
Multi-Target Neural Differentiation (MTND) Therapeutic Cocktail to Suppress Brain Tumor
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
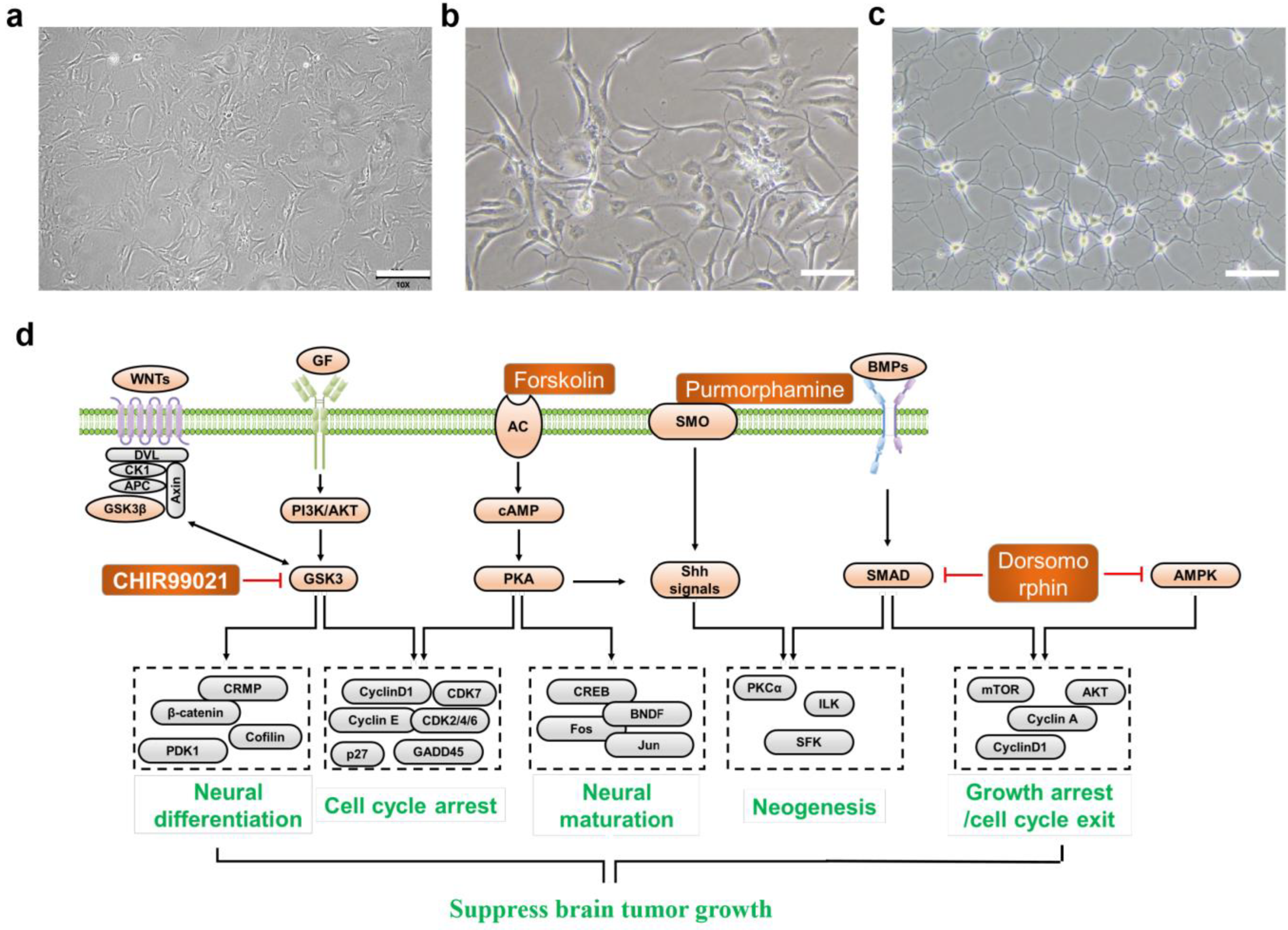
2.1. Design of MTND
2.2. Selection and Mechanism of MTND
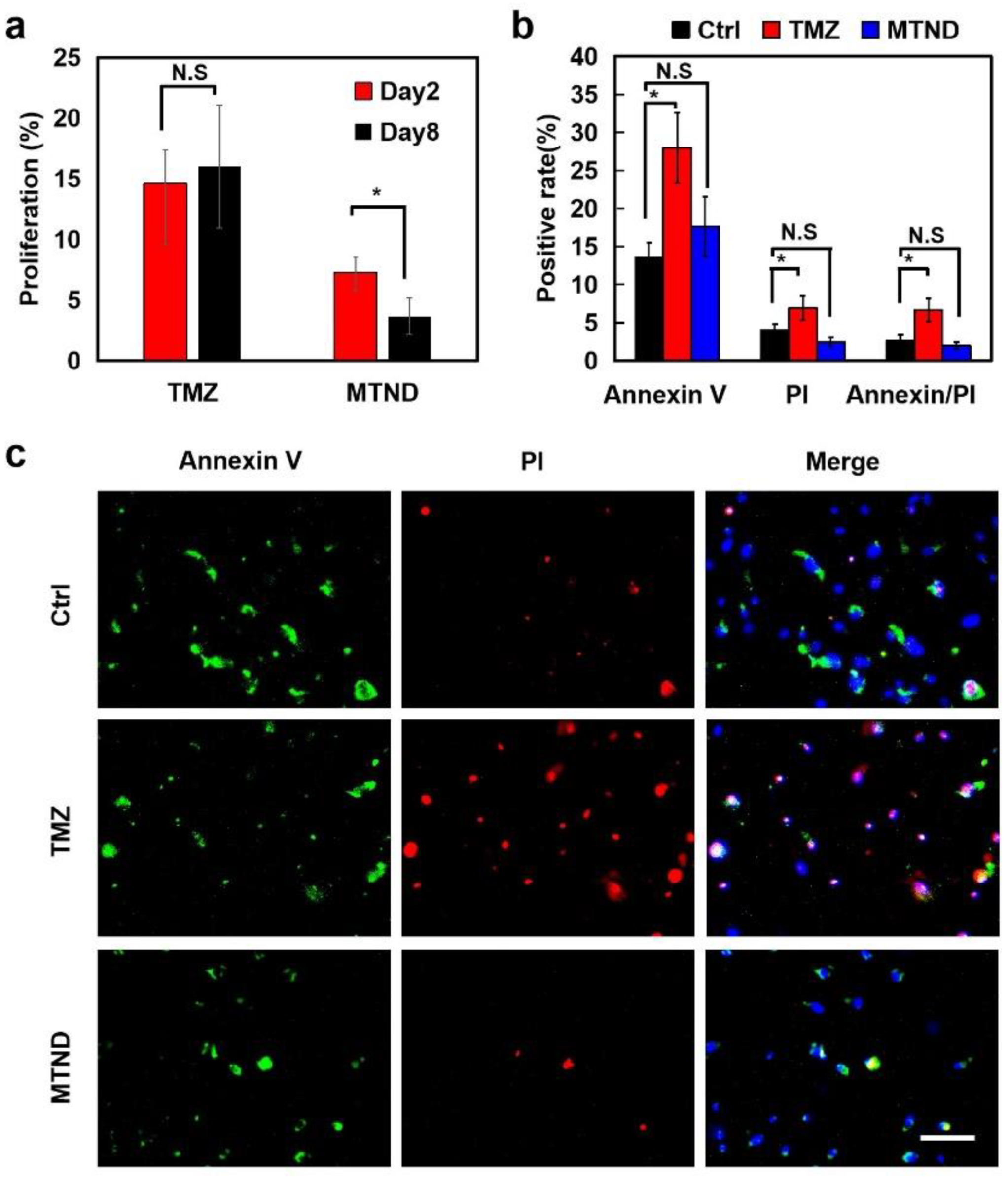
2.3. MTND Showing Enhanced Therapeutic Efficacy in In Vitro and In Vivo Experiments
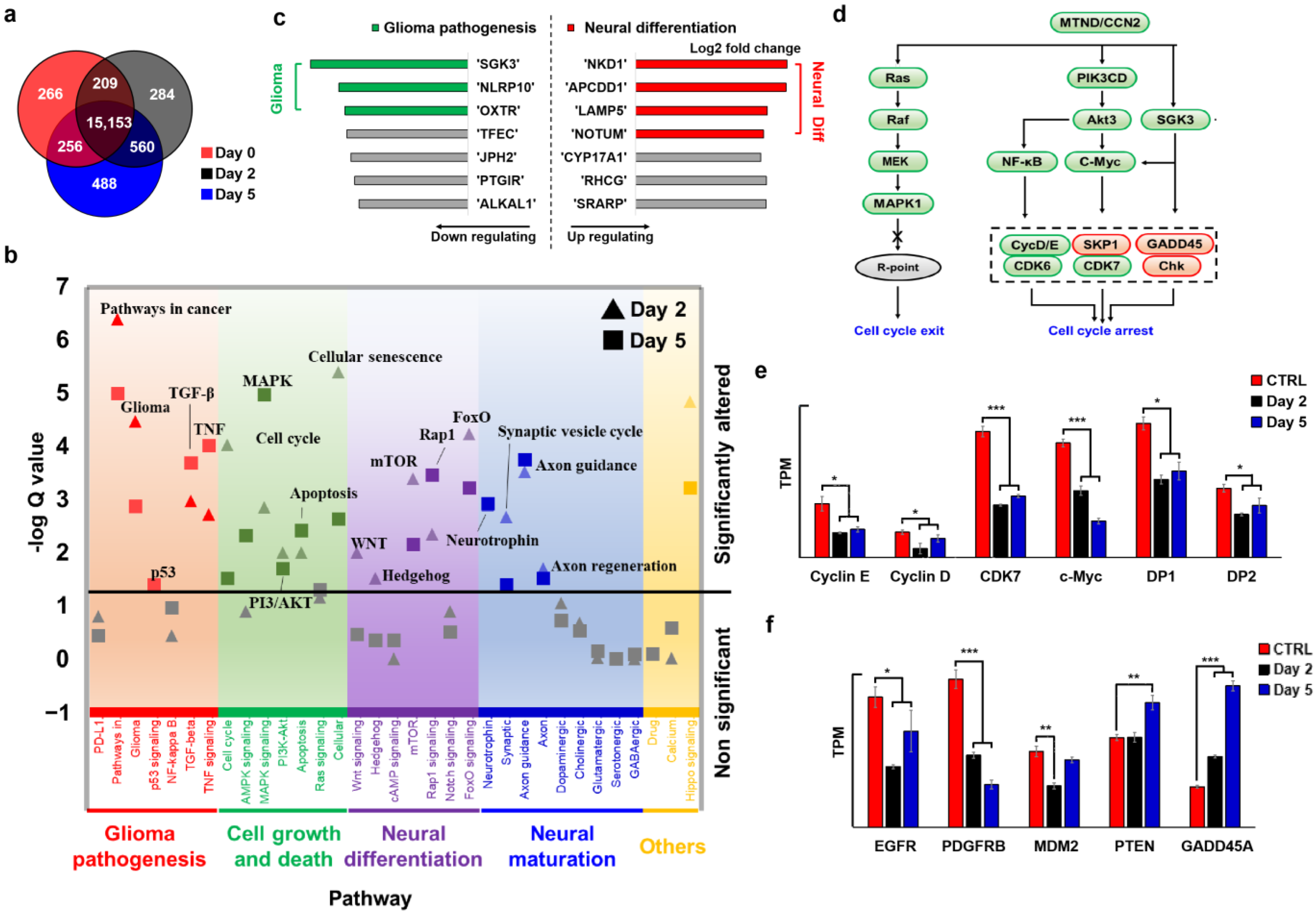
2.4. MTND Induces Multi-Target Therapeutic Genotype Alterations to Achieve Cell Cycle Exit/Arrest
2.5. MTND Facilitates Neural Regeneration
2.6. MTND Induces Low Resistance Development and Lower Toxicity Stress
3. Materials and Methods
3.1. Cell Culture
3.2. MTND Treatment
3.3. Immunofluorescence Staining
3.4. EdU Cell Proliferation Assays
3.5. Cell Migration Assays
3.6. In-Vivo Experiments
3.7. RNA Sequencing and Data Analysis
3.8. Cell Apoptosis Assays
3.9. Information of Clinical Cell Lines
- T-26;
- WHO grade: III;
- Histology: Anaplastic astrocytoma;
- Molecular background: IDH1(-); MGMT gene promoter methylation(+); PTEN, TSC2 deletion; CDK4, CDK6, EGFR, MET, SMO, MDM4, TERT amplification; FGFR3–TACC3, PIK3CAp.K111E, PTENp.I101T, BCORp.A606fs cancer driver genes mutation;
- Prior treatment with radiation: There was no radiotherapy before the tumor was surgically removed.
- T-36;
- WHO grade: IV;
- Histology: Glioblastoma;
- Molecular background: IDH1(-); TERT promoter region mutation; MGMT gene promoter methylation(+); BRCA2, CDKN2A, CDKN2B, PTEN deletion; CDK6, SMO amplification, PIK3CAp.H1047Y, STAG2p.E470 cancer driver gene mutation;
- Prior treatment with radiation: There was no radiotherapy before the tumor was surgically removed.
- T-51;
- WHO grade: I;
- Histology: Meningiomas;
- Molecular background: NF2p.A451fs cancer driver gene mutation;
- Prior treatment with radiation: There was no radiotherapy before the tumor was surgically removed.
- T-59;
- WHO grade: IV;
- Histology: Glioblastoma;
- Molecular background: IDH1(-); TERT promoter region mutation; MGMT gene promoter methylation(+); SMO amplification; TP53p.R280K, BRCA2p.N289H, CARD11p.K83M, NOTCH2p.H107P cancer driver genes mutation;
- Prior treatment with radiation: There was no radiotherapy before the tumor was surgically removed.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahles, T.A.; Saykin, A.J. Candidate mechanisms for chemotherapy-induced cognitive changes. Nat. Rev. Cancer 2007, 7, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Huang, Z.; Zhai, K.; Huang, Q.; Tao, W.; Kim, L.; Wu, Q.; Almasan, A.; Yu, J.S.; Li, X.; et al. Inhibiting DNA-PK induces glioma stem cell differentiation and sensitizes glioblastoma to radiation in mice. Sci. Transl. Med. 2021, 13, abc7275. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Bigner, D.D. Development of novel targeted therapies in the treatment of malignant glioma. Nat. Rev. Drug Discov. 2004, 3, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Reardon, D.A. Neuro-oncology in 2015: Progress in glioma diagnosis, classification and treatment. Nat. Rev. Neurol. 2016, 12, 69–70. [Google Scholar] [CrossRef]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Antiangiogenic therapies for high-grade glioma. Nat. Rev. Neurol. 2009, 5, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, T.Y.-T.; Hou, Y.; Bartom, E.; Lu, X.; Shilatifard, A.; Yue, F.; Saratsis, A. Epigenomic landscape and 3D genome structure in pediatric high-grade glioma. Sci. Adv. 2021, 7, abg4126. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef]
- Sharifzad, F.; Ghavami, S.; Verdi, J.; Mardpour, S.; Sisakht, M.M.; Azizi, Z.; Taghikhani, A.; Łos, M.J.; Fakharian, E.; Ebrahimi, M.; et al. Glioblastoma cancer stem cell biology: Potential theranostic targets. Drug Resist. Updat. 2019, 42, 35–45. [Google Scholar] [CrossRef]
- Bonavita, E.; Pelly, V.S.; Zelenay, S. Resolving the dark side of therapy-driven cancer cell death. J. Exp. Med. 2018, 215, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Yan, V.C.; Butterfield, H.E.; Poral, A.H.; Yan, M.J.; Yang, K.L.; Pham, C.-D.; Muller, F.L. Why Great Mitotic Inhibitors Make Poor Cancer Drugs. Trends Cancer 2020, 6, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Brada, M.J.; van den Bent, M.; Tonn, J.-C.; Pentheroudakis, G.; on behalf of the ESMO Guidelines Working Group. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), iii93–iii101. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Bredel, M.; Zentner, J. Brain-tumour drug resistance: The bare essentials. Lancet Oncol. 2002, 3, 397–406. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; De Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Chung, J.; Lee, K.; Balaj, L.; Min, C.; Carter, B.S.; Hochberg, F.H.; Breakefield, X.O.; Lee, H.; Weissleder, R. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun. 2015, 6, 6999. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Lu, C.; Zhou, P.; Zhao, L.; Lyu, X.; Yin, J.; Shi, Z.; You, Y. EIF4A3-induced circular RNA ASAP1 promotes tumorigenesis and temozolomide resistance of glioblastoma via NRAS/MEK1/ERK1-2 signaling. Neuro Oncol. 2021, 23, 611–624. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiro, S.; Tsugu, H.; Komatsu, F.; Ohmura, T.; Ohta, M.; Sakamoto, S.; Fukushima, T.; Inoue, T. Efficacy of temozolomide treatment in patients with high-grade glioma. Anticancer. Res. 2009, 29, 911–917. [Google Scholar] [PubMed]
- Van Gool, S.W.; Makalowski, J.; Bitar, M.; Van de Vliet, P.; Schirrmacher, V.; Stuecker, W. Synergy between TMZ and individualized multimodal immunotherapy to improve overall survival of IDH1 wild-type MGMT promoter-unmethylated GBM patients. Genes Immun. 2022, 23, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial–mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Liu, Q. Differentiation therapy: A promising strategy for cancer treatment. Chin. J. Cancer 2016, 35, 3. [Google Scholar] [CrossRef] [Green Version]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic Acid and Arsenic Trioxide for Acute Promyelocytic Leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Renaud, J.-P.; Rochel, N.; Ruff, M.; Vivat, V.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal structure of the RAR-γ ligand-binding domain bound to all-trans retinoic acid. Nature 1995, 378, 681–689. [Google Scholar] [CrossRef]
- Stubbins, R.J.; Karsan, A. Differentiation therapy for myeloid malignancies: Beyond cytotoxicity. Blood Cancer J. 2021, 11, 193. [Google Scholar] [CrossRef]
- Waxman, S. Differentiation therapy in acute myelogenous leukemia (non-APL). Leukemia 2000, 14, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Bowen, D.; Kell, J.; Knapper, S.; Morgan, Y.G.; Lok, J.; Grech, A.; Jones, G.; et al. Arsenic trioxide and all-trans retinoic acid treatment for acute promyelocytic leukaemia in all risk groups (AML17): Results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2015, 16, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Avvisati, G.; Cicconi, L.; Thiede, C.; Paoloni, F.; Vignetti, M.; Ferrara, F.; Divona, M.; Albano, F.; Efficace, F.; et al. Improved Outcomes With Retinoic Acid and Arsenic Trioxide Compared With Retinoic Acid and Chemotherapy in Non–High-Risk Acute Promyelocytic Leukemia: Final Results of the Randomized Italian-German APL0406 Trial. J. Clin. Oncol. 2017, 35, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallentin, C.-J.; Nguyen, J.D.; Finkbeiner, P.; Stephenson, C.R.J. Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts. J. Am. Chem. Soc. 2012, 134, 8875–8884. [Google Scholar] [CrossRef]
- Dow, L.E.; O’rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.W.; Brady, J.J.; Han, M.; Cai, H.; Tsai, M.K.; Pierce, S.E.; Cheng, R.; Demeter, J.; Feldser, D.M.; Jackson, P.K.; et al. LKB1 drives stasis and C/EBP-mediated reprogramming to an alveolar type II fate in lung cancer. Nat. Commun. 2022, 13, 1090. [Google Scholar] [CrossRef]
- Storm, E.E.; Durinck, S.; de Sousa E Melo, F.; Tremayne, J.; Kljavin, N.; Tan, C.; Ye, X.; Chiu, C.; Pham, T.; Hongo, J.-A.; et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 2016, 529, 97–100. [Google Scholar] [CrossRef]
- Thiele, C.J.; Reynolds, C.P.; Israel, M.A. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 1985, 313, 404–406. [Google Scholar] [CrossRef]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of High-Risk Neuroblastoma with Intensive Chemotherapy, Radiotherapy, Autologous Bone Marrow Transplantation, and 13-cis-Retinoic Acid. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef]
- Reynolds, C.; Matthay, K.K.; Villablanca, J.G.; Maurer, B.J. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 2003, 197, 185–192. [Google Scholar] [CrossRef]
- Piacentini, M.; Annicchiarico-Petruzzelli, M.; Oliverio, S.; Piredda, L.; Biedler, J.L.; Melino, G. Phenotype-specific “tissue” transglutaminase regulation in human neuroblastoma cells in response to retinoic acid: Correlation with cell death by apoptosis. Int. J. Cancer 1992, 52, 271–278. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastjerdi, F.V.; Zeynali, B.; Tafreshi, A.P.; Shahraz, A.; Chavoshi, M.S.; Najafabadi, I.K.; Vardanjani, M.M.; Atashi, A.; Soleimani, M. Inhibition of GSK-3β enhances neural differentiation in unrestricted somatic stem cells. Cell Biol. Int. 2012, 36, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zuo, X.; Jing, J.; Ma, Y.; Wang, J.; Liu, D.; Zhu, J.; Du, X.; Xiong, L.; Du, Y.; et al. Small-Molecule-Driven Direct Reprogramming of Mouse Fibroblasts into Functional Neurons. Cell Stem Cell 2015, 17, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Zhu, W.; Zhou, Y.; Huang, Y.; Ou, Y.; Li, Y.; Yan, G. Transcriptional and post-transcriptional down-regulation of cyclin D1 contributes to C6 glioma cell differentiation induced by forskolin. J. Cell. Biochem. 2011, 112, 2241–2249. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Li, S.; Li, H.; Yang, C.; Lin, J. The role of Shh signalling pathway in central nervous system development and related diseases. Cell Biochem. Funct. 2021, 39, 180–189. [Google Scholar] [CrossRef]
- Bahrami, N.; Bayat, M.; Mohamadnia, A.; Khakbiz, M.; Yazdankhah, M.; Ai, J.; Ebrahimi-Barough, S. Purmorphamine as a Shh Signaling Activator Small Molecule Promotes Motor Neuron Differentiation of Mesenchymal Stem Cells Cultured on Nanofibrous PCL Scaffold. Mol. Neurobiol. 2017, 54, 5668–5675. [Google Scholar] [CrossRef]
- Torrisi, F.; Alberghina, C.; Furno, D.L.; Zappalà, A.; Valable, S.; Volti, G.L.; Tibullo, D.; Vicario, N.; Parenti, R. Connexin 43 and Sonic Hedgehog Pathway Interplay in Glioblastoma Cell Proliferation and Migration. Biology 2021, 10, 767. [Google Scholar] [CrossRef]
- Li, H.; Hu, Z.; Jiang, H.; Pu, J.; Selli, I.; Qiu, J.; Zhang, B.; Feng, J. TET1 Deficiency Impairs Morphogen-free Differentiation of Human Embryonic Stem Cells to Neuroectoderm. Sci. Rep. 2020, 10, 10343. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK Inhibitor Compound C Is a Potent AMPK-Independent Antiglioma Agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef] [Green Version]
- Pieper, A.A.; McKnight, S.L.; Ready, J.M. P7C3 and an unbiased approach to drug discovery for neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6716–6726. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Greenall, S.A.; Lim, Y.C.; Mitchell, C.B.; Ensbey, K.S.; Stringer, B.W.; Wilding, A.L.; O’Neill, G.M.; McDonald, K.L.; Gough, D.J.; Day, B.W.; et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017, 6, e336. [Google Scholar] [CrossRef]
- Meng, W.; Wang, J.; Wang, B.; Liu, F.; Li, M.; Zhao, Y.; Zhang, C.; Li, Q.; Chen, J.; Zhang, L.; et al. CDK7 inhibition is a novel therapeutic strategy against GBM both in vitro and in vivo. Cancer Manag. Res. 2018, 10, 5747–5758. [Google Scholar] [CrossRef] [Green Version]
- Cantanhede, I.G.; de Oliveira, J.R.M. PDGF Family Expression in Glioblastoma Multiforme: Data Compilation from Ivy Glioblastoma Atlas Project Database. Sci. Rep. 2017, 7, 15271. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Leche, C.A.; Kiyatkin, A.; Yu, Z.; Stayrook, S.E.; Ferguson, K.M.; Lemmon, M.A. Glioblastoma mutations alter EGFR dimer structure to prevent ligand bias. Nature 2022, 602, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Mark. 2018, 33, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermark, B. Platelet-derived growth factor in glioblastoma-driver or biomarker? Upsala J. Med. Sci. 2014, 119, 298–305. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, X.; Xie, J.; Yang, Y.; Qiu, Z.; Lu, W.; Lin, X.; Xu, B. Multi-Target Neural Differentiation (MTND) Therapeutic Cocktail to Suppress Brain Tumor. Int. J. Mol. Sci. 2023, 24, 12329. https://doi.org/10.3390/ijms241512329
Hu X, Xie J, Yang Y, Qiu Z, Lu W, Lin X, Xu B. Multi-Target Neural Differentiation (MTND) Therapeutic Cocktail to Suppress Brain Tumor. International Journal of Molecular Sciences. 2023; 24(15):12329. https://doi.org/10.3390/ijms241512329
Chicago/Turabian StyleHu, Xiaoping, Jingdun Xie, Yilin Yang, Ziyi Qiu, Weicheng Lu, Xudong Lin, and Bingzhe Xu. 2023. "Multi-Target Neural Differentiation (MTND) Therapeutic Cocktail to Suppress Brain Tumor" International Journal of Molecular Sciences 24, no. 15: 12329. https://doi.org/10.3390/ijms241512329