
Forced Hepatic Expression of NRF2 or NQO1 Impedes Hepatocyte Lipid Accumulation in a Lipodystrophy Mouse Model

Abstract
:1. Introduction
2. Results
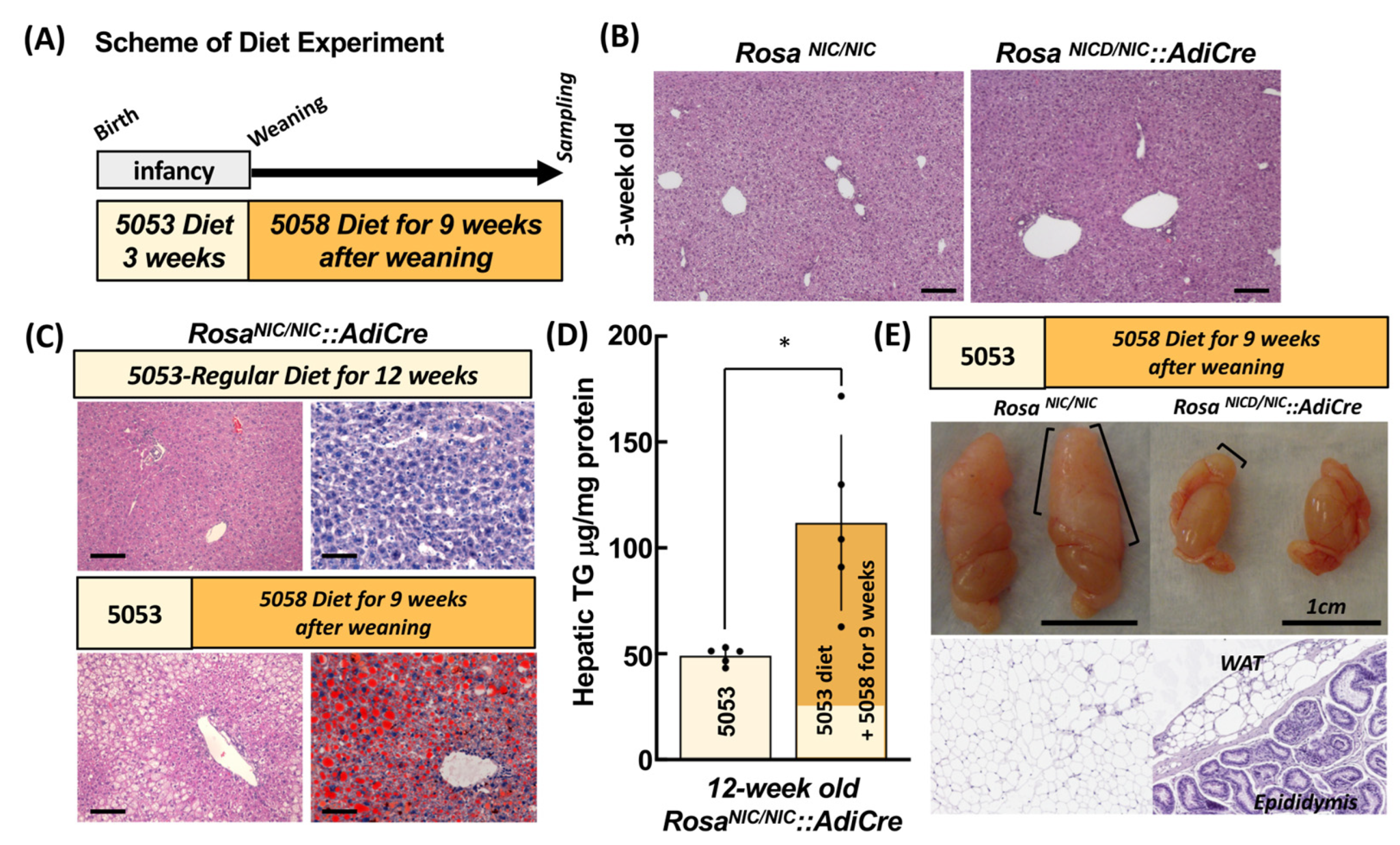
2.1. Characterization of RosaNIC/NIC::AdiCre Mice
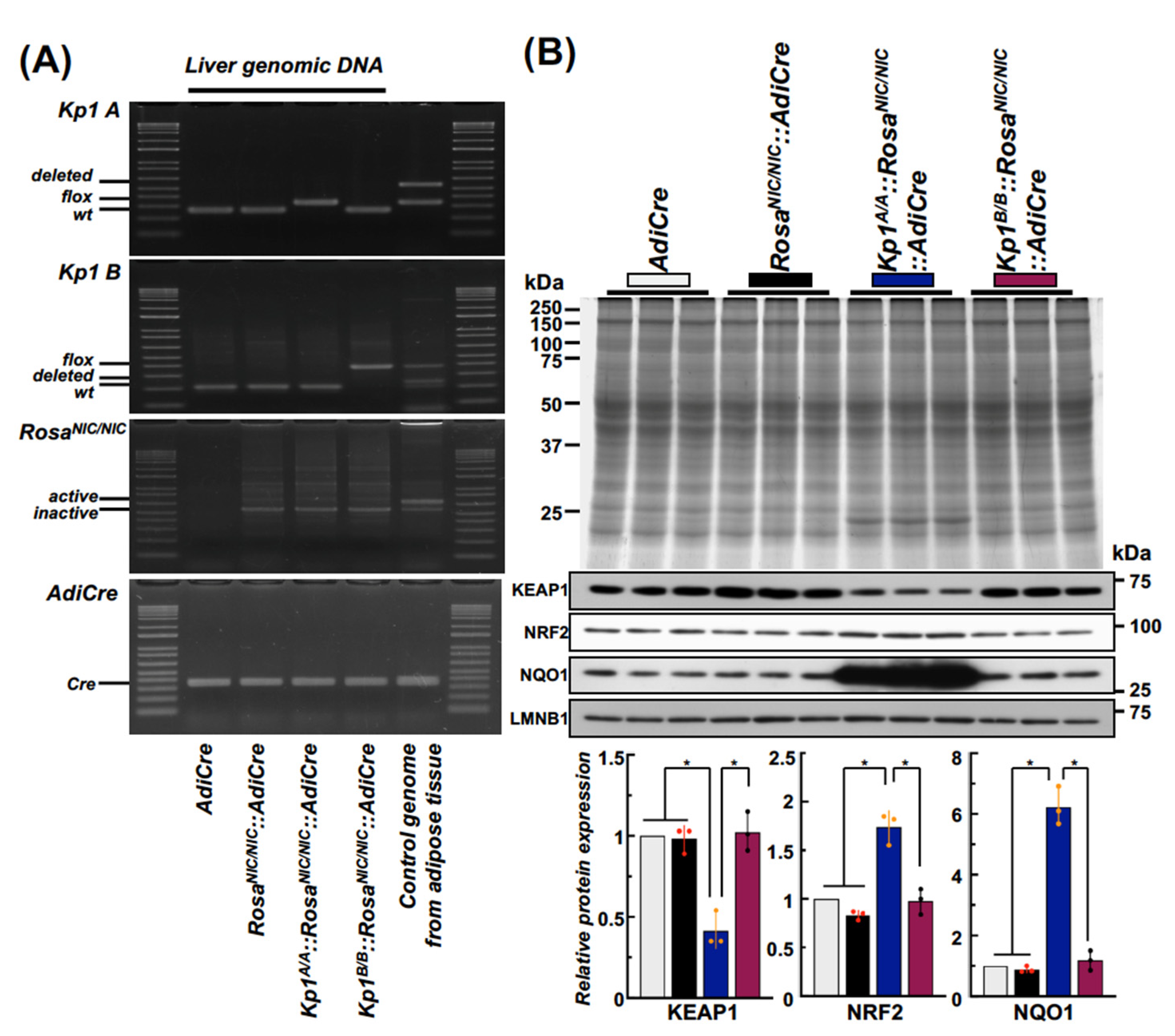
2.2. Establishment of Kp1A/A::RosaNIC/NIC::AdiCre and Kp1B/B::RosaNIC/NIC::AdiCre Mice and the Comparison of Hepatic NRF2 Signaling
2.3. Differential Hepatic Lipid Accumulation among the Genetic Mutant Mice
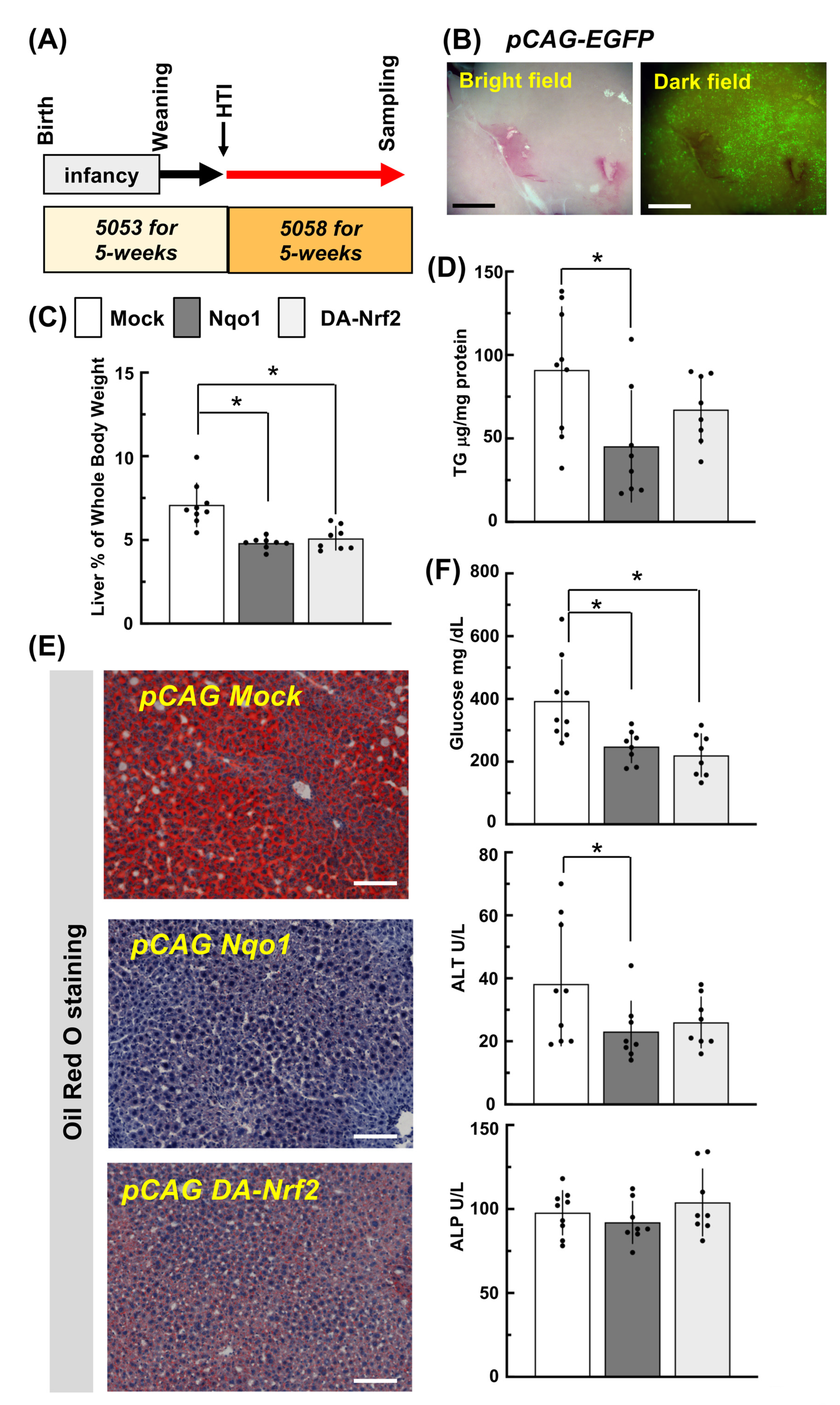
2.4. The Application of DNA-HTI into Mice
2.5. Development of a Dominant Active-Nrf2 Expression Vector
2.6. Forced Expression of Nrf2 and Its Target Gene Nqo1 in Liver by HTI and Its Effects One Week Later on NRF2 Target Gene Expression
2.7. Forced Hepatic Expression of Nrf2 and Its Target Gene Nqo1 of RosaNIC/NIC::AdiCre Lipodystrophy Model Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Blood Collection for Biochemical Analyses and Dissection of Liver
4.3. Triglyceride Assay
4.4. NQO1 Enzyme Assay
4.5. In Vivo Imaging and Stereo-Microscopic Observation of Livers Targeted with Luciferase or EGFP Reporter Genes by Hydrodynamic Tail Vein Injection
4.6. HTI of pCAG DA-Nrf2 and pCAG Nqo1 Vectors
4.7. Mutagenesis for Construction of DA-Nrf2 cDNA
4.8. Construction of the Expression Vectors
4.9. Cell Culture, Transfection and Reporter Gene Assay
4.10. Immune Blotting Analyses
4.11. Histology
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Patni, N.; Garg, A. Congenital generalized lipodystrophies—New insights into metabolic dysfunction. Nat. Rev. Endocrinol. 2015, 11, 522–534. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Khoo, N.K.; Schoiswohl, G.; O’Doherty, R.M.; Kensler, T.W. Notch intracellular domain overexpression in adipocytes confers lipodystrophy in mice. Mol. Metab. 2015, 4, 543–550. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Yagishita, Y.; Fazzari, M.; Palliyaguru, D.L.; Rao, U.N.; Zaravinos, A.; Khoo, N.K.; Schopfer, F.J.; Weiss, K.R.; Michalopoulos, G.K.; et al. Nrf2 prevents Notch-induced insulin resistance and tumorigenesis in mice. JCI Insight 2018, 3, e97735. [Google Scholar] [CrossRef]
- Yagishita, Y.; Chartoumpekis, D.V.; Kensler, T.W.; Wakabayashi, N. NRF2 and the Moirai: Life and Death Decisions on Cell Fates. Antioxid. Redox Signal. 2023, 38, 684–708. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Maher, J.M.; Suzuki, T.; Kawatani, Y.; Motohashi, H.; Yamamoto, M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell. Biol. 2010, 30, 3016–3026. [Google Scholar] [CrossRef]
- Blake, D.J.; Singh, A.; Kombairaju, P.; Malhotra, D.; Mariani, T.J.; Tuder, R.M.; Gabrielson, E.; Biswal, S. Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am. J. Respir. Cell Mol. Biol. 2010, 42, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Waddington, J.C.; Tailor, A.; Lister, A.; Hamlett, J.; Berry, N.; Park, B.K.; Sporn, M.B. CDDO-imidazolide Targets Multiple Amino Acid Residues on the Nrf2 Adaptor, Keap1. J. Med. Chem. 2020, 63, 9965–9976. [Google Scholar] [CrossRef]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef]
- Slocum, S.L.; Skoko, J.J.; Wakabayashi, N.; Aja, S.; Yamamoto, M.; Kensler, T.W.; Chartoumpekis, D.V. Keap1/Nrf2 pathway activation leads to a repressed hepatic gluconeogenic and lipogenic program in mice on a high-fat diet. Arch. Biochem. Biophys. 2016, 591, 57–65. [Google Scholar] [CrossRef]
- Zhou, T.; Kamimura, K.; Zhang, G.; Liu, D. Intracellular gene transfer in rats by tail vein injection of plasmid DNA. AAPS J. 2010, 12, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, A.; Long, D.J., II; Stringer, J.L.; Jaiswal, A.K. In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J. Biol. Chem. 2001, 276, 22559–22564. [Google Scholar] [CrossRef]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Higuchi, N.; Nishikawa, Y.; Kameda, S.; Iino, N.; Kazama, J.J.; Takahashi, N.; Sugawa, M.; Hanawa, H.; Tada, N.; et al. High-level expression of naked DNA delivered to rat liver via tail vein injection. J. Gene Med. 2002, 4, 333–341. [Google Scholar] [CrossRef]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Tong, K.I.; Kobayashi, A.; Yamamoto, M.; Yokoyama, S. Structural insights into the similar modes of Nrf2 transcription factor recognition by the cytoplasmic repressor Keap1. J. Synchrotron Radiat. 2008, 15, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Zhang, S.; Chellian, J.; Mohd, Z.; Epemolu, O.; Dinkova-Kostova, A.T.; Balijepalli, M.K.; Pichika, M.R. Swietenine Alleviates Nonalcoholic Fatty Liver Disease in Diabetic Mice via Lipogenesis Inhibition and Antioxidant Mechanisms. Antioxidants 2023, 12, 595. [Google Scholar] [CrossRef]
- Akinci, B.; Sahinoz, M.; Oral, E. Lipodystrophy Syndromes: Presentation and Treatment; MDText.com, Inc.: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Fazzari, M.; Khoo, N.K.H.; Schopfer, F.J.; Sipula, I.; Yagishita, Y.; Michalopoulos, G.K.; O’Doherty, R.M.; et al. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E180–E195. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Murakami, S.; Biswal, S.S.; Sakaguchi, S.; Harigae, H.; Yamamoto, M.; Motohashi, H. Systemic Activation of NRF2 Alleviates Lethal Autoimmune Inflammation in Scurfy Mice. Mol. Cell. Biol. 2017, 37, e00063-17. [Google Scholar] [CrossRef]
- Kong, X.; Thimmulappa, R.; Craciun, F.; Harvey, C.; Singh, A.; Kombairaju, P.; Reddy, S.P.; Remick, D.; Biswal, S. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am. J. Respir. Crit. Care Med. 2011, 184, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.; Martina, M.N.; Bandapalle, S.; Racusen, L.C.; Potteti, H.R.; Hamad, A.R.; Reddy, S.P.; Rabb, H. T Lymphocyte-Specific Activation of Nrf2 Protects from AKI. J. Am. Soc. Nephrol. 2015, 26, 2989–3000. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, A.; Choi, Y.; Bernier, M.; Zhang, Y.; Diaz-Ruiz, A.; Aon, M.A.; Kalafut, K.; Ehrlich, M.R.; Murt, K.; Ali, A.; et al. NQO1 protects obese mice through improvements in glucose and lipid metabolism. NPJ Aging Mech. Dis. 2020, 6, 13. [Google Scholar] [CrossRef]
- Suda, T.; Liu, D. Hydrodynamic gene delivery: Its principles and applications. Mol. Ther. 2007, 15, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef]
- Cortes, V.A.; Curtis, D.E.; Sukumaran, S.; Shao, X.; Parameswara, V.; Rashid, S.; Smith, A.R.; Ren, J.; Esser, V.; Hammer, R.E.; et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 2009, 9, 165–176. [Google Scholar] [CrossRef]
- Moitra, J.; Mason, M.M.; Olive, M.; Krylov, D.; Gavrilova, O.; Marcus-Samuels, B.; Feigenbaum, L.; Lee, E.; Aoyama, T.; Eckhaus, M.; et al. Life without white fat: A transgenic mouse. Genes Dev. 1998, 12, 3168–3181. [Google Scholar] [CrossRef]
- Cortes, V.A.; Cautivo, K.M.; Rong, S.; Garg, A.; Horton, J.D.; Agarwal, A.K. Leptin ameliorates insulin resistance and hepatic steatosis in Agpat2-/- lipodystrophic mice independent of hepatocyte leptin receptors. J. Lipid Res. 2014, 55, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox. Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Moscovitz, O.; Tsvetkov, P.; Hazan, N.; Michaelevski, I.; Keisar, H.; Ben-Nissan, G.; Shaul, Y.; Sharon, M. A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol. Cell 2012, 47, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Kang, E.S.; Kim, H.J.; Kim, S.H.; Chun, S.W.; Ahn, C.W.; Cha, B.S.; Nam, M.; Lee, H.C. The C609T variant of NQO1 is associated with carotid artery plaques in patients with type 2 diabetes. Mol. Genet. Metab. 2009, 97, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Mehndiratta, M.; Gupta, S.; Kalra, O.P.; Shukla, R.; Gambhir, J.K. Genetic association of NAD(P)H quinone oxidoreductase (NQO1*2) polymorphism with NQO1 levels and risk of diabetic nephropathy. Biol. Chem. 2016, 397, 725–730. [Google Scholar] [CrossRef]
- Hwang, J.H.; Kim, D.W.; Jo, E.J.; Kim, Y.K.; Jo, Y.S.; Park, J.H.; Yoo, S.K.; Park, M.K.; Kwak, T.H.; Kho, Y.L.; et al. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes 2009, 58, 965–974. [Google Scholar] [CrossRef]
- Choi, W.H.; Ahn, J.; Jung, C.H.; Jang, Y.J.; Ha, T.Y. beta-Lapachone Prevents Diet-Induced Obesity by Increasing Energy Expenditure and Stimulating the Browning of White Adipose Tissue via Downregulation of miR-382 Expression. Diabetes 2016, 65, 2490–2501. [Google Scholar] [CrossRef]
- Eguchi, J.; Wang, X.; Yu, S.; Kershaw, E.E.; Chiu, P.C.; Dushay, J.; Estall, J.L.; Klein, U.; Maratos-Flier, E.; Rosen, E.D. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011, 13, 249–259. [Google Scholar] [CrossRef]
- Murtaugh, L.C.; Stanger, B.Z.; Kwan, K.M.; Melton, D.A. Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 14920–14925. [Google Scholar] [CrossRef] [PubMed]
- Parasuraman, S.; Raveendran, R.; Kesavan, R. Blood sample collection in small laboratory animals. J. Pharmacol. Pharmacother. 2010, 1, 87–93. [Google Scholar] [CrossRef]
- Prochaska, H.J.; Santamaria, A.B. Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: A screening assay for anticarcinogenic enzyme inducers. Anal. Biochem. 1988, 169, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Shin, S.; Slocum, S.L.; Agoston, E.S.; Wakabayashi, J.; Kwak, M.K.; Misra, V.; Biswal, S.; Yamamoto, M.; Kensler, T.W. Regulation of notch1 signaling by nrf2: Implications for tissue regeneration. Sci. Signal. 2010, 3, ra52. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Clarke, P.E.; Martino, P.A.; Deng, P.S.; Yeh, C.H.; Lee, T.D.; Prochaska, H.J.; Talalay, P. Mouse liver NAD(P)H:quinone acceptor oxidoreductase: Protein sequence analysis by tandem mass spectrometry, cDNA cloning, expression in Escherichia coli, and enzyme activity analysis. Protein Sci. 1994, 3, 1296–1304. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Kanagaratham, C.; Jancik, S.; Radzioch, D. Promoter deletion analysis using a dual-luciferase reporter system. Methods Mol. Biol. 2013, 977, 79–93. [Google Scholar] [CrossRef]
- Peach, M.; Marsh, N.; Miskiewicz, E.I.; MacPhee, D.J. Solubilization of proteins: The importance of lysis buffer choice. Methods Mol. Biol. 2015, 1312, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Höger, T.H.; Krohne, G.; Franke, W.W. Amino acid sequence and molecular characterization of murine lamin B as deduced from cDNA clones. Eur. J. Cell Biol. 1988, 47, 283–290. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Utility/Feature |
|---|---|
| AdiCre | Adipose tissue specific Cre expression: research control |
| RosaNIC/NIC | Research control |
| Rosa NIC/NIC::AdiCre | Lipodystrophy model mouse |
| Kp1A/A:: Rosa NIC/NIC::AdiCre | Constitutive hypomorphic Keap1 expression compound mouse |
| Kp1A/A:: Rosa NIC/NIC | Control of Kp1A/A::Rosa NIC/NIC::AdiCre mouse |
| Kp1B/B:: Rosa NIC/NIC::AdiCre | Adipose tissue specific Keap1 depleted compound mouse |
| Kp1B/B:: Rosa NIC/NIC | Control of Kp1B/B:: Rosa NIC/NIC::AdiCre mouse |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wakabayashi, N.; Yagishita, Y.; Joshi, T.; Kensler, T.W. Forced Hepatic Expression of NRF2 or NQO1 Impedes Hepatocyte Lipid Accumulation in a Lipodystrophy Mouse Model. Int. J. Mol. Sci. 2023, 24, 13345. https://doi.org/10.3390/ijms241713345
Wakabayashi N, Yagishita Y, Joshi T, Kensler TW. Forced Hepatic Expression of NRF2 or NQO1 Impedes Hepatocyte Lipid Accumulation in a Lipodystrophy Mouse Model. International Journal of Molecular Sciences. 2023; 24(17):13345. https://doi.org/10.3390/ijms241713345
Chicago/Turabian StyleWakabayashi, Nobunao, Yoko Yagishita, Tanvi Joshi, and Thomas W. Kensler. 2023. "Forced Hepatic Expression of NRF2 or NQO1 Impedes Hepatocyte Lipid Accumulation in a Lipodystrophy Mouse Model" International Journal of Molecular Sciences 24, no. 17: 13345. https://doi.org/10.3390/ijms241713345