org/10.3390/ijms241411399
Chicago/Turabian StyleWichmann, Max, Cecile L. Maire, Niklas Nuppenau, Moataz Habiballa, Almut Uhde, Katharina Kolbe, Tanja Schröder, Katrin Lamszus, Boris Fehse, and Dawid Głów. 2023. "Deep Characterization and Comparison of Different Retrovirus-like Particles Preloaded with CRISPR/Cas9 RNPs" International Journal of Molecular Sciences 24, no. 14: 11399. https://doi.org/10.3390/ijms241411399